

Significance
Appropriate development of inflammation is essential to protect hosts from microbial infections, but inflammation can occasionally overshoot and cause collateral damage in hosts. Such hyperinflammation happens during endotoxemia and sepsis, and is attributed to excessive production of proinflammatory cytokines by host cells, such as macrophages. In this study, we demonstrate an unconventional mechanism by which T cells regulate cytokine production of macrophages. It is our general understanding that fast-acting immune cells (such as macrophages) “instruct” how T cells should behave through the step termed antigen presentation. However, what we show here is that T cells instruct macrophages to down-regulate a key proinflammatory cytokine, TNF, within hours after the initiation of endotoxemia.
Keywords: OPN, CD40L, CD154, simultaneous stimulation, T cell migration
Abstract
Endotoxemia is caused by excessive inflammation, but the immune system has various mechanisms to avoid collateral organ damage in endotoxemia. A handful of reports have shown that innate immune responses are suppressed by the adaptive immune system. However, the molecular mechanism by which adaptive immune cells suppress innate inflammatory responses is not clear. Here, we report that T cells are shown to interact with macrophages at the early stage of enodotoxemia and to prolong survival of mice through controlling TNF and IL-10 levels by macrophage CD40 stimulation. The cross-talk between CD40 and toll-like receptor (TLR4) signaling first mediates IL-1 receptor-associated kinase 1 (IRAK1) nuclear translocation and its binding to the IL-10 gene promoter in macrophages, without interfering with the NFκB pathway. IL-10 is then detected by macrophages in an autocrine fashion to destabilize Tnfa mRNA. To induce IRAK1-mediated IL-10 expression, signals from both CD40 and TLR4 are essential. CD40 signaling induces IRAK1 sumoylation in the presence of TNF receptor-associated factor 2 (TRAF2) and intracellular isoform of osteopontin (iOPN) whereas TLR4 signaling provides IFN regulatory factor 5 (IRF5) as a chaperone for sumoylated IRAK1 nuclear translocation. Interaction of T cells with macrophages was observed in the spleen in vivo after endotoxemia induction with LPS injection. Our study demonstrates a mechanistic basis for the immunosuppressive role of macrophage CD40 in LPS endotoxemia.
When acute infections occur, hosts will respond to invading microbes by recruiting immune cells and allowing these cells to produce copious proinflammatory cytokines, such as TNF, to eliminate the microbes from the hosts. This acute inflammatory response needs to be well-regulated because excessive inflammation is harmful for the host itself. To prevent hyperinflammation, innate immune cells are equipped with various inhibitory mechanisms. The majority of inhibitory mechanisms that are currently understood are innate cell-intrinsic., i.e., they involve inhibition by epigenetics (1) and intracellular inhibitory proteins, such as TRAF family member-associated NFκB activator (TANK) (2), A20 (3, 4), and proteins involved in autophagy (5, 6). On the other hand, a handful of reports have shown that innate immune responses are suppressed by the adaptive immune system (7–9). For example, T cells suppress macrophage IL-1β expression by down-regulating nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activity by cell–cell interaction (8). T cells protect mice from poly-I:C–induced endotoxemia by suppressing production of proinflammatory cytokines (7). Another study showed a role for type I IFN–activated B cells in protective early innate immune responses during bacterial sepsis (9). However, molecular mechanisms of innate suppression by the adaptive immune system are not clear. In addition, it was elusive whether direct contact between innate and adaptive immune cells actually occurs in vivo.
In this study, we demonstrated the in vivo T cell–macrophage interaction at the early stage of endotoxemia. Macrophage CD40 ligation by CD40L (CD154) on T cells is shown to down-regulate excessive TNF production by LPS-stimulated macrophages and to control LPS-induced endotoxemia. The mechanism does not interfere with the NFκB pathway but is achieved by an unconventional mechanism by which IRAK1 works as a transcription factor of the Il10 gene in the presence of the intracellular isoform of osteopontin (iOPN).
Results
T Cells Have a Protective Role in LPS Endotoxemia by Down-Regulating TNF.
Rag2−/− mice, which lack T and B cells, were significantly susceptible to LPS-induced endotoxemia compared with wild-type (WT) mice (Fig. 1A). Rag2−/− mice had higher serum levels of IL-10 but were lower in serum TNF levels compared with WT mice at 17 h after LPS infection (Fig. 1B). We took a time course of serum IL-10 levels, which had the peak at 3 h after LPS injection for both WT and Rag2−/− mice, but Rag2−/− mice failed to increase IL-10 levels as WT did at the peak time and beyond (Fig. S1A). Serum levels of TNF, a main causal factor of sepsis and endotoxemia (10–15), had the peak at 1 h after LPS treatment to the same levels in WT and Rag2−/− mice (Fig. S1A). TNF levels quickly went down after the peak, but Rag2−/− mice did not reduce TNF serum levels as much as WT mice did and had twice more TNF serum concentration at 12 h and beyond (Fig. S1A and Fig. 1B). Thus, levels of TNF had an inverse correlation with those of IL-10 at 3h and beyond (Fig. 1B and Fig. S1A). Other major proinflammatory cytokines and chemokines, such as IL-6 and chemokine (C–X–C motif) ligand 1 (CXCL1), did not show difference between Rag2−/− and WT mice (Fig. S1B), ruling out the general alteration in expressing cytokines and chemokines in Rag2−/− mice. Similar to the protein levels, levels of Tnfa and Il10 mRNA, but not Il6 and Il12p40 mRNA, were significantly different between Rag2−/− and WT mice in splenic CD11b+ cells (Fig. S1C), suggesting that the presence of T or B cells decreases TNF and increases IL-10 at the stage before translation.
Fig. 1.

CD4+ T cells protect mice from LPS endotoxemia and suppress macrophage TNF production via interaction with macrophages. (A) Endotoxemia induced by i.p. LPS (40 mg/kg mouse weight) injection in WT mice (n = 10), Rag2−/− (n = 5). (B) Serum levels of IL-10 and TNF in mice at 17 h after LPS injection. (C) LPS endotoxemia in T-cell–reconstituted Rag2−/− mice. Data were obtained from Rag2−/− (n = 8), T-cell–reconstituted Rag2−/− (n = 6), and CD4+ T-cell–reconstituted Rag2−/− mice (n = 7). (D) Macrophages were cultured in the medium containing LPS (100 ng/mL) for 24 h. CD4+ T cells were added either directly or separately with a transwell (TW) insert as indicated. (E) Localization of CD4+ T cells (red) and red-pulp macrophages (RPM) (F4/80) (green) in the spleen isolated from naive mice and LPS-treated mice at indicated time points. Staining to localize CD4+ T cells and RPM is shown with red and green, respectively. (Scale bar, 25 μm.) (F) Localization of CD4+ T cells (red) and subsets of macrophages (green); RPM (F4/80), marginal metallophilic macrophages (MMM) (MOMA-1), or marginal zone macrophages (MZM) (SIGN-R1) in the spleen from mice 3 h after LPS treatment. (Scale bar, 15 μm.) All of the experiments are representatives from at least two similar experiments for each. *P < 0.05.
Reconstitution of total T cells from naive WT mice successfully improved survival of the Rag2−/− recipients against endotoxemia (Fig. 1C), suggesting that T cells protected Rag2−/− recipients. A similar protection effect against endotoxemia was observed with reconstitution of either CD4+ T cells or CD8+ T cells in Rag2−/− recipients (Fig. 1C and Fig. S1D).
T Cell–Macrophage Direct Contact Reduces TNF-Mediated Inflammation.
To ask whether T cell–macrophage contact is necessary for altering TNF and IL-10 levels, we first carried out ex vivo coculture of T cells and macrophages in the presence of LPS with or without transwells. Addition of CD4+ T cells to the macrophage culture in the presence of LPS reduced changed TNF and IL-10 production without transwells, but the changes were abrogated by separating T cells from macrophages by transwells (Fig. 1D). Again, the change in cytokine expression was not universal because production of IL-6 and CXCL1 levels was not altered (Fig. S1E). The data suggested that direct contact between CD4+ T cells and macrophages is required for the T-cell–mediated function.
Next, we asked whether T cell–macrophage interaction actually occurs in LPS-injected mice by harvesting spleens at various time points and analyzing spleen sections by confocal microscopy. CD4+ T cells and macrophages were separately localized in the spleen of naive mice until 1 h after LPS treatment (Fig. 1E and Fig. S1F). However, 3 h after LPS injection, CD4+ T cells moved into the region where red-pulp macrophages (RPMs) were abundant (Fig. 1E and Fig. S1F) and interacted with RPMs (Fig. 1F and Movie S1). Real-time movement of T cells toward macrophages was also observed with two-photon microscopy (Movie S2). In addition to RPMs, CD4+ T cells seemed to interact with marginal metallophilic macrophages (MMMs), but not with marginal zone macrophages (MZMs)(Fig. 1F and Fig. S1G). CD8+ T cells, but not B cells, also showed localized proximity to RPMs after LPS treatment (Fig. S1 H and I). In summary, direct interaction between T cells and macrophages suppressed TNF production by macrophages, and the interaction started to be observed in vivo 3 h after LPS treatment.
CD40 Is Required for the T-Cell–Mediated Attenuation of TNF Production.
Because attenuation of macrophage TNF by T cells occurred without T-cell cognate antigens (Fig. 1D), we focused on non-MHC molecules and selected CD40 on macrophages as a candidate mediator. CD40L expression was identified on CD62L+CD44−CD4+ T cells from LPS-treated mice, as well as in CD62L−CD44+CD4+ T cells, Tregs, and CD8+ T cells from naive and LPS-treated mice (Fig. S2 A and B). However, CD62L+CD44−CD4+ T cells from naive mice did not express significantly higher CD40L compared with an isotype control (Fig. S2 A and B). Indeed, our ex vivo T cell–macrophage coculture experiments showed that CD62L+CD44−CD4+ T cells from naive mice failed to suppress TNF production by macrophages, but various subsets of CD4+ T cells (naive, effector, regulatory) and CD8+ T cells from LPS-treated mice successfully suppressed TNF production (Fig. S2C). CD40L blocking antibody (Ab) successfully abrogated CD4+ T-cell–mediated TNF attenuation (Fig. 2A). When cocultured with Cd40−/− macrophages, CD4+ T cells failed to show TNF attenuation and IL-10 up-regulation (Fig. 2B). Furthermore, CD40 agonistic Ab attenuated LPS-induced production of TNF, but not IL-6, in a dose-dependent manner (Fig. 2C and Fig. S2D). (CD40 Ab used throughout this study was agonistic Ab, not antagonistic Ab.) On the other hand, CD40 Ab enhanced IL-10 production in LPS-stimulated macrophages (Fig. 2D). Next, we tested the protective role of CD40 in endotoxemia. Cd40−/− mice were indeed more susceptible than WT mice in LPS-induced endotoxemia (Fig. 2E), and showed higher and lower expression levels of TNF and IL-10 than WT mice, respectively (Fig. 2F). Furthermore, reconstitution of Cd40l−/− T cells did not protect Rag2−/− recipients against endotoxemia (Fig. 2G). Taken together, CD40L–CD40 interaction between T cells and macrophages seems to protect mice by negatively regulating toll-like receptor (TLR4)-mediated TNF production.
Fig. 2.

CD40 is required for the T-cell–mediated attenuation of TNF production. (A) In the presence of LPS, macrophages were cultured with CD40L blocking Ab or control IgG. (B) WT or Cd40−/− macrophages were cultured with or without CD4+ T cells in the presence of LPS for 24 h. (C) Macrophages were cultured in the medium containing LPS and CD40 agonistic Ab of indicated concentrations for 24 h. (D) Macrophages were treated with LPS in the presence CD40 Ab or control IgG. (E) LPS endotoxemia induced by i.p. LPS injection (40 mg/kg mouse weight) in WT mice (n = 5) and Cd40−/− mice (n = 5). (F) TNF and IL-10 levels in serum from mice 17 h after LPS injection. (G) LPS endotoxemia in Rag2−/− mice with (n = 5) or without (n = 5) Cd40l−/− T-cell reconstitution. All of the experiments here are representative of at least two similar experiments for each. Final concentrations of LPS and Ab/IgG in tissue culture were 100 ng/mL and 10 µg/mL, respectively, unless otherwise noted. *P < 0.05. ns, no significance.
CD40 Signaling Negatively Regulates the Stabilization of Tnfa mRNA Through IL-10 Production.
Next, we tried to clarify how CD40 stimulation inhibited LPS-induced cytokine production. A major pathway responsible for activating cytokine expression by TLR4 is through NFκB. However, stimulating macrophages with CD40 agonistic Ab had no impact on NFκB activity whereas NFκB in B cells responded to CD40 treatment (Fig. S3A). NFκB p65 was not translocated to the nucleus with CD40 stimulation alone in macrophages (Fig. 3A and Fig. S3B). CD40 Ab also failed to alter NFκB activity and nuclear translocation in LPS-stimulated macrophages (Fig. 3A and Fig. S3 A–C). These results suggested that the NFκB pathway is not involved in regulating LPS-mediated cytokine production by CD40 stimulation (Fig. 2 A–C). We therefore sought the possibility of posttranscriptional regulation by evaluating Tnfa mRNA stability. The half-life of Tnfa mRNA in macrophages with LPS alone was 3.7 h, but CD40 stimulation shortened the half-life to 1.5 h (Fig. 3B), suggesting that CD40-mediated TNF attenuation is attributed to the destabilization of Tnfa mRNA.
Fig. 3.
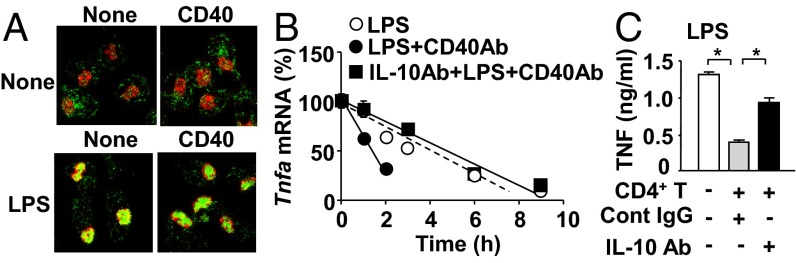
Costimulation of CD40 and TLR4 destabilizes Tnfa mRNA through IL-10 but does not down-regulate NFκB. (A) Confocal microscopic images of NFκB nuclear translocation in macrophages. Nuclear and NFκB staining was shown with red and green, respectively. Shown is representative data from three independent experiments. Quantitative analysis of this experiment is shown in Fig. S3B. (B) Stability of Tnfa mRNA in macrophages. α-Amanitin was added to stop transcription (time = 0). Shown are relative Tnfa mRNA levels based on those at time = 0 as 100%. Cells were pretreated with LPS or CD40 Ab and LPS 12 h before the addition of α-amanitin. IL-10 neutralization Ab was added to an indicated group with LPS plus CD40 Ab. Data are presented as mean ± SEM (n = 4 per group). (C) IL-10 neutralization Ab or isotype control was added to macrophage/CD4+ T-cell coculture. TNF concentrations in the 24-h culture supernatants were determined by ELISA. Data are presented as mean ± SEM (n = 4 per group). Final concentrations of LPS and Ab/IgG in tissue culture were 100 ng/mL and 10 µg/mL, respectively. All of the experiments here are representative of at least two similar experiments. *P < 0.05.
As previously reported that IL-10 receptor signaling destabilizes Tnfa mRNA (16, 17), recombinant (r) IL-10 treatment reduced LPS-induced Tnfa mRNA levels in macrophages (Fig. S4). Neutralization of IL-10 indeed reverted the effect of CD40 treatment in LPS-stimulated macrophages (Fig. 3B). In addition, IL-10 neutralization prevented TNF attenuation by CD4+ T cells (Fig. 3C). In conclusion, CD40-mediated attenuation of TNF production by macrophages is attributed to the destabilization of Tnfa mRNA mediated by IL-10.
Simultaneous Stimulation of TLR4 and CD40 Induces IRAK1 Nuclear Translocation, Which Leads to IL-10 Production in Macrophages.
Here, we sought to understand the molecular mechanism by which CD40 and TLR4 costimulation enhances IL-10 production. Because CD40 stimulation did not alter NFκB activation in macrophages (Fig. 3A and Fig. S3), mechanisms that require NFκB were ruled out. Instead, we looked into IRAK1 because IRAK1 directly binds to the Il10 promoter as a transcription factor (18). Simultaneous stimulation with LPS plus CD40 Ab, but not LPS or CD40 Ab alone, successfully translocated IRAK1 to the nucleus (Fig. 4A). Immunoblotting analysis detected IRAK1 in a nuclear lysate by costimulation of TLR4 and CD40 (Fig. 4B). The size of the nuclear IRAK1 (∼100 kDa) indicated posttranslational modifications (18). Furthermore, binding of IRAK1 to the Il10 promoter in TLR4/CD40-stimulated macrophages was detected by a ChIP assay (Fig. 4C). Irak1 shRNA reduced IL-10 production in macrophages stimulated with LPS and CD40 Ab (Fig. 4D). Taken together, simultaneous stimulation of TLR4 and CD40 induces IRAK1 nuclear translocation to enhance IL-10 production, which leads to down-regulation of TNF expression (Fig. 3 B and C).
Fig. 4.

Costimulation of CD40 and TLR4 induces IRAK1-mediated Il10 gene expression. (A, Left) Representative confocal microscopic images of IRAK1 after LPS, CD40 Ab, or LPS plus CD40 Ab for 60 min. (Right) Fluorescent intensities of IRAK1 in nuclei were calculated relative to the average value of unstimulated cells using the Fiji software. One circle or square denotes a result from one cell. (B) Detection of IRAK1 in nuclear lysates from macrophages harvested 30 min after cell treatment with LPS alone or with LPS plus CD40 Ab. Proliferating cell nuclear antigen (PCNA) and GAPDH are markers of the nuclei and cytosol, respectively. Images came from the identical blot. Order of bands was rearranged from the original film for presentation. (C) ChIP assay for the IRAK1 binding to the Il10 promoter. IgG was used as an isotype control of IRAK1 Ab. (D) IL-10 expression by macrophages stimulated with LPS plus CD40 Ab for 24 h with or without Irak1 shRNA. Data are presented as mean ± SEM (n = 4 per group). (E and F) Evaluating IRAK1 sumoylation (E), and coimmunoprecipitation (co-IP) between IRF5 and IRAK1 (F) in macrophages with indicated stimulation. Hereafter, we used β-actin as the internal input control for some immunoprecipitation (IP) experiments, in which Western detection of molecules targeted for IP was considered to show many bands due to posttranslational modifications. (G) Nuclear translocation of IRF5 and IRAK1 after cell stimulation with LPS, CD40 Ab, or LPS plus CD40 Ab for 60 min. DAPI (blue) for nuclear staining and fluorescent intensities of IRF5 in nucleus. Final concentrations of LPS and Ab/IgG in tissue culture were 100 ng/mL and 10 µg/mL, respectively. (Scale bars in A and G, 5 μm.) *P < 0.05.
CD40 Signaling Induces IRAK1 Sumoylation; and TLR4 Signaling Provides IFN Regulatory Factor 5 as a Chaperone for IRAK1 Nuclear Translocation.
As previously reported (19, 20), we confirmed that IRAK1 was indeed sumoylated—but not with LPS treatment alone (Fig. 4E and Fig. S5). Because CD40 signaling induces IRAK1 sumoylation (Fig. 4E) but does not translocate IRAK1 to the nucleus (Fig. 4A), we expected the involvement of TLR4 signaling to achieve IRAK1 nuclear translocation. Because IRAK1 does not have a nuclear localization signal (Predict Protein software; www.predictprotein.org), a chaperone protein may be involved in IRAK1 nuclear translocation. Based on the report showing that TLR4 signaling causes nuclear translocation of IFN regulatory factor 5 (IRF5) (21), we asked whether IRF5 could be a chaperone of IRAK1. Indeed, IRF5/IRAK1 association was enhanced with TLR4/CD40 costimulation (Fig. 4F). Although LPS stimulation alone is sufficient for IRF5 nuclear translocation, IRAK1 nuclear translocation required TLR4/CD40 costimulation (Fig. 4G). In vivo colocalization and nuclear translocation of IRF5 and IRAK1 were observed in splenic macrophages of mice 3 h after LPS treatment (Fig. S6). The data suggested that CD40 signaling was required for IRAK1 sumoylation and that TLR4 signaling was required for nuclear translocation of IRF5, which serves as a chaperone of IRAK1.
TRAF2 Mediates IRAK1 Sumoylation in Macrophages Simultaneously Stimulated Through TLR4 and CD40.
Because sumoylation is generally exerted by E3 ubiquitin ligases (22), we sought a role played by E3 ubiquitin ligases, TRAFs. Because CD40 stimulation was sufficient for IRAK1 sumoylation (Fig. 4E and Fig. S5), we focused on TNF receptor-associated factor 2 (TRAF2), which is involved in the CD40-signaling pathway but not in the TLR4 pathway (23). We first found that CD40 stimulation enhanced association between TRAF2 and IRAK1 in LPS-stimulated macrophages (Fig. 5A). Traf2 shRNA treatment, which reduced TRAF2 expression (Fig. 5B), resulted in the attenuation in the following events; IRAK1 sumoylation (Fig. 5C), interaction between IRAK1 and IRF5 (Fig. 5D), IRAK1 nuclear translocation (Fig. 5E), and IL-10 production (Fig. 5F). The data demonstrated the critical role of TRAF2 in IRAK1 sumoylation and nuclear translocation to induce IL-10 production.
Fig. 5.

TRAF2 is a key molecule for CD40-mediated IRAK1 sumoylation and nuclear translocation. (A) Co-IP of TRAF2 and IRAK1 after LPS plus CD40 Ab treatment. (B–E) TRAF2 expression (B), IRAK1 sumoylation (C), IRAK1-IRF5 co-IP (D), and IRAK1 nuclear translocation (E) in Traf2 shRNA-transfected macrophages stimulated with LPS plus CD40 Ab for 60 min. (Data in B and C were obtained from the same set of lysates. Therefore, we used the same β-actin blots for the internal control of B and C. Scale bar in E, 5 μm.) (F) IL-10 production in macrophages stimulated with LPS plus CD40Ab for 24 h was detected by ELISA. Final concentrations of LPS and Ab/IgG in tissue culture were 100 ng/mL and 10 µg/mL, respectively. *P < 0.05. Data are presented as mean ± SEM (n = 4 per group).
iOPN Is Essential for CD40-Derived Signal Transduction in T-Cell–Mediated Control of Hyperinflammation in Endotoxemia.
We have previously found interaction between IRAK1 and iOPN (24); thus, an iOPN-specific mechanism is possibly involved in the CD40-mediated IRAK1 modification. We first found that IRAK1 colocalized with OPN in the cytoplasm after simultaneous stimulation of TLR4 and CD40 (Fig. 6A) and was coimmunoprecipitated with OPN after TLR4/CD40 costimulation (Fig. 6B). In Opn−/− macrophages, IRAK1 showed attenuated phenotypes in sumoylation (Fig. 6C and Fig. S7A), association with TRAF2 (Fig. 6D), and nuclear translocation (Fig. 6E and Fig. S7 B and C) with TLR4/CD40 costimulation. CD40 stimulation did not promote IRAK1 binding to the Il10 promoter in Opn−/− macrophages (Fig. S7D), which ended up to show no increase of IL-10 production (Fig. 6F), with significantly less IL-10 production than WT macrophages (Fig. 6G). Taken together, iOPN is involved in TRAF2-mediated sumoylation and nuclear translocation of IRAK1 to enhance Il10 gene expression.
Fig. 6.

iOPN is essential for CD40-mediated host protection from LPS endotoxemia by TNF attenuation. (A) Confocal microscopic image showing colocalization of IRAK1 and iOPN. Cells were treated with LPS plus CD40 Ab for 2 h. (Scale bar, 5 μm.) (B) Co-IP of OPN and IRAK1 from macrophages stimulated with LPS and CD40 Ab for indicated durations. (C–E) IRAK1 sumoylation (C), IRAK1 and TRAF2 co-IP (D), and IRAK1 nuclear translocation and IRAK1 fluorescent intensity in nucleus (E) in WT and Opn−/− macrophages stimulated with LPS and CD40 Ab for 60 min. Staining intensity histogram, along with the thin transverse lines, is shown in Fig. S7B. (F and G) IL-10 levels in culture supernatants of Opn−/− macrophages (F), and WT vs. Opn−/− macrophages stimulated with LPS plus CD40 Ab (G). (H) Stability of Tnfa mRNA in WT or Opn−/−macrophages after α-amanitin treatment followed by LPS plus CD40 Ab treatment. (I) TNF levels in WT vs. Opn−/− macrophage culture supernatants with or without CD4+ T cells in the presence of LPS for 24 h. (J) TNF levels in WT or Opn−/− macrophage culture supernatants after CD40 Ab plus LPS stimulation for 24 h. (K) TNF levels in culture supernatants with Opn−/− macrophages lentivirally transfected with either iOPN or control GFP expression vector. (L) LPS endotoxemia induced as indicated in Fig. 1A in WT mice (n = 9), Opn−/− mice (n = 9), and Opn−/−Rag2−/− (n = 5). (M) TNF and IL-10 levels in serum isolated from mice 12 h after LPS injection. All experimental data are representative of at least two independent experiments. Final concentrations of LPS and Ab/IgG in tissue culture were 100 ng/mL and 10 µg/mL, respectively, unless otherwise noted. *P < 0.05.
Absence of OPN in macrophages resulted in a long half-life of Tnfa mRNA (Fig. 6H) and in failed suppression of TNF production (Fig. 6 I and J). However, OPN is not involved in the IL-10–mediated Tnfa mRNA destabilization because rIL-10 still down-regulated Tnfa mRNA levels without OPN (Fig. S7E). LPS-induced IL-6 production was not altered between WT and Opn−/− macrophages (Fig. S7F), again suggesting that the impact of OPN is specific for IL-10 and TNF. Notice that WT and Opn−/− macrophages express comparable levels of TNF when stimulated with LPS alone (Fig. 6I), suggesting the involvement of OPN in the CD40 pathway, not in the TLR4 pathway. Importantly, Opn−/− macrophages have normal cell-surface CD40 expression (Fig. S7G), ruling out the issue of CD40 signal intensity into macrophages.
There are two isoforms of OPN, iOPN and the secreted type of OPN (sOPN) (25, 26). We confirmed that iOPN expression by lentivirus in Opn−/− macrophages (27) restored the T-cell–mediated effect of TNF attenuation (Fig. 6K). On the other hand, sOPN was not involved here because treatment either with OPN neutralization Ab or with rOPN to macrophage culture failed to alter TNF production by LPS plus CD40 stimulation (Fig. S7H). These results confirmed that iOPN, but not sOPN, is critical for T-cell–mediated TNF attenuation.
Finally, we found that Opn−/− mice were susceptible to endotoxemia regardless of their Rag2 genotype (Fig. 6L). Serum levels of TNF and IL-10 in LPS-injected Opn−/− mice showed no significant difference from those in Opn−/−Rag2−/− mice (Fig. 6M) whereas more TNF and less IL-10 levels were observed in the absence of OPN, compared with WT mice (Fig. 6M). Macrophages isolated from LPS-treated Opn−/− mice showed elevated Tnfa and reduced Il10 mRNA expression, but no change in mRNA expression of Il6 and Cxcl1 (Fig. S7I). The data here suggested that OPN is critically involved in CD40 signaling to attenuate TNF levels in LPS endotoxemia.
Discussion
Our in vivo and ex vivo data showed that CD40 ligation of macrophages is essential for the suppressive function by adaptive immune cells. It seems that T cells expressing CD40L play a role in the suppression. As demonstrated by a previous report (28) and our results here (Fig. S2 A and B), Foxp3+ Tregs constitutively express CD40L and may provide CD40L to macrophages. CD62L+CD4+ T cells from LPS-treated mice expressed CD40L and seemed to be CD40L providers whereas CD62L+CD4+ T cells from naive mice did not express CD40L (28) (Fig. S2 A and B). In our experiment, we used T cells obtained from naive mice and found them to suppress TNF production by macrophages. This suppression seems to be attributed to the inclusion of a detectable population of CD62L−CD44+CD4+ (effector) T cells, which express CD40L (Fig. S2 A and B) in naive mice. Such a population appears in naive cells due to activation by commensal bacteria, environmental antigens, and homeostatic proliferation. In fact, all these CD40L-expressing CD4+ T subpopulation can suppress macrophage TNF production (Fig. S1D). In addition to CD4+ T cells, CD8+ T cells protected mice from LPS endotoxemia, suppressed macrophage TNF production, expressed CD40L in endotoxemic mice, and colocalized with macrophages soon after LPS treatment. Therefore, CD8+ T cells also contribute to the suppression of early innate inflammatory responses by adaptive immune cells in vivo. On the other hand, B cells did not seem to migrate toward macrophages. Because B cells were reported to play a role in protective early innate immune responses during bacterial sepsis (9), B cells use a different mechanism from that which T cells use for controlling hyperinflammation.
Microscopic analyses demonstrated that T cells physically interact with macrophages in the spleen after, but not before, 3-h LPS treatment. Consistent with the timing of the T cell–macrophage in vivo interaction, WT and Rag2−/− mice showed different serum levels of TNF and IL-10 after, but not before, 3-h LPS treatment. Therefore, T-cell–mediated suppression of the proinflammatory response in innate immunity seems to start only 3 h after LPS injection. The data suggest that the proinflammatory response in the innate immune system is at full throttle soon after infection. However, to minimize tissue damage, hosts have to start applying the break. Using T cells for this purpose may be one of multiple ways for hosts to negatively control the full-blown proinflammatory response. Here, future studies are needed to determine what makes T cells start migrating toward macrophages 3 h after LPS injection.
A recent study showed that IRAK1-deficient mice showed reduced susceptibility to sepsis induced by cecal ligation puncture (CLP) (29). The result does not conflict with our results here; rather, it shows that the primary role of IRAK1 is to induce proinflammatory responses mainly through NFκB in the TLR/IL-1R signaling. In fact, the report demonstrated that IRAK1-deficient mice with sepsis resulted in the general down-regulation of cytokines, including IL-6, IL-10, and IL-1β, as shown by the CLP study with IRAK1-deficint mice (29). In contrast, our study showed an NFκB-independent pathway in the presence of IRAK1, which elicits a distinct role as a transcription factor downstream of CD40.
We showed that CD40 agonistic Ab induced NFκB activity in B cells, but not in macrophages (Fig. S3A). A review article (30) mentioned that macrophages have weaker sensitivity to CD40 stimulation than B cells, and that CD40 agonistic Ab and soluble CD40L are not potent enough to elicit macrophage proinflammatory responses. Therefore, it is possible that biological responses triggered by CD40 stimulation widely vary depends on cell types and the intensity of CD40 signaling. Our data suggested that CD40 Ab stimulation (i.e., weak CD40 stimulation) elicits the anti-inflammatory role of CD40. But immune responses by weak vs. strong CD40 stimulation are still not clear, particularly in vivo. This question brought us to the conflicting results in previous studies regarding the role of CD40 in sepsis and LPS endotoxemia. Some studies demonstrated the protective role of CD40, as we did. Those include the strong correlation between CD40 expression on peripheral blood monocytes and improved outcome in septic patients (31), and the protection of mice from CLP sepsis and acute Salmonella infection by agonistic CD40 Ab treatment (32, 33). In contrast, another study showed that Cd40−/− mice were more resistant to sepsis by CLP although Cd40l−/− mice were as susceptible as WT mice (34). The study (34), in which a CLP model was used, also demonstrated that bacterial heat shock protein 70 (HSP70) directly stimulates CD40, suggesting that distinct responses between Cd40−/− mice and Cd40l−/− mice to CLP may be attributed to CD40 stimulation by bacterial components. One possibility to explain the discrepancy in the role of CD40 is that various CD40 ligands (bacterial ligands, CD40L on cells, therapeutic CD40 agonistic Ab) have different affinity to CD40, enough to differentially activate CD40 signaling. Different intensity of CD40 stimulation may have generated different biological outcomes.
iOPN was previously established as a proinflammatory molecule, but we found in this study that iOPN served as a negative regulator of inflammatory responses in concert with adaptive immune cells. iOPN faithfully behaves as a proinflammatory molecule when hosts are comprised of the innate immune system alone (24). However, we have shown that iOPN could behave as an anti-inflammatory molecule when the adaptive immune system is present.
We summarized the proposed molecular mechanism downstream of CD40 and TLR4 in Fig. 7. The CD40 signal provides TRAF2-mediated sumoylation to IRAK1 in the presence of iOPN. Sumoylated IRAK1 also interacts with IRF5, which is activated by TLR4 signaling and chaperones IRAK1 to the nucleus. It is possible that sumoylated IRAK1 uses other IRFs than IRF5 as a chaperone. Here, we suggest that this mechanism, triggered by dual activation of the TLR4 and CD40 pathways, plays a role in attenuating excessive TNF expression by myeloid cells in the presence of T cells.
Fig. 7.

Schematic diagram of CD40-mediated suppression of TNF production. CD40 signal controls excessive TNF production by LPS-stimulated macrophages. First, CD40L on T cells stimulates CD40 on macrophages. On the other hand, LPS stimulates TLR4. CD40 signaling induces IRAK1 sumoylation in the presence of TRAF2 and intracellular isoform of osteopontin (iOPN). Sumoylation of IRAK1 allows its interaction with IRF5, which is activated by TLR4 signaling and works as an IRAK1 chaperone to the nucleus. Nuclear IRAK1 then binds to the Il10 promoter in macrophages. In an autocrine fashion, IL-10 activates IL-10R signaling, which destabilizes Tnfa mRNA. Therefore, dual signaling from CD40 and TLR4 are essential to control TNF in this mechanism.
In this study, we elucidated a molecular mechanism by which adaptive immune cells play a protective role, in an antigen-independent fashion, by controlling hyperinflammation in the innate immunity at the early stage of septic endotoxemia.
Materials and Methods
To induce endotoxemia, purified Escherichia coli LPS (serotype 055:B5; Sigma-Aldrich) was administered i.p. into mice (40 mg/kg). Sex-matched (male or female) and age-matched (5- to 6-wk old) animals were used for all of the experiments. Detailed descriptions of animals, T-cell adoptive transfer, confocal microscopy and two-photon analyses, coimmunoprecipitation, cell preparation and culture setup, cDNA preparation and real-time PCR, measurements of NFκB activity and Tnfa mRNA stabilization, and ChIP assays are found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Drs. Lee Reinhardt, Sam Johnson, and Benjamin Carlson for help setting up two-photon microscopy analyses and Drs. Tso-Pang Yao and Sophia Sarafova for critical reading of the manuscript. This work was supported by National Institutes of Health Grants R01AI088100 (to M.L.S.), R21AI103584 (to M.L.S.), and R01AI073896 (to J.R.P.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321427111/-/DCSupplemental.
References
- 1.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 2.Kawagoe T, et al. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol. 2009;10(9):965–972. doi: 10.1038/ni.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heyninck K, Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-kappaB activation at the level of TRAF6. FEBS Lett. 1999;442(2-3):147–150. doi: 10.1016/s0014-5793(98)01645-7. [DOI] [PubMed] [Google Scholar]
- 4.Heyninck K, et al. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol. 1999;145(7):1471–1482. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 6.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim KD, et al. Adaptive immune cells temper initial innate responses. Nat Med. 2007;13(10):1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guarda G, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460(7252):269–273. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]
- 9.Kelly-Scumpia KM, et al. B cells enhance early innate immune responses during bacterial sepsis. J Exp Med. 2011;208(8):1673–1682. doi: 10.1084/jem.20101715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock: A review of laboratory models and a proposal. J Surg Res. 1980;29(2):189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 11.Michie HR, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318(23):1481–1486. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]
- 12.Choo-Kang BS, et al. TNF-blocking therapies: An alternative mode of action? Trends Immunol. 2005;26(10):518–522. doi: 10.1016/j.it.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Lorente JA, Marshall JC. Neutralization of tumor necrosis factor in preclinical models of sepsis. Shock. 2005;24(Suppl 1):107–119. doi: 10.1097/01.shk.0000191343.21228.78. [DOI] [PubMed] [Google Scholar]
- 14.Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18(3-4):335–343. doi: 10.1016/j.cytogfr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Exley AR, Leese T, Holliday MP, Swann RA, Cohen J. Endotoxaemia and serum tumour necrosis factor as prognostic markers in severe acute pancreatitis. Gut. 1992;33(8):1126–1128. doi: 10.1136/gut.33.8.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajasingh J, et al. IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. FASEB J. 2006;20(12):2112–2114. doi: 10.1096/fj.06-6084fje. [DOI] [PubMed] [Google Scholar]
- 17.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. 2005;102(24):8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y, Li T, Sane DC, Li L. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem. 2004;279(49):51697–51703. doi: 10.1074/jbc.M410369200. [DOI] [PubMed] [Google Scholar]
- 19.Su J, Richter K, Zhang C, Gu Q, Li L. Differential regulation of interleukin-1 receptor associated kinase 1 (IRAK1) splice variants. Mol Immunol. 2007;44(5):900–905. doi: 10.1016/j.molimm.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 20.Gottipati S, Rao NL, Fung-Leung WP. IRAK1: A critical signaling mediator of innate immunity. Cell Signal. 2008;20(2):269–276. doi: 10.1016/j.cellsig.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Takaoka A, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434(7030):243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 22.Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 23.Häcker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11(7):457–468. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 24.Inoue M, et al. Cutting edge: Critical role of intracellular osteopontin in antifungal innate immune responses. J Immunol. 2011;186(1):19–23. doi: 10.4049/jimmunol.1002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinohara ML, Kim HJ, Kim JH, Garcia VA, Cantor H. Alternative translation of osteopontin generates intracellular and secreted isoforms that mediate distinct biological activities in dendritic cells. Proc Natl Acad Sci USA. 2008;105(20):7235–7239. doi: 10.1073/pnas.0802301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoue M, Shinohara ML. Intracellular osteopontin (iOPN) and immunity. Immunol Res. 2011;49(1-3):160–172. doi: 10.1007/s12026-010-8179-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinohara ML, et al. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol. 2006;7(5):498–506. doi: 10.1038/ni1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guiducci C, Valzasina B, Dislich H, Colombo MP. CD40/CD40L interaction regulates CD4+CD25+ T reg homeostasis through dendritic cell-produced IL-2. Eur J Immunol. 2005;35(2):557–567. doi: 10.1002/eji.200425810. [DOI] [PubMed] [Google Scholar]
- 29.Chandra R, et al. IRAK1-dependent signaling mediates mortality in polymicrobial sepsis. Inflammation. 2013;36(6):1503–1512. doi: 10.1007/s10753-013-9692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suttles J, Stout RD. Macrophage CD40 signaling: A pivotal regulator of disease protection and pathogenesis. Semin Immunol. 2009;21(5):257–264. doi: 10.1016/j.smim.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 31.Sugimoto K, et al. Monocyte CD40 expression in severe sepsis. Shock. 2003;19(1):24–27. doi: 10.1097/00024382-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Schwulst SJ, et al. Agonistic monoclonal antibody against CD40 receptor decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2006;177(1):557–565. doi: 10.4049/jimmunol.177.1.557. [DOI] [PubMed] [Google Scholar]
- 33.Marriott I, Thomas EK, Bost KL. CD40-CD40 ligand interactions augment survival of normal mice, but not CD40 ligand knockout mice, challenged orally with Salmonella dublin. Infect Immun. 1999;67(10):5253–5257. doi: 10.1128/iai.67.10.5253-5257.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nolan A, Weiden MD, Hoshino Y, Gold JA. Cd40 but not CD154 knockout mice have reduced inflammatory response in polymicrobial sepsis: A potential role for Escherichia coli heat shock protein 70 in CD40-mediated inflammation in vivo. Shock. 2004;22(6):538–542. doi: 10.1097/01.shk.0000143416.20649.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.