

Significance
One of the important puzzles in virology is how viruses assemble protective protein containers for their genomes rapidly and efficiently during an infection. Recent advances in the field of RNA viruses suggests that multiple specific contacts between the genomic RNA and the proteins in these containers play crucial roles in this process, but the detailed molecular mechanisms by which this occurs are largely obscure. We describe here a mathematical model of virus assembly that incorporates these contacts and other details of real virus infections. It demonstrates how such contacts act collectively to reduce the complexity of virus formation, ensuring efficient and selective packaging of the viral genomes. These competitive advantages shed new light on viral assembly and evolution and open up unique avenues for antiviral therapy.
Abstract
One of the important puzzles in virology is how viruses assemble the protein containers that package their genomes rapidly and efficiently in vivo while avoiding triggering their hosts’ antiviral defenses. Viral assembly appears directed toward a relatively small subset of the vast number of all possible assembly intermediates and pathways, akin to Levinthal’s paradox for the folding of polypeptide chains. Using an in silico assembly model, we demonstrate that this reduction in complexity can be understood if aspects of in vivo assembly, which have mostly been neglected in in vitro experimental and theoretical modeling assembly studies, are included in the analysis. In particular, we show that the increasing viral coat protein concentration that occurs in infected cells plays unexpected and vital roles in avoiding potential kinetic assembly traps, significantly reducing the number of assembly pathways and assembly initiation sites, and resulting in enhanced assembly efficiency and genome packaging specificity. Because capsid assembly is a vital determinant of the overall fitness of a virus in the infection process, these insights have important consequences for our understanding of how selection impacts on the evolution of viral quasispecies. These results moreover suggest strategies for optimizing the production of protein nanocontainers for drug delivery and of virus-like particles for vaccination. We demonstrate here in silico that drugs targeting the specific RNA–capsid protein contacts can delay assembly, reduce viral load, and lead to an increase of misencapsidation of cellular RNAs, hence opening up unique avenues for antiviral therapy.
The formation of a protective protein container is a vital step in most viral life cycles (1). It is a prime example of molecular self-assembly, the fundamental principle underlying the formation of protein nanocontainers that are important in virology, cell biology, and bionanotechnology (2). Whereas the molecular mechanisms of capsid formation and genome encapsulation vary across viral families, there are a number of common features that can be characterized collectively. Procapsid formation may occur via the self- or assisted assembly of coat protein subunits (CPs) and be followed by the introduction of the genomic material via a packaging motor, as seen in many double-stranded DNA viruses (3). Capsid assembly may also follow a coassembly process involving protein subunits and the viral genome, a phenomenon occurring in many single-stranded RNA (ssRNA) viruses (4). These latter are one of the largest viral families, including major pathogens (5).
The study of virus assembly in vitro has been characterized by a reductive approach, focusing on the isolation of a small number of previously disassembled components of the system and an analysis of the reactions between them. In these studies genome encapsidation is often nonspecific with noncognate RNA and even anionic polymers being encapsidated (6–10), leading to an interpretation that CPs alone have the ability to form capsids identical to those formed in vivo. However, these studies fail to address the efficiency, fidelity, and speed of assembly in vivo and also the observation that in cells genome encapsidation is highly specific (11). Additionally, they neglect the exquisite timing of events following infection, genome uncoating, gene expression, and creation of progeny genomes and eventually virions. Only late in each infection cycle is capsid assembly required and this is triggered by an increase in CP concentration.
Whereas many earlier studies have examined the assembly of protein subunits alone (12–17), we have recently emphasized the important cooperative roles played by the protein subunits interacting with the viral genomes of ssRNA viruses (4, 18). These have been shown to control the specification of the quasiequivalent conformations of the CPs, e.g., via dynamic allostery in bacteriophage MS2 (4, 19) and the assembly pathways each system follows. It appears that many RNA viruses have multiple sites within their genomes, called packaging signals (PSs) that contact CPs during assembly (18, 20, 21). The sequences/secondary structures of PSs are degenerate and they cannot be identified by sequence analysis alone. They have a range of CP affinities that have been tuned by evolution to promote productive assembly of the protein shell. We have identified such PSs in a number of ssRNA viruses and demonstrated the molecular basis of some of their effects, including in the RNA phages (4, 21) and for the plant satellite virus satellite tobacco necrosis virus (STNV) (22, 23). Single molecule assays of assembly of these viruses in vitro recapitulate the specificity of genome encapsidation seen in vivo (21), presumably because they are done at very low concentrations, approximating those in vivo, and directly demonstrate how CP–PS interactions impact on assembly as a collective. Single copy high-affinity PSs have been reported for a wide range of viruses (24, 25). Such genomes are very likely to also contain unrecognized lower affinity sites, suggesting that CP–PS interactions are more common than widely believed. Although these studies provide important insights into the mechanism of capsid assembly, the fundamental reasons for the observed assembly efficiency in vivo remain largely opaque. In particular, it is still unclear how viruses manage to overcome the limitations of the viral equivalent of a Levinthal’s paradox, i.e., the selection of the assembly intermediates and pathways best suited to enhance viral load and the navigation among the vast numbers of possible pathways and potential kinetic traps in a limited amount of time. Even for a relatively small virus, e.g., bacteriophage MS2 with a capsid formed from 90 protein dimers (26), there are on the order of 1015 different possible intermediate species. If each of these were explored, capsid assembly would be excessively slow, or even worse, potentially result in kinetic traps where capsid proteins form stable off-pathway intermediates. This contrasts with what is observed in nature; assembly of capsids is highly efficient and robust to kinetic trapping. Previous models that exemplify the roles of CP–CP interactions and, where applicable, interactions with genomic RNAs, only partially address these issues (7–10). We show here that a vital step in resolving the viral assembly paradox lies in the timing of viral CP production, the protein “ramp,” i.e., the accumulation of CP that would naturally occur in the in vivo process. In particular, we demonstrate that the protein ramp directs assembly to only a limited number of assembly intermediates and pathways, and that it also ensures packaging specificity in a cellular environment. These results suggest a paradigm shift in our approach to studying capsid formation toward in vitro and in silico assembly studies that mimic important regulatory mechanisms that occur in vivo, such as the protein ramp.
Results
The Model System.
In previous modeling studies of capsid formation, it has proven fruitful to demonstrate the physical principles underlying the assembly process using the simplest possible capsid geometry, a dodecahedron formed from 12 pentagonal protein building blocks (12, 13, 27). Such a system is capable of exemplifying the assembly of capsids with more complex geometries, while still being amenable to an in depth analysis of the assembly pathways. Following this strategy, we consider in silico assembly of pentagonal building blocks into dodecahedral containers mediated by contacts with PSs in a hypothetical RNA molecule (Fig. 1). The PSs bind to the centers of the pentagons with varying affinities, and the bound CPs can then form additional CP–CP interactions, thus modeling a coassembly process during which the RNA is packaged as the capsid container forms. In this system, assembly is based on two local rules (27): (i) the attachment or detachment of CP units to or from the PSs present on the RNA and (ii) the association or disassociation of CP units bound to neighboring PSs (Fig. 1B). Our model defines assembly as starting via nucleation of the first CP–CP interaction on the RNA. This is consistent with known in vitro assembly processes (21, 28). So that the simulation reflects what may happen in reality, the range of affinities for these intermolecular contacts are drawn from experimental values or inferred by us and others (18, 29, 30) for the PS–CP contacts in bacteriophage MS2 and from ref. 13 for CP–CP contacts, respectively.
Fig. 1.

The dodecahedral model of virus assembly. (A) The building blocks of the system are: pentagons as proxies for capsid protein (CP) and viral RNA sequences, each with 12 CP binding sites modeled as RNA stem loops (PSs), with positions labeled −6 to 6 from the 5′ to the 3′ end. Each PS can interact with CP with a different affinity. The distribution of PS affinities, here and throughout is indicated as binding free energies as a string of color-coded beads. ΔGb values (Kd values) of −9.5 > ΔGb > −12.0 kcal/M (100 nM > Kd > 1.5 nM) are shown as green, −5.4 > ΔGb > −9.5 kcal/M (100 μM > Kd > 100 nM) as blue, and −3.0 > ΔGb > −5.4 kcal/M (6.2 mM > Kd > 100 μM) as red, respectively. (B) The two basic reactions of the assembly model: CP binding to RNA and subsequent formation of CP–CP contacts.
The exclusion of CP–CP interactions independent of RNA binding reflects experimental results showing that PS interactions with CP significantly accelerate protein–protein association. For example, in bacteriophage MS2, PS–CP interaction results in an allosteric conformational change, displacing a preexisting conformational equilibrium in the CP that lies heavily in favor of just one conformer (4, 19, 31, 32). As a result, assembly in vitro goes to completion in less than 1 h (21), as opposed to a largely incomplete assembly reaction taking many days in the absence of RNA (33, 34). Similarly, STNV CP will not assemble in vitro in the absence of RNA because PS–CP interactions are essential for overcoming electrostatic repulsions at the N termini of the CP (20, 22).
At each step of the simulation, all possible reactions between the assembly intermediates that are present are calculated and their relative probabilities of reacting determined. The reaction probabilities depend on the concentrations of the intermediates involved in the reaction and either the CP–RNA or CP–CP affinities, expressed in terms of binding free energies, as explained in Methods. The resulting assembly kinetics is sampled via a Gillespie-type algorithm (27, 35), i.e., at each step of the simulation the next reaction that occurs is sampled in a stochastic manner based on relative probabilities of the reactions. Time is incremented in units of seconds based on the reaction rates, which capture the average time for a single reaction to occur.
We use a Gillespie-type algorithm (35), because modeling assembly by numerically solving differential equations for the assembly kinetics is not practicable due to the complexity of the problem. The Gillespie method is scalable to real viral systems and allows us to record the assembly pathway for each RNA in the simulation. Gillespie algorithms are designed for well-mixed systems. Assembly in the presence of a linear polymer is not neccessarily so, but because PSs are located on the periphery of an otherwise compact RNA molecule and are in constant exchange with CP, we assume this to be a good approximation. We also expect local concentration effects due to cooperative binding of CPs to PSs (36) and binding accessibility of PSs due to steric effects to be negligible, based on data for MS2 (18, 21).
The Effects of the Protein Ramp on the Kinetics of Assembly.
Previous work on capsid formation in silico considered the assembly of CPs in isolation, essentially mimicking an in vitro assembly scenario in which the rate of CP expression from the genomic RNAs via ribosomes is neglected. We provide here an in silico model of virus assembly in which the speed of protein subunit synthesis (the protein ramp) is included. To achieve this, we modeled a CP ramp following Eigen’s example used to discuss a viral hypercycle with respect to bacteriophage Qβ (37), which was based on experimental determination of the amounts of CP, RNA templates, and infectious phage throughout the complete viral lifecycle. The experiment and Eigen’s model suggest that enough CP is produced in roughly the last 5–10 min before cell lysis to assemble ∼10,000–20,000 Qβ progeny. We have also shown that RNA phages assemble using multiple PSs to improve efficiency and fidelity (18, 21, 27). Using a relative low estimate for the progeny produced per round of infection, we implement a linear rate of CP addition to 3000 hypothetical RNAs (Fig. 2), where enough CP would be titrated into the system over 5 min to complete assembly on every RNA (Methods). The RNAs used in this analysis are identical copies of the most/least efficient assembly template identified (RNA1/2; Fig. 2A), from analogous simulations of RNAs where each of the PS affinities for the RNA were randomly chosen to be a negative value above −12 kcal/M, the value of the highest-affinity PS in bacteriophage MS2 (29).
Fig. 2.

The impact of the protein ramp. (A) A population of 3000 RNAs, each containing a random selection of PS affinities was constructed, each RNA copied 3000 times, and the percentage of RNA packaged at a CP:RNA ratio (12:1) after 1000 s calculated. The histogram shows the frequency of occurrence of RNA sequences resulting in a particular yield of capsids. The highest (99%, RNA1) and lowest (75%, RNA2) yielding RNAs in the sample were used in further simulations. A representative cellular RNA was constructed by assigning a low affinity (−3 kcal/M) to all 12 PSs. (B) Assembly kinetics of RNA1 determined from the average of 100 assembly simulations with 3000 RNAs at the ratio of CP:RNA (12:1). The graph shows time-dependent assembly of (i) assembly intermediates (protein arrangements shown as cartoons) that are on pathway and complete to capsid; (ii) correctly assembled capsids; and (iii) kinetically trapped malformed species (Left). In the presence of the protein ramp (CP concentration shown as a dashed line, here and throughout) all of the RNAs are successfully encapsidated. The percentage of successfully packaged RNAs that nucleate at each pair of PSs is shown below each graph. In the presence of a ramp, nucleation is confined to the two highest affinity PSs.
Assembly of 3000 copies of RNA1 in the presence of 36,000 CP subunits was simulated with and without the protein ramp. An intercapsomere bond strength of −3.75 kcal/M (corresponding to a Kd of 1.7 mM for a single CP–CP contact; note that by contrast the Kd for CP–RNA contacts is between 1.1 mM and 1.5 nM, i.e., between −4 and −12 kcal/M) was assumed following representative values given in the literature for other viral CP–CP contacts (12–15, 38). In the absence of the ramp (Fig. 2B), i.e., when all 36,000 CPs are present at the start, mimicking many in vitro reassembly assays, a number of lower-order on-pathway intermediates form initially and are then used up, but a large fraction (∼40%) of the CP becomes kinetically trapped in malformed RNA–CP complexes, reducing the yield of correctly formed capsids. In sharp contrast, 100% of the RNAs get packaged in the presence of the CP ramp. The kinetics of assembly also differ between the two situations. Without a ramped protein gradient capsid assembly occurs far earlier than with it. Because assembly removes genomes that would be acting as templates in a real infection, earlier assembly in vivo is potentially counterproductive. Moreover, the capsid assembly kinetics suggest a strong nucleation event, which is then followed by a rapid completion phase in which capsids are assembled faster than the linear production of CP. This mimics the observed assembly kinetics for Qβ infections (37) and is consistent with the results of single molecule assembly assays (21).
To investigate the reasons for this difference, we analyzed the PSs on the RNA at which nucleation takes place (Fig. 2B). We define nucleation as the point when formation of the initial CP–CP complex on the RNA occurs. In the absence of the ramp, nucleation may occur anywhere across the RNA. In the in vivo simulation, i.e., a CP concentration that starts low and increases, nucleation occurs principally on the adjacent, highest affinity pair of PSs (−12 and −11) (89.2% with the other pathways involving just one of these PS sites). These results show that the protein ramp facilitates coordinated nucleation, allowing assembly to proceed in a more controlled manner, as exemplified by the higher particle yield and absence of kinetic traps. To explore whether these effects were unique to RNA1, we carried out analogous simulations with the worst assembling sequence from the distribution in Fig. 2A, namely RNA2. The results, shown in Fig. S1, reveal formation of the same on-pathway intermediates with similar differences in assembly efficiency between the scenarios including or excluding the protein ramp. However, in this case not all RNA2s are packaged, even in the presence of the ramp. Analysis of assembly nucleation with RNA2 shows a distribution of nucleation sites both with and without ramp, albeit at multiple stronger nucleation sites in its presence. This demonstrates that RNA1 has an advantage due to its single adjacent pair of strong PSs and their role in nucleation, compared with three such sites in RNA2. These results exemplify Eigen’s comment (37) that replication, which is widely used as the main criterion for fitness of viral populations in an evolutionary context, is, although important, only one of the criteria of overall performance in a viral infection. In particular, Fig. 2 shows that the interplay of the protein ramp and virus assembly has a significant impact on fitness that has not been appreciated previously.
To examine the effects of capsid geometry on these results, we also carried out a simulation for a 60mer capsid formed from 60 identical building blocks that each bind to a PS in a genome, as is the case of bacteriophage MS2. In this more complex setting, addition of the protein ramp has the effect of dramatically raising assembly efficiency (from 1 to 75%), suggesting that the 12mer model discussed here is indeed representative of more complicated viruses. We also explored the consequences of disrupting the highest affinity PSs, which control nucleation under these conditions. To do this, we reduced the affinities of these two sites to a low value (creating RNA1′; Fig. S2), hence mimicking mutagenesis experiments that impact on PS structure and affinity. Nucleation becomes uncoordinated, and assembly is incomplete (∼80%), resulting in the formation of a significant fraction of misassembled species, and also in the misencapsidation of cellular RNAs.
A Reduction in Complexity.
The complexity of the virus assembly process can be understood in terms of the number of possible pathways from a single protein subunit to the fully formed capsid. Because the model requires proteins to bind the RNA before attaching to other protein subunits on the growing capsid shell, assembly occurs effectively along the RNA molecule, starting at a nucleation site and then continuing toward the 5′ and 3′ ends. Exclusion of association of protein subunits that are not bound to neighboring PSs on the genomic RNA is motivated by experimental results for MS2 (21, 31). Assembly pathways can therefore be characterized by the different possible packaging arrangements of the RNA molecule within the fully formed capsid, together with specification of the initiation site. From a mathematical point of view, a superposition of all possible pathways connecting the putative RNA–CP contacts looks like a polyhedral shell, the icosahedron shown in red within the dodecahedral capsid (Fig. 3A). Any connected path visiting every RNA–CP contact precisely once describes a possible layout of the packaged RNA. Connected paths of this type are called Hamiltonian paths (39), and there are 1264 such paths for the icosahedron shown in Fig. 3A. For larger viruses, such as bacteriophage MS2 with 90 building blocks and 60 PSs, the situation is significantly more complex with 40,678 such paths, but the principle of enumerating distinct pathways is similar. Hamiltonian paths are in a one-to-one correspondence with the organizations of the genomic RNA in the vicinity of the capsid, i.e., they specify which PS binds to which CP building block (capsomere). Because a Hamiltonian path specifies neither where assembly nucleates nor in which order the recruitment of CP subunits occurs toward the 5′ and 3′ ends, each Hamiltonian path represents an ensemble of possible assembly pathways. However, because in our simulations nucleation occurs at the two adjacent high-affinity PSs located at the 5′ end of the test RNA1, there is a unique assembly scenario for each Hamiltonian path in this case, i.e., assembly occurs linearly toward the 3′ end.
Fig. 3.

Impact of the ramp on assembly pathway selection and intermediate stability. (A) In assembled capsids the PSs contact the midpoint of every pentagonal face of the dodecahedron and hence the RNA describes a connected path on the inscribed icosahedron (red). One such Hamiltonian path is shown on the Right; the icosahedron is the superposition of all possible Hamiltonian paths. (B) The frequency of occurrence of different Hamiltonian paths across a sample of 150,000 RNA1s determined by tracking the addition of CP on individual RNAs. The distribution in the presence of the ramp (red) shows a bias toward a small number of different paths, whereas in its absence, all possible paths occur (black). Dashed lines indicate the number of paths used by ∼90% of the 150,000 capsids. (C) Assembly intermediates contain more or less stable species depending on the number of CP–CP contacts. (D) For each assembly pathway taken the number of deviations in protein configurations from the most stable structure during assembly are shown. In the absence of the ramp (Left), up to six deviations can occur and their frequencies are relatively similar. In its presence (Right) assembly is strongly biased toward the most stable assembly intermediates.
In Fig. 3B we plot the number of particles in the ensemble of assembled material with RNA1 with a specific Hamiltonian path for their packaged RNAs, with the y-axis showing the frequency of occurrence and the x-axis labeling the possible Hamiltonian paths, ordered from most to least frequently used. The protein ramp results in a dramatic reduction in the number of different Hamiltonian paths followed, demonstrating its role in reducing the complexity of the assembly process. As a second complexity measure, we compute for every assembly pathway, the number of intermediates in that pathway that do not correspond to the most stable intermediate (i.e., the intermediates that have less than the maximal possible number of CP–CP contacts; Fig. 3C). We then plot the percentage of pathways that have a given number of deviations from the most stable ones, i.e., from those on which every intermediate forms the maximum number of intersubunit bonds possible. In contrast to an assembly process in the absence of RNA, the most stable pathway is not unique because there can be different RNA configurations for any given protein configuration. Once again it appears that the protein ramp biases assembly toward the small subset of the most stable intermediates. These features provide a solution to the viral analog of Levinthal’s paradox (40) for capsid assembly and illustrate how assembly in such systems is greatly aided by the natural timing of viral CP production.
The Protein Ramp and Selective Packaging.
Assembly efficiency is one aspect of in vivo viral life cycles. It is intimately linked with packaging specificity, because cellular RNAs may act as competitors for assembling CPs, leading to subpopulations of nonviable particles and reducing the overall viral load (11). Because observations suggest that such competition is avoided, we examined whether the protein ramp plays a role in specificity. We used the same in silico assembly experiment, but introduced an element of competition between RNA1, the viral RNA, and potential cellular competitors. The latter were assigned identical, low affinities for 12 potential PSs creating RNA3 (Fig. 2). We then used an ensemble of 2000 RNA1s and differing ratios of RNA3 excess, simulating the situation in vivo. The reactions were run with just sufficient CP to fully assemble the aliquot of RNA1. The results of a simulation at a RNA1:RNA3 ratio of 1:300, in agreement with experimental estimates for the number of mRNAs in a mammalian cell (105 to 106) (41), are shown in Fig. 4. The effects of the ramp are again very spectacular. With ramped protein addition very little (<1%) of RNA3 becomes misincorporated into capsids, whereas in the absence of the ramp the competitor “cellular” RNA3s become the dominant species incorporated and this effect is robust across a wide range of concentration ratios. RNA2, the worst performing RNA from the initial random distribution in Fig. 2, is also very sensitive to the CP ramp, but less able to prevent misincorporation by RNA3 (Fig. S1B). In light of these results, we have examined the threshold affinities of the PSs in RNA3 required to make it an effective competitor (Fig. S1C). For a wide range of affinities, only the viral RNA1 is packaged, but for PS values of less than −5.5 kcal/M, RNA3 becomes a successful competitor. Note, it is unlikely that any cellular RNAs will contain an ensemble of such high-affinity sites leading to successful competition in vivo (21, 42).
Fig. 4.
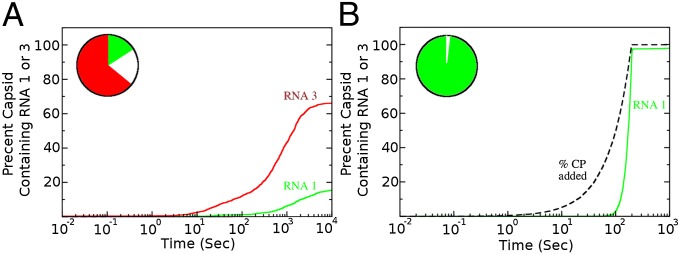
Impact of the protein ramp on packaging selectivity. An in silico competition experiment between 2000 copies of RNA1 and 600,000 copies of RNA3 at a CP:RNA1 ratio (8:1) just sufficient to assemble 2000 capsids. (A) In the absence of the ramp, a significant fraction of RNA3 is encapsulated and the assembly yield of RNA1 is much lower due to the production of malformed species. The circular Inset indicates the percentages of encapsulated RNA1 (green), RNA3 (red), and malformed species (white) at the end of the simulation. (B) With the ramp assembly of RNA1 occurs almost to 100% with no encapsidation of RNA3, i.e., there is high packaging specificity.
The protein ramp also dramatically improves the efficiency of protein assembly in the absence of RNA (Fig. S3). This raises the interesting issue that viruses coassembling around their genomes must assemble efficiently enough to compete with a protein-only reaction. As we have shown previously, RNAs with high affinities at every PS site are poor at assembly, because they nucleate at many places and become kinetically trapped (27). Here we have shown that in the case of low affinities at every PS, e.g., as in RNA3, they are also poor substrates for encapsidation in the presence of protein ramp. It appears therefore that a combination of mixed PS affinities is the most successful strategy of encapsidation for RNAs in a coassembly process.
The simulation with ramp reflects the observed specificity of viral infections in vivo (11) and has major implications for our understanding of quasispecies evolution. The differential specificity for different members of a viral quasispecies, as exemplified here for RNA1 and RNA2, suggests that the presence of a protein ramp impacts differentially on the fitness of different members of a quasispecies (due to their distinct PS distributions). This has important consequences for the evolution of the quasispecies as a whole, which have not been appreciated previously. Finally we note that these results also provide an explanation for outcomes of in vitro assembly experiments in which the CP subunits are all added at the start of the reaction. These routinely show that it is possible to encapsidate noncognate RNAs (43), whereas our results show that in the presence of the ramp, i.e., in an in vivo scenario, this would be much less likely to occur.
A Route to Antiviral Therapy.
These results suggest that drugs targeting CP interactions with the higher affinity PSs within a viral genome should have a highly disruptive effect on capsid assembly. We carried out simulations with 2000 RNA1s and the required CP concentration to form 2000 viral particles in the presence of drug molecules binding strong PSs with a Kd of 10 nM and weak PSs with a Kd of 50 μM. The protein concentration is again modeled as a ramp, while the drug is administered in a single dose at the start of the simulation for different concentrations cD = 1, 10, 20, and 50 μM. We observe an increasing delay in the onset of assembly with increasing drug concentration in the absence of cellular RNAs, whereas in their presence at a concentration ratio of 1:300 for RNA1:RNA3 the drug results in a significant difference in viral load (e.g., a drop of 20% at 1 mM; Fig. 5B) and increased amounts of misincorporated cellular RNA. Similar simulations for a number of concentration ratios of RNA1:RNA3 show: (i) a time delay in capsid assembly, (ii) a reduction in overall viral load, (iii) a significant population of malformed species, and (iv) an increase in the number of packaged cellular RNAs. In a real viral infection, malformed species or noninfectious capsids carrying cellular RNA would trigger natural antiviral defenses (immune responses and/or RNA silencing) potentially resulting in clearance of the infection. These results demonstrate that targeting PSs with drugs that inhibit their functions, especially high-affinity ones, is a promising new avenue for antiviral therapy.
Fig. 5.

Impact of PS-binding drugs on assembly efficiency. (A) Amounts of encapsidated viral RNAs (RNA1) in the absence of RNA3 (shown as percentages of RNA1 (green) and malformed species (white) in the presence of the ramp at differing drug concentrations. (B) As in A in the presence of cellular RNAs (RNA3, red) at a ratio RNA1:RNA3 of 1:300.
Discussion
Viruses are major pathogens of people, animals, and plants, yet options for treatment and prevention are currently limited. Vaccination is the most successful medical intervention, but is likely to be restricted to a severely limited set of these pathogens. The rate at which emerging viruses develop or existing ones mutate requires urgent development of novel means to control their spread and ameliorate their effects (44, 45). Viral capsid assembly is a central process in most cases, but is as yet a largely unexploited drug target (7, 46, 47). It has strong potential because malformed virus-like particles make good immunogens that expose conserved protein epitopes, or in the case of ssRNA plant viruses, the genomic RNA would trigger host gene silencing. Previous attempts to probe the detailed molecular mechanism of viral assembly have relied on in vitro assays in which stoichiometric amounts of the assembly components have been used from the start, and many attempts to simulate assembly have used the same starting conditions. The results of such assays and simulations do not match virus production in vivo (11) and therefore fail to encompass all of the features of “real” infections. This implies the need for a more holistic view that mimics important features of the in vivo assembly process.
Here, we have simulated assembly of ssRNA viruses that coassemble their CP shells around their genomes (23, 48). Recently, we have provided compelling evidence that, at least in a number of model viruses, such assembly reactions depend on specific interactions between viral CPs and dispersed, sequence-degenerate segments of the genome (PSs) (4, 18–23), reflecting both in vitro and in vivo data for the RNA phages (49–51) and plant satellite viruses (20, 52). Given this background, the intricate interdependence of the distribution of PSs and their affinities in the viral RNA becomes an important feature to model. In addition, we have explored the consequences of having the CP concentration increase over time, mimicking what happens in a real infection. The results are dramatic. This simple effect alters the kinetics and yield of capsids produced. It does so by controlling nucleation, limiting the number of assembly pathways each particle follows, ensuring that intermediates are stable and that the virus avoids the pitfalls of the viral analog of Levinthal’s paradox. Remarkably, it also has significant advantages in ensuring specificity of genome incorporation. Thus, a number of features corresponding to observed virus biology are predicted as a consequence of the PS-mediated assembly mechanism. Such predictions do not arise from more coarse-grained models of assembly pathways (7–9).
The ssRNA viruses are just one class of many viral families, but the protein ramp could potentially also have an influence on the assembly of CP subunits in isolation or that use scaffold proteins to play a role similar to the RNA, i.e., those that assemble procapsids (53). In silico experiments suggest that assembly of protein subunits in isolation is highly prone to kinetic trapping (15, 38). This has led to the suggestions that assembly involves two phases of CP–CP interactions, a slower nucleation rate, and a faster elongation rate, which takes over once a critical nucleus is formed (15). In the case of assembly of empty capsids from protein alone (Fig. S3), the protein ramp results in both the elimination of kinetic traps, as well as a reduction in the assembly pathways without the need to invoke a special nucleation rate. This result addresses the kinetic trapping puzzle observed in earlier studies of virus assembly (15, 38) by mimicry of the CP accumulation that would occur in an in vivo situation.
Our understanding of RNA biology has recently been transformed by the realization of the extent of the genome that is transcribed. Many of these transcripts interact with proteins and self-assemble into complex molecular machines controlling everything from gene expression to protein synthesis to splicing and development. Each of these complexes needs to assemble sequence specifically, and some of them are known to interact via multiple RNA motifs of varying affinity akin to viral PSs (54). Our results with this virus simulation highlight the importance of modeling the dynamic concentrations of RNAs and their protein partners in such cases. More widely, they have far reaching consequences for our understanding of one of the most fundamental processes in nature, molecular self-assembly, and also important technological implications. Protein containers play a key role in nanotechnology applications, for example as drug delivery vehicles or in the context of vaccine design. Insights on how to improve the yield of these containers and have tighter control over their assembly in vitro are therefore of particular interest. Conversely, these results open up unique avenues for antiviral therapies targeting specific groups of PSs. As shown here, such drugs would reduce viral load, i.e., the number of infectious particles formed during an infection, and at the same time increase formation of noninfectious particles containing cellular competitor RNAs that trigger the host’s antiviral defenses. Given that PSs are generic features shared across large classes of viral families, including major human pathogens (25), this is a form of drug therapy that is applicable widely and could therefore have significant impact on the health sector.
Methods
The in silico assembly experiments were performed using the Gillespie algorithm (35), which has been modified to incorporate PS–CP interactions as described in ref. 27. For the protein ramp simulations, the computational model adds CP to the simulation following a Markov chain procedure where the time interval until the next addition of a single CP is computed from ΔT = ln(r−1)/a0, with r a randomly chosen number between 0 and 1. For the dodecahedral model, we use a CP synthesis rate of a0 = 120 CP per second. Reactions between CP and RNA are then allowed to take place until the next CP is added, which mimics the gradual synthesis of protein subunits characteristic of in vivo assembly. For the competition simulations, 2000 RNA1 (or RNA2) were combined with 600 k RNA3 for a total of 602 k RNAs. Enough CP to assemble the aliquot of RNA1(2) was either ramped in following the above procedure or added at the beginning of the simulation. Further details about the general Gillespie procedure and specific rate choices for CP–CP and CP–PS interactions can be found in SI Methods and in ref. 27.
Supplementary Material
Acknowledgments
We thank UK’s Engineering and Physical Sciences Research Council (EP/K028286/1 and EP/K027689/1), the Biotechnology and Biological Sciences Research Council (BB/J00667X/1), and the University of York for funding.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. P.P. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319479111/-/DCSupplemental.
References
- 1.Rossmann MG, Johnson JE. Icosahedral RNA virus structure. Annu Rev Biochem. 1989;58:533–573. doi: 10.1146/annurev.bi.58.070189.002533. [DOI] [PubMed] [Google Scholar]
- 2.Steinmetz NF, et al. Buckyballs meet viral nanoparticles: Candidates for biomedicine. J Am Chem Soc. 2009;131(47):17093–17095. doi: 10.1021/ja902293w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun S, et al. Structure and function of the small terminase component of the DNA packaging machine in T4-like bacteriophages. Proc Natl Acad Sci USA. 2012;109(3):817–822. doi: 10.1073/pnas.1110224109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stockley PG, et al. A simple, RNA-mediated allosteric switch controls the pathway to formation of a T=3 viral capsid. J Mol Biol. 2007;369(2):541–552. doi: 10.1016/j.jmb.2007.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneemann A. The structural and functional role of RNA in icosahedral virus assembly. Annu Rev Microbiol. 2006;60:51–67. doi: 10.1146/annurev.micro.60.080805.142304. [DOI] [PubMed] [Google Scholar]
- 6.Comas-Garcia M, Cadena-Nava RD, Rao AL, Knobler CM, Gelbart WM. In vitro quantification of the relative packaging efficiencies of single-stranded RNA molecules by viral capsid protein. J Virol. 2012;86(22):12271–12282. doi: 10.1128/JVI.01695-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kivenson A, Hagan MF. Mechanisms of capsid assembly around a polymer. Biophys J. 2010;99(2):619–628. doi: 10.1016/j.bpj.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elrad OM, Hagan MF. Encapsulation of a polymer by an icosahedral virus. Phys Biol. 2010;7(4):045003. doi: 10.1088/1478-3975/7/4/045003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forrey C, Muthukumar M. Electrostatics of capsid-induced viral RNA organization. J Chem Phys. 2009;131:105101. [Google Scholar]
- 10.Belyi VA, Muthukumar M. Electrostatic origin of the genome packing in viruses. Proc Natl Acad Sci USA. 2006;103(46):17174–17178. doi: 10.1073/pnas.0608311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Routh A, Domitrovic T, Johnson JE. Host RNAs, including transposons, are encapsidated by a eukaryotic single-stranded RNA virus. Proc Natl Acad Sci USA. 2012;109(6):1907–1912. doi: 10.1073/pnas.1116168109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Wang JC, Taylor MW, Zlotnick A. In vitro assembly of an empty picornavirus capsid follows a dodecahedral path. J Virol. 2012;86(23):13062–13069. doi: 10.1128/JVI.01033-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zlotnick A. To build a virus capsid. An equilibrium model of the self assembly of polyhedral protein complexes. J Mol Biol. 1994;241(1):59–67. doi: 10.1006/jmbi.1994.1473. [DOI] [PubMed] [Google Scholar]
- 14.Zlotnick A, Aldrich R, Johnson JM, Ceres P, Young MJ. Mechanism of capsid assembly for an icosahedral plant virus. Virology. 2000;277(2):450–456. doi: 10.1006/viro.2000.0619. [DOI] [PubMed] [Google Scholar]
- 15.Endres D, Zlotnick A. Model-based analysis of assembly kinetics for virus capsids or other spherical polymers. Biophys J. 2002;83(2):1217–1230. doi: 10.1016/S0006-3495(02)75245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweeney B, Zhang T, Schwartz R. Exploring the parameter space of complex self-assembly through virus capsid models. Biophys J. 2008;94(3):772–783. doi: 10.1529/biophysj.107.107284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamalyaria F, Rohlfs R, Schwartz R. Queue-based method for efficient simulation of biological self-assembly systems. J Comput Phys. 2005;204:100–120. [Google Scholar]
- 18.Dykeman EC, Stockley PG, Twarock R. Packaging signals in two single-stranded RNA viruses imply a conserved assembly mechanism and geometry of the packaged genome. J Mol Biol. 2013;425(17):3235–3249. doi: 10.1016/j.jmb.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Dykeman EC, Stockley PG, Twarock R. Dynamic allostery controls coat protein conformer switching during MS2 phage assembly. J Mol Biol. 2010;395(5):916–923. doi: 10.1016/j.jmb.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Bunka DH, et al. Degenerate RNA packaging signals in the genome of Satellite Tobacco Necrosis Virus: Implications for the assembly of a T=1 capsid. J Mol Biol. 2011;413(1):51–65. doi: 10.1016/j.jmb.2011.07.063. [DOI] [PubMed] [Google Scholar]
- 21.Borodavka A, Tuma R, Stockley PG. Evidence that viral RNAs have evolved for efficient, two-stage packaging. Proc Natl Acad Sci USA. 2012;109(39):15769–15774. doi: 10.1073/pnas.1204357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ford RJ, et al. Sequence-specific, RNA-protein interactions overcome electrostatic barriers preventing assembly of satellite tobacco necrosis virus coat protein. J Mol Biol. 2013;425(6):1050–1064. doi: 10.1016/j.jmb.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stockley PG, Ranson NA, Twarock R. A new paradigm for the roles of the genome in ssRNA viruses. Future Virology. 2013;8:531–543. [Google Scholar]
- 24.Qu F, Morris TJ. Encapsidation of turnip crinkle virus is defined by a specific packaging signal and RNA size. J Virol. 1997;71(2):1428–1435. doi: 10.1128/jvi.71.2.1428-1435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim DY, Firth AE, Atasheva S, Frolova EI, Frolov I. Conservation of a packaging signal and the viral genome RNA packaging mechanism in alphavirus evolution. J Virol. 2011;85(16):8022–8036. doi: 10.1128/JVI.00644-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golmohammadi R, Valegård K, Fridborg K, Liljas L. The refined structure of bacteriophage MS2 at 2.8 A resolution. J Mol Biol. 1993;234(3):620–639. doi: 10.1006/jmbi.1993.1616. [DOI] [PubMed] [Google Scholar]
- 27.Dykeman EC, Stockley PG, Twarock R. Building a viral capsid in the presence of genomic RNA. Phys Rev E Stat Nonlin Soft Matter Phys. 2013;87(2):022717. doi: 10.1103/PhysRevE.87.022717. [DOI] [PubMed] [Google Scholar]
- 28.Rolfsson O, Toropova K, Ranson NA, Stockley PG. Mutually-induced conformational switching of RNA and coat protein underpins efficient assembly of a viral capsid. J Mol Biol. 2010;401(2):309–322. doi: 10.1016/j.jmb.2010.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carey J, Uhlenbeck OC. Kinetic and thermodynamic characterization of the R17 coat protein-ribonucleic acid interaction. Biochemistry. 1983;22(11):2610–2615. doi: 10.1021/bi00280a003. [DOI] [PubMed] [Google Scholar]
- 30.Lago H, Parrott AM, Moss T, Stonehouse NJ, Stockley PG. Probing the kinetics of formation of the bacteriophage MS2 translational operator complex: Identification of a protein conformer unable to bind RNA. J Mol Biol. 2001;305(5):1131–1144. doi: 10.1006/jmbi.2000.4355. [DOI] [PubMed] [Google Scholar]
- 31.Morton VL, et al. The impact of viral RNA on assembly pathway selection. J Mol Biol. 2010;401(2):298–308. doi: 10.1016/j.jmb.2010.05.059. [DOI] [PubMed] [Google Scholar]
- 32.Knapman TW, Morton VL, Stonehouse NJ, Stockley PG, Ashcroft AE. Determining the topology of virus assembly intermediates using ion mobility spectrometry-mass spectrometry. Rapid Commun Mass Spectrom. 2010;24(20):3033–3042. doi: 10.1002/rcm.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beckett D, Wu HN, Uhlenbeck OC. Roles of operator and non-operator RNA sequences in bacteriophage R17 capsid assembly. J Mol Biol. 1988;204(4):939–947. doi: 10.1016/0022-2836(88)90053-8. [DOI] [PubMed] [Google Scholar]
- 34.Beckett D, Uhlenbeck OC. Ribonucleoprotein complexes of R17 coat protein and a translational operator analog. J Mol Biol. 1988;204(4):927–938. doi: 10.1016/0022-2836(88)90052-6. [DOI] [PubMed] [Google Scholar]
- 35.Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
- 36.McGhee JD, von Hippel PH. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J Mol Biol. 1974;86(2):469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 37.Eigen M, Biebricher CK, Gebinoga M, Gardiner WC. The hypercycle. Coupling of RNA and protein biosynthesis in the infection cycle of an RNA bacteriophage. Biochemistry. 1991;30(46):11005–11018. doi: 10.1021/bi00110a001. [DOI] [PubMed] [Google Scholar]
- 38.Endres D, Miyahara M, Moisant P, Zlotnick A. Designing two self-assembly mechanisms into one viral capsid protein. Protein Sci. 2005;14:1518–1525. doi: 10.1110/ps.041314405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton WR. 1858. An account of the Icosian calculus. Proc R Ir Acad 6:415–416.
- 40.Levinthal C. 1969. How to fold graciously. Mossbauer Spect Biol Syst 22–24.
- 41.Islam S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11(2):163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 42.Borodavka A, Tuma R, Stockley PG. A two-stage mechanism of viral RNA compaction revealed by single molecule fluorescence. RNA Biol. 2013;10(4):481–489. doi: 10.4161/rna.23838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bancroft JB. The self-assembly of spherical plant viruses. Adv Virus Res. 1970;16:99–134. doi: 10.1016/s0065-3527(08)60022-6. [DOI] [PubMed] [Google Scholar]
- 44.Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA. 1999;96(24):13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daszak P, Cunningham AA, Hyatt AD. Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science. 2000;287(5452):443–449. doi: 10.1126/science.287.5452.443. [DOI] [PubMed] [Google Scholar]
- 46.Zlotnick A, Porterfield JZ, Wang JC. To build a virus on a nucleic acid substrate. Biophys J. 2013;104(7):1595–1604. doi: 10.1016/j.bpj.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagan MF, Elrad OM, Jack RL. Mechanisms of kinetic trapping in self-assembly and phase transformation. J Chem Phys. 2011;135(10):104115. doi: 10.1063/1.3635775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stockley PG, et al. Packaging signals in single-stranded RNA viruses: nature’s alternative to a purely electrostatic assembly mechanism. J Biol Phys. 2013;39(2):277–287. doi: 10.1007/s10867-013-9313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ling CM, Hung PP, Overby LR. Independent assembly of Qbeta and MS2 phages in doubly infected Escherichia coli. Virology. 1970;40(4):920–929. doi: 10.1016/0042-6822(70)90138-8. [DOI] [PubMed] [Google Scholar]
- 50.Peabody DS. Role of the coat protein-RNA interaction in the life cycle of bacteriophage MS2. Mol Gen Genet. 1997;254(4):358–364. doi: 10.1007/s004380050427. [DOI] [PubMed] [Google Scholar]
- 51.Dent KC, Barr JN, Hiscox JA, Stockley PG, Ranson NA. The asymmetric structure of an icosahedral virus bound to its receptor suggests a mechanism for genome release. Structure. 2013;21(7):1225–1234. doi: 10.1016/j.str.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larson SB, McPherson A. Satellite tobacco mosaic virus RNA: Structure and implications for assembly. Curr Opin Struct Biol. 2001;11(1):59–65. doi: 10.1016/s0959-440x(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 53.Rao VB, Black LW. Structure and assembly of bacteriophage T4 head. Virol J. 2010;7:356. doi: 10.1186/1743-422X-7-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valley CT, et al. Patterns and plasticity in RNA-protein interactions enable recruitment of multiple proteins through a single site. Proc Natl Acad Sci USA. 2012;109(16):6054–6059. doi: 10.1073/pnas.1200521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.