

Abstract
The description of aggregation processes with molecular dynamics simulations is a playground for testing biomolecular force fields, including a new generation of force fields that explicitly describe electronic polarization. In this work, we study a system consisting of 50 glycyl-l-alanine (Gly-Ala) dipeptides in solution with 1001 water molecules. Neutron diffraction experiments have shown that at this concentration, Gly-Ala aggregates into large clusters. However, general-purpose force fields in combination with established water models can fail to correctly describe this aggregation process, highlighting important deficiencies in how solute–solute and solute–solvent interactions are parametrized in these force fields. We found that even for the fully polarizable AMOEBA force field, the degree of association is considerably underestimated. Instead, a fixed point-charge approach based on the newly developed IPolQ scheme [Cerutti et al. J. Phys. Chem.2013, 117, 2328] allows for the correct modeling of the dipeptide aggregation in aqueous solution. This result should stimulate interest in novel fitting schemes that aim to improve the description of the solvent polarization effect within both explicitly polarizable and fixed point-charge frameworks.
1. Introduction
Molecular dynamics (MD) is a well-established computational tool to model the behavior of molecular systems in chemistry, biology, and material sciences. To allow atomistic computer simulations to access relevant system sizes and time-scales, a common approximation is to describe electrostatic interactions using fixed point-charges that are centered on the atoms. This computationally inexpensive approach leads to satisfactory results for a wide variety of applications that do not require detailed knowledge of the electronic structure. However, an intrinsic limitation of the fixed point-charge model is that it captures many-body effects, such as electronic polarization, only in a mean-field way. Electronic polarization is caused by the rearrangement of electron density in response to changes in its environment and is often considered to be the main current challenge for reaching chemical accuracy (errors <1 kcal/mol) in biomolecular simulations.1,2 For this reason, a new generation of force fields is emerging with functional forms that are more complex and explicitly include a polarization energy term. A successful example is the AMOEBA (atomic multipole optimized energetics for biomolecular applications) polarizable force field developed by Ponder and Ren,3 which replaces the fixed partial charge model with polarizable atomic multipoles through quadrupole moments. AMOEBA has been demonstrated to outperform nonpolarizable force fields, for example, in describing solvation free energies of drug-like small molecules and dynamical properties away from ambient conditions,2 as well as active sites of metalloenzymes.4
In view of the availability and continued development of polarizable force fields, it is a good time to assess whether the fixed point-charge model of nonpolarizable force fields remains a viable alternative. When choosing between a polarizable and nonpolarizable force field, one has to consider to what extent the simplified point-charge model will be able to properly describe the system of interest which by nature is quantum mechanical. For water, for example, it is possible through force matching5 to produce a nonpolarizable model that accurately reproduces bulk properties obtained from a fully polarizable water model.6 As another example, pairwise additive potentials were shown to accuractely describe the dissociation profile of Na–Cl in water, as calculated by ab initio MD.7 In addition, nonpolarizable force fields may remain an attractive option simply due to their simplicity and efficiency.
The most common approach to determine atomic charges for a nonpolarizable force field is based on quantum mechanical (QM) calculations and a procedure termed restrained electrostatic potential (RESP) fit.8 RESP point-charges are optimized to reproduce the electrostatic potential at regions in space that are relevant to reproducing typical intermolecular interactions (such as the surface of the molecules defined by the van der Waals (vdW) radii). The reference QM calculations are often performed in the gas phase, while the RESP charges are then used for condensed phase simulations within an effective two-body additive model that is supposed to implicitly represent polarization. In an attempt to capture the polarizing effect of the condensed phase environment, it is thus common to choose a QM method that leads to overpolarization and overestimates the gas phase charges.1 Instead of this ad hoc approximation, it would be better to derive RESP charges from a condensed phase QM reference.9 However, in this case, the electrostatic energy of the system would be overestimated due to the lack of a “self-polarization” energy term in nonpolarizable force fields. Self-polarization is sometimes called the missing energy term in conventional force fields,10 and it corresponds to an energy penalty that is required to rearrange the electron density and create the polarized system.
An alternative to RESP fits is the supermolecular approach in which the charges are optimized to reproduce geometries and interaction energies of reference QM calculations for the model compound interacting with individual solvent molecules, usually a single water molecule.11 In this approach, the interaction with the solvent molecule in the QM calculation leads to local electronic polarization, which is thus implicitly included in the obtained charges. Similar to the RESP approach, it is common to select a QM method that leads to overpolarization in order to implicitly account for bulk polarization in the condensed phase.
Recently, there has been some discussion in the literature about the best strategy to obtain polarized charges for condensed phase simulations.12−15 Leontyev and Stuchebrukhov proposed a charge scaling approach based on a continuum electrostatics argument for ions and ionized groups.15 Scaling the charges by a factor of 0.7 was shown to improve the agreement of MD simulations with neutron scattering experiments for systems that otherwise incorrectly aggregate in the simulations, including concentrated solutions of atomic and small molecular ions.16,17 However, it remains to be seen if this simple rescaling approach will also work for complex systems that show too little aggregation in MD simulations, such as the one studied here.
Karamertzanis et al. have proposed a different approach in which the self-polarization penalty is approximated by using charges that are an average between the gas phase and the condensed phase values. This effectively spreads the energy penalty throughout all interacting pairs of atoms.14 This idea was used in a recent contribution by Cerutti et al.12 to derive charges for a new AMBER18 force field. The novel scheme, termed IPolQ (for “implicitly polarized charges”), includes the polarizing effect of the solvent by performing an iterative, self-consistent optimization of the charges on a molecular fragment that is embedded in a potential generated by an ensemble of surrounding water molecules. Classical MD simulations are used to sample both the ligand and solvent degrees of freedom in order to obtain a representative average. The missing self-polarization energy term is implicitly included in IPolQ charges by calculating an average charge between the QM RESP values in solution and in the gas phase. In this scheme, the electrostatic potential produced by the new charges is no longer exact; however, the electrostatic energy of the system is likely to be improved. Finally, the authors also proposed to reoptimize nonbonded vdW parameters to optimize hydration free energies. An important feature of the IPolQ charges is that they are derived in the presence of the solvent and are therefore fully consistent with the water model used during the parametrization procedure.
In this contribution, we focus on simulating the aggregation process of the zwitterionic dipeptide glycyl-l-alanine (Gly-Ala) in aqueous solution with different force fields. A balanced description of the interactions of the dipeptides among themselves and with the water molecules is important to accurately model the aggregation behavior and by extension is of high relevance for describing protein–ligand binding and protein–protein interactions. Previously, McLain et al.19,20 used neutron scattering to determine the aggregation behavior of Gly-Ala in combination with empirical potential structure refinement (EPSR). EPSR involves MD simulations with a force field that is empirically modified until the simulations are able to reproduce experimental diffraction patterns at which point the molecular structure and aggregation pattern can be extracted from the simulations. Parameters from the AA-OPLS21 force field were used for the dipeptide as a starting point of the refinement process to simulate the aggregation process in combination with the SPC/E10 water model. While the final agreement with experiments shows that a pairwise additive force field can in principle accurately describe the aggregation process, general-purpose force fields in their default parametrization are not necessarily able to do so. For instance, Tulip and Bates analyzed results from MD simulations with the CHARMM2222 force field and three different water models and observed less aggregation than derived from the experiment.23 Similarly, Kucukkal and Stuart24 found a disappointing correlation with experiments when conducting MD simulations of the Gly-Ala, Gly-Pro, and Ala-Pro dipeptides, with either the polarizable CHARMM3025,26 or the fixed point-charge CHARMM22 force fields for the dipeptides, in combination with the polarizable TIP4P-FQ27 and fixed point-charge TIP3P28 water models, respectively. In the present contribution, we show that a similar problem exists for the AMBER ff12SB force field, which differs from the AMBER ff99SB18,29 force field only in backbone and side chain torsion parameters but uses the same RESP derived charges. We also show that the fully polarizable AMOEBA2 force field is not able to describe the aggregation process. We find that results obtained with the CHARMM3630 force field, which uses a supermolecular approach for the charge derivation, are in better agreement with the EPSR model. Finally, we discuss results from simulations with the AMBER force field in combination with IPolQ charges that show a clear improvement in the description of the system—for the aggregation process and other structural properties such as ensemble-averaged site–site radial distribution functions, g(r)—when compared to the EPSR model that is fitted to neutron scattering data.
2. Methods
All fixed point-charge force field simulations were performed with release 12.3 of the GPU accelerated version31−33 of the AMBER34,35 software package. The AMBER ff12SB force field, which includes revised backbone and side chain torsion parameters over ff99SB,18,29 as well as the CHARMM3630 force field were used for the dipeptides and the TIP3P28 and TIP4P-Ew36 models for water. For the IPolQ and IPolQ+vdW simulations, the charges and selected vdW parameters of the AMBER ff12SB force field were modified as described in the corresponding publication12 and in the Supporting Information. The CHAMBER program37 was used to convert CHARMM topology, parameter, and coordinate files into AMBER format.
A total of 50 glycyl-l-alanine dipeptide molecules were solvated with 1001 water molecules in a cubic box of 34.2 Å side length, representing the experimental density of 0.1 atoms per Å3. All simulations were performed in the isothermal–isobaric ensemble (NpT) using Langevin dynamics38 with a collision frequency of 5 ps–1 and a target temperature of 300 K and the Berendsen barostat39 with a target pressure of 1 bar and a pressure relaxation time of 1 ps. A time step of 2.0 fs was used with bond distances to hydrogen atoms constrained using the SHAKE40,41 algorithm. A cutoff of 9 Å was used for the real-space nonbonded interactions, and the particle mesh Ewald (PME) algorithm42 was used to account for long-range electrostatics beyond the cutoff. Simulations were run for a total of 100 ns, and the first 10 ns were considered equilibration time and discarded from any analysis.
All AMOEBA force field2 MD simulations were performed in the NpT ensemble at 300 K and 1.0 bar, using the OpenMM43,44 Python-based application layer. Periodic boundary conditions were used, along with a nonbonded interaction cutoff of 10 Å. A time step of 1.0 fs was used for the AMOEBA simulations while no constraints were used. Mutual polarization was used along with an induced target epsilon of 0.01 after checking convergence of the simulations with respect to epsilon. Energy conservation was monitored, and none of the simulations showed energy drift. Simulations were run for a total of 10 ns simulation time and the first 1 ns discarded from any analysis. The convergence of the simulations (see Supporting Information for details) was carefully monitored and tested by means of comparison to accelerated MD45 (aMD) simulations using the AMOEBA force field as implemented in OpenMM.4 The acceleration levels of accelerated MD have been chosen on the basis of the empirical equation presented by Lindert et al.46
For validation, all simulations were also performed in the canonical ensemble (NVT) at the experimental density. These results are collected in the Supporting Information. It is worth mentioning that the results for both ensembles are very similar for all force fields, with NVT simulations leading to slightly increased structure in the relevant radial distribution functions as compared to NpT simulations.
3. Results and Discussion
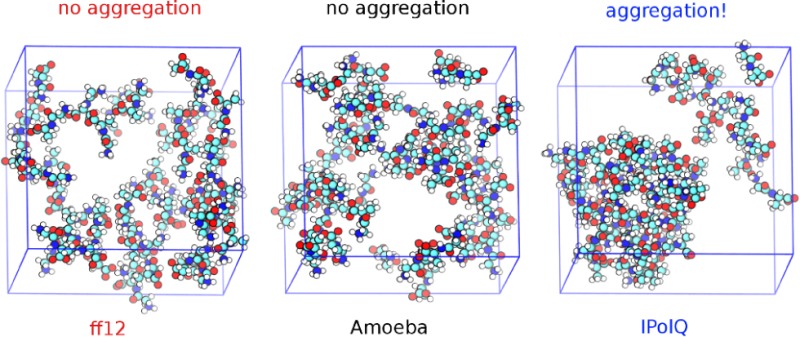
Neutron scattering experiments by McLain et al. suggest that Gly-Ala dipeptides aggregate in concentrated solutions with high probabilities for the formation of large dipeptide clusters.19 A probability of around 90% was found for a fully percolating cluster that contains all 50 Gly-Ala dipeptides of the experimentally derived EPSR model. Single Gly-Ala dipeptides and dimers were also observed while clusters of sizes 5 to 45 were essentially absent. Figure 1 shows representative snapshots from our MD trajectories of Gly-Ala dipeptides at the experimental concentration in water, highlighting the difference in aggregation behavior between the different force fields. Both the standard AMBER ff12SB force field and the polarizable AMOEBA force field lead to an unstructured solution with little cluster formation, while using IPolQ charges in combination with the AMBER ff12SB force field leads to clustering as observed experimentally. This suggests that the balance of solute–solute and solute–water interactions is improved in the IPolQ charge model, while the more complex many-body AMOEBA potential does not lead to a qualitatively better description of the interactions governing (de)solvation and peptide aggregation.
Figure 1.
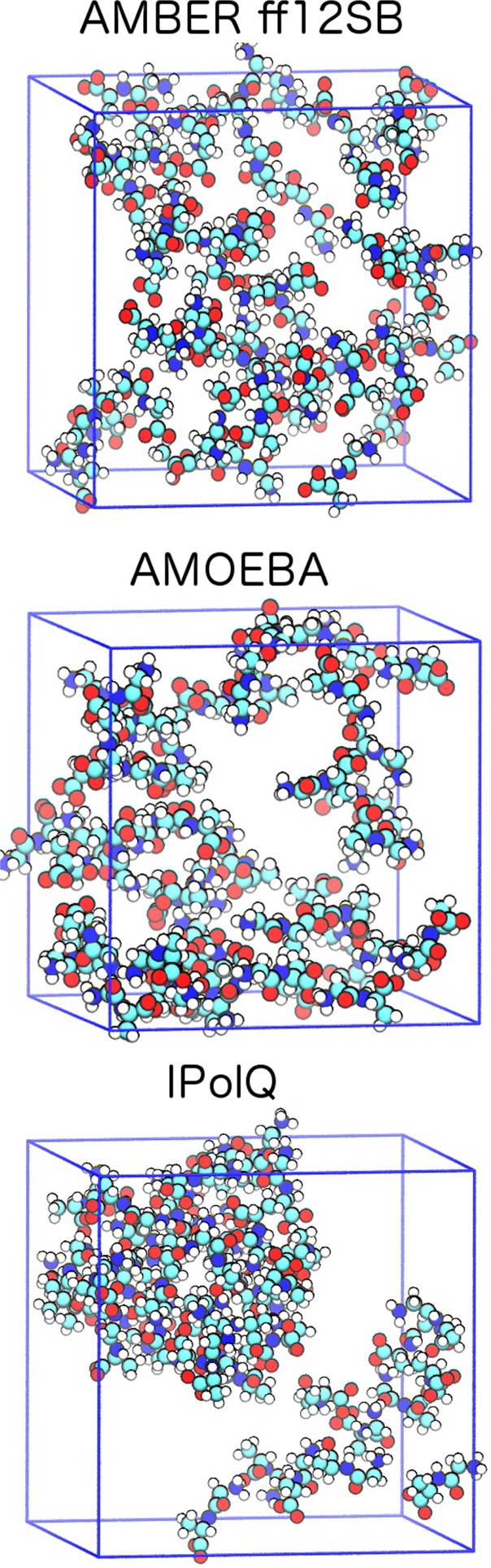
Representative snapshots from MD trajectories showing the aggregation behavior of glycyl-l-alanine dipeptides. The standard AMBER ff12SB force field (top) and the polarizable AMOEBA force field (center) lead to unstructured solutions, while the use of IPolQ charges with the AMBER ff12SB force field leads to experimentally observed clustering. Water molecules are omitted for clarity.
The cluster formation of the Gly-Ala dipeptides is believed to be mainly driven by interactions between charged, hydrophilic groups as opposed to contacts between hydrophobic groups.19 The most pronounced interactions are between the charged NH3 and CO2 end groups of the peptide fragments which can be characterized by the radial distribution function g(rOC–HX). The interactions between the hydrophobic side chain methyl groups in the Gly-Ala dipeptides can be characterized by g(rCB–CB). For the nomenclature used for the labeling of atoms in this work, see Figure 2.
Figure 2.

Molecular structure of the dipeptide glycyl-l-alanine with the labeling scheme used in this work.
Experimental results for the radial distribution functions g(rOC–HX) and g(rCB–CB) from the work of McLain et al.19 are shown in Figures 3 and 4, respectively, along with results from our NpT simulations.
Figure 3.
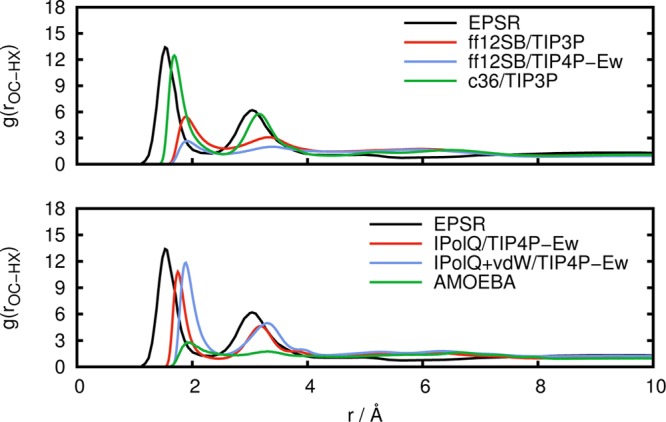
Radial distribution function g(rOC–HX) between carboxylate oxygen atoms and amine hydrogen atoms obtained from experiment (EPSR) in comparison to simulations with standard fixed point-charge force fields (top) and with IPolQ derived charges and the polarizable AMOEBA force field (bottom).
Figure 4.
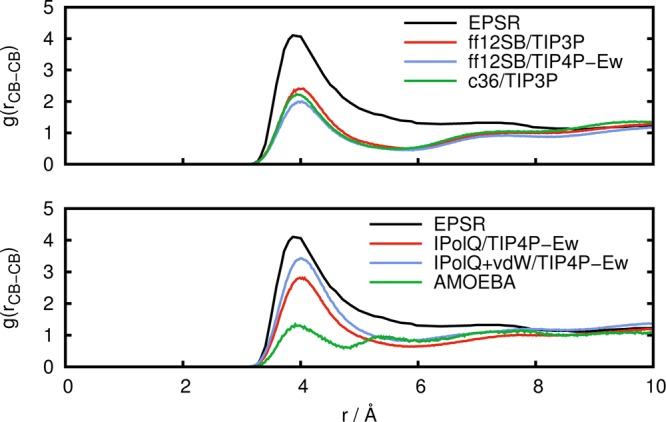
Radial distribution function g(rCB–CB) between alanine side chain methyl carbon atoms obtained from experiment (EPSR) in comparison to simulations with standard fixed point-charge force fields (top) and with IPolQ derived charges and the polarizable AMOEBA force field (bottom).
The experimental EPSR results in Figure 3 show that pronounced interactions occur between the carboxy and amine functional groups, underlining that the association is driven by interactions between these charged groups. The standard fixed point-charge AMBER ff12SB force field predicts a significantly understructured g(rOC–HX), both with the TIP3P water model and to an even larger extent with the TIP4P-Ew water model, which is in line with the lack of cluster formation with this force field. In comparison to the EPSR model, the peaks in the radial distribution function are also shifted to larger values. Similar observations were made by others for the nonpolarizable AA-OPLS and CHARMM22 force fields19,23,24 with different water models, indicating that commonly employed procedures to derive parameters for fixed point-charge force fields are in general not appropriate to describe aggregation of peptides in water. Figure 3 shows that the CHARMM36 force field (c36) in combination with the TIP3P water model leads to results that are in good agreement with the EPSR reference, predicting a somewhat understructured radial distribution function. The use of IPolQ charges that were optimized with the TIP4P-Ew water model leads to a significant improvement of the results as compared to the underlying AMBER ff12SB force field, bringing g(rOC–HX) into closer agreement with the EPSR results. Thus, the IPolQ charges, which can be considered to be the optimal fixed point-charges for the hydrated Gly-Ala peptide, do indeed lead to an improved description of the balance between solute–solute and solute–water interactions. The IPolQ charges lead to amino acids with increased polarity as compared to the standard AMBER ff12SB force field with significantly larger charges on the atoms of the amino end group (details of the differences between IPolQ and ff12SB are discussed in the Supporting Information). This observation alone can explain the increased tendency for aggregation; however, it is important to stress that it is the subtle balance between the interactions driving (de)solvation and aggregation that is required to obtain a realistic description of the cluster formation. Using the modified vdW terms that were optimized to improve hydration free energies in combination with the IPolQ charges12 (denoted as IPolQ+vDW) has a relatively modest effect on g(rOC–HX). Despite successes in other cases,2,4 the polarizable AMOEBA force field fails to reproduce any structure in this radial distribution function, predicting an unstructured solution of Gly-Ala dipeptides without aggregation. The first peak in the radial distribution function g(rOC–HX) is barely present with AMOEBA, and the second peak is absent. This is surprising since one would hope that the inclusion of polarizability in the force field would improve the agreement with experimental results. Instead, the results are worse than those of common fixed point-charge force fields. A similar observation was made for the polarizable CHARMM30 force field in combination with the polarizable TIP4P-FQ water model,24 suggesting that current polarizable force fields are not yet a safe general replacement of pairwise additive force fields.
Figure 4 shows results for the radial distribution function g(rCB–CB) between alanine side chain methyl atoms. In agreement with the results for g(rOC–HX) from Figure 3, the AMBER ff12SB force field predicts less structure than determined from experimental results; however, the CHARMM36 force field does not perform better here. The agreement improves when IPolQ charges are employed, and additional use of the vdW terms that were optimized for use with IPolQ charges further improves the agreement with the EPSR model. The lack of dipeptide aggregation with AMOEBA results in an understructured g(rCB–CB) which is in line with the results for g(rOC–HX) that showed a disagreement between AMOEBA and the experimental EPSR results.
We now turn to an analysis of the structure of the water surrounding the Gly-Ala dipeptide molecules, focusing on the carboxylate and amino groups of the Gly-Ala dipeptides since interactions between these groups are the dominant driving force for aggregation. Results for the radial distribution functions g(rOC–HW) and g(rOC–OW) are shown in Figures 5 and 6, respectively. These correspond to the distances between the carboxylate oxygen atoms and the water hydrogen or oxygen atoms, respectively. Results for the radial distribution function g(rHX–OW) between the amino hydrogen atoms and water oxygen atoms are shown in Figure 7.
Figure 5.
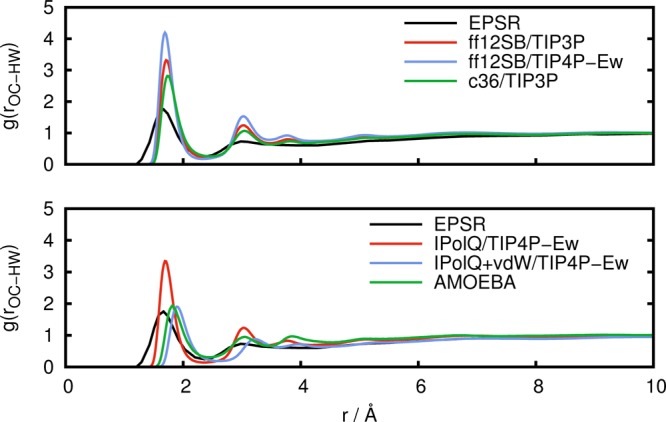
Radial distribution function g(rOC–HW) between carboxylate oxygen atoms and water hydrogen atoms obtained from experimental results (EPSR) in comparison to simulations with standard fixed point-charge force fields (top) and with IPolQ derived charges and the polarizable AMOEBA force field (bottom).
Figure 6.
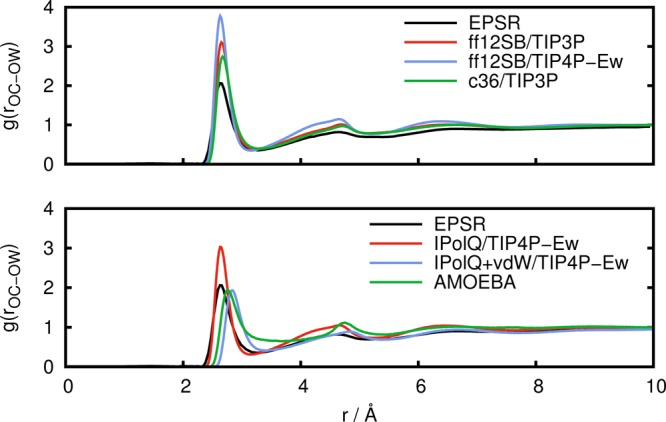
Radial distribution function g(rOC–OW) between carboxylate oxygen atoms and water oxygen atoms obtained from experimental results (EPSR) in comparison to simulations with standard fixed point-charge force fields (top) and with IPolQ derived charges and the polarizable AMOEBA force field (bottom).
Figure 7.
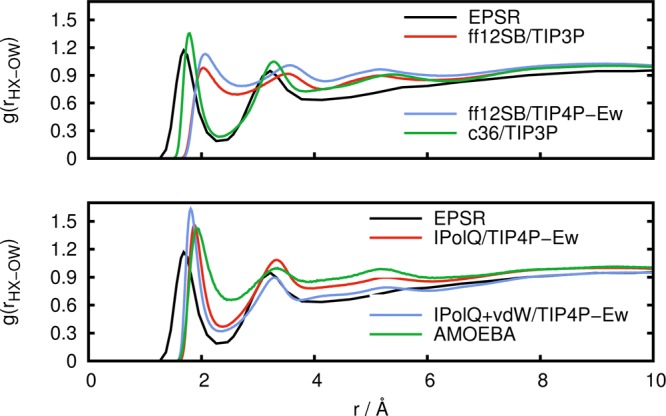
Radial distribution function g(rHX–OW) between amine hydrogen atoms and water oxygen atoms obtained from experimental results (EPSR) in comparison to simulations with standard fixed point-charge force fields (top) and with IPolQ derived charges and the polarizable AMOEBA force field (bottom).
From Figures 5 and 6, we can see that in comparison to the experimental data, the CHARMM36 force field predicts a coordination of water to the carboxylate group that is too large. The same holds for the AMBER ff12SB force field, both with the TIP3P and to a larger extent with the TIP4P-Ew water model. This indicates that the solute–solvent interactions are too strong, making desolvation unfavorable and hindering aggregation. Again, the use of IPolQ charges improves the agreement with experimental results, and the combination of IPolQ charges with the optimized vdW terms leads to further improvements. The AMOEBA force field also shows good agreement with the experimental results for the structure of water surrounding the carboxylate group, with results that are very similar to IPolQ+vdW. This good agreement between AMOEBA and experimental results for the water structure around the carboxylate group is in contrast with the inability of AMOEBA to predict Gly-Ala aggregation manifested through the lack of structure in the radial distribution functions g(rOC–HX) and g(rCB–CB), see Figures 3 and 4. Since the solute–solvent interactions seem to be accurately modeled by AMOEBA, this suggests that AMOEBA underestimates the solute–solute interactions, that is, the electrostatic interactions between the zwitterionic dipeptides.
Equally important is the water structure around the charged amino group. From Figure 7, we can see that the CHARMM36 force field predicts a radial distribution function g(rHX–OW) that is very close to the reference EPSR data. The AMBER ff12SB force field, however, is not able to reproduce the experimental solvent structure around the amino group, neither with the TIP3P nor with the TIP4P-Ew water model. The peaks in the radial distribution function are shifted to the right, and the minimum around 2.3 Å is barely present. In contrast, both IPolQ and IPolQ+vdW are in better agreement with the experiment, only slightly overestimating the first peak but improving the position of both peaks and the density minimum. AMOEBA leads to a clear improvement over the AMBER ff12SB force field but leads to a local water density that is too high over the whole range as compared to experimental results, CHARMM36, IPolQ, or IPolQ+vdW.
4. Conclusion
In this work, MD simulations of a concentrated solution of the zwitterionic glycyl-l-alanine dipeptide highlight areas of possible improvement in the polarizable AMOEBA force field, which fails to describe the aggregation behavior of Gly-Ala dipeptides into large clusters at high concentrations in aqueous solutions as determined from neutron scattering experiments by McLain et al.19 The cluster formation is determined by a subtle balance between solute–solvent interactions governing the desolvation process and solute–solute interactions. AMOEBA is able to describe the hydration structure of the peptides rather well. Thus, it appears that the current parametrization of AMOEBA properly describes the solute–solvent interactions while underestimating the interactions among the peptides.
Our simulations show that the AMBER ff12SB force field, which is widely used for biomolecular simulations in explicit solvent, is also unable to describe the experimentally observed aggregation of Gly-Ala. This result has potential implications for many studies that involve (de)solvation and charged proteins and ligands (including drug design and the study of aggregation processes occurring in diseases such as Creutzfeldt–Jakob or Alzheimer diseases). We find that the ff12SB force field overestimates the coordination number of water around the carboxylate and amino groups, both with the TIP3P water model and to an even larger extent with the TIP4P-Ew water model. This overestimation of the peptide–water interactions makes desolvation unfavorable and contributes to the observed lack of cluster formation. This observation is in agreement with results from other groups for the established AA-OPLS and CHARMM nonpolarizable force fields.19,23,24
Finally, a key result of our study is that the aggregation can be described with a fixed point-charge model. We find that the latest nonpolarizable CHARMM36 force field results in a radial distribution function between the oppositely charged amino and carboxylate groups of the zwitterionic peptide that closely resembles the experimental reference, although this force field clearly overhydrates the carboxylate group. The use of IPolQ charges leads to the correct aggregation behavior and also improves the structure of water around carboxylate and amino groups, thus representing an improvement over the standard AMBER ff12 SB force field. In our opinion, the main reason for the success of the IPolQ scheme12 as compared to the regular RESP derived charges is that it includes the polarizing effect of the solvent in a self-consistent fashion (that is consistent with the solvent model), while implicitly correcting for the missing self-polarization energy term. Thus, the IPolQ charges are possibly the optimal nonpolarizable point-charges for describing the electrostatic energy of a hydrated solute in condensed phase MD simulations. Problems may of course still arise if parts of a system parametrized with IPolQ charges experience a completely different dielectric environment, for example by getting buried in the hydrophobic core of a protein during the course of a simulation, which should be addressed by an explicitly polarizable force field. Nevertheless, it seems that the IPolQ protocol is a significant step forward which should stimulate continued interest in fitting schemes that aim to improve the description of the solvent polarization effect within a fixed point-charge framework. It will be interesting to see how these new charges perform for important biological processes that involve desolvation and solute–solute interactions. Optimized point-charges could offer significant improvements for free energies that describe binding of drug-like molecules to proteins.47,48 Furthermore, IPolQ may help model highly charged biological systems, such as ion channels, that have so far been challenging to accurately model with pairwise additive force fields.49,50
Acknowledgments
A.W.G. would like to acknowledge discussions with William Swope and Julia Rice that planted the seed for the idea to use IPolQ for the simulations presented here and David Cerutti for clarifications about the IPolQ parameters. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (NSF) grant number OCI-1053575. Computer time was provided through an NSF XSEDE award TG-CHE120054 to A.W.G. The J.A.M. group is supported by NSF, NIH, HHMI, NBCR, and CTBP.
Supporting Information Available
A description of the modified charges and vdW terms used with IpolQ and IPolQ+vdW, respectively. Results obtained from simulations in the canonical ensemble (NVT) at the experimental density. Plots of radial distribution functions demonstrating the convergence of the simulations. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Author Contributions
⊥ Contributed equally to this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ponder J. W.; Case D. A. Adv. Protein Chem. 2003, 66, 27–85. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.; Wu C.; Ren P.; Pande V. S.; Chodera J. D.; Schnieders M. J.; Haque I.; Mobley D. L.; Lambrecht D. S.; DiStasio R. A. Jr.; Head-Gordon M.; Clark G. N.; Johnson M. E.; Head-Gordon T. J. Phys. Chem. B 2010, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P.; Ponder J. W. J. Comput. Chem. 2002, 23, 1497–506. [DOI] [PubMed] [Google Scholar]
- Lindert S.; Bucher D.; Eastman P.; Pande V.; McCammon J. A. J. Chem. Theory Comput. 2013, 9, 4684–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izvekov S.; Parrinello M.; Burnham C. J.; Voth G. A. J. Chem. Phys. 2004, 120, 10896–913. [DOI] [PubMed] [Google Scholar]
- Iuchi S.; Izvekov S.; Voth G. A. J. Chem. Phys. 2007, 126, 124505. [DOI] [PubMed] [Google Scholar]
- Timko J.; Bucher D.; Kuyucak S. J. Chem. Phys. 2010, 132, 114510. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W. D.; Kollman P. A. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar]
- Bucher D.; Guidoni L.; Maurer P.; Röthlisberger U. J. Chem. Theory Comput. 2009, 5, 2173–2179. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar]
- Mackerell A. D. J. Comput. Chem. 2004, 25, 1584–604. [DOI] [PubMed] [Google Scholar]
- Cerutti D. S.; Rice J. E.; Swope W. C.; Case D. A. J. Phys. Chem. B 2013, 117, 2328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope W. C.; Horn H. W.; Rice J. E. J. Phys. Chem. B 2010, 114, 8621–30. [DOI] [PubMed] [Google Scholar]
- Karamertzanis P. G.; Raiteri P.; Galindo A. J. Chem. Theory Comput. 2010, 6, 1590–1607. [DOI] [PubMed] [Google Scholar]
- Leontyev I.; Stuchebrukhov A. Phys. Chem. Chem. Phys. 2011, 13, 2613–26. [DOI] [PubMed] [Google Scholar]
- Mason P. E.; Wernersson E.; Jungwirth P. J. Phys. Chem. B 2012, 116, 8145–8153. [DOI] [PubMed] [Google Scholar]
- Vazdar M.; Jungwirth P.; Mason P. E. J. Phys. Chem. B 2013, 117, 1844–1848. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- McLain S. E.; Soper A. K.; Daidone I.; Smith J. C.; Watts A. Angew. Chem., Int. Ed. 2008, 47, 9059–62. [DOI] [PubMed] [Google Scholar]
- McLain S. E.; Soper A. K.; Watts A. Eur. Biophys. J. 2008, 37, 647–55. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. J. Am. Chem. Soc. 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; et al. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Tulip P. R.; Bates S. P. J. Chem. Phys. 2009, 131, 015103. [DOI] [PubMed] [Google Scholar]
- Kucukkal T. G.; Stuart S. J. J. Phys. Chem. B 2012, 116, 8733–40. [DOI] [PubMed] [Google Scholar]
- Patel S.; Brooks C. L. III. J. Comput. Chem. 2004, 25, 1–15. [DOI] [PubMed] [Google Scholar]
- Patel S.; MacKerell A. D. Jr.; Brooks C. L. III. J. Comput. Chem. 2004, 25, 1504–14. [DOI] [PubMed] [Google Scholar]
- Rick S. W.; Stuart S. J.; Berne B. J. J. Chem. Phys. 1994, 101, 6141–6156. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. J. Comput. Chem. 2013, 34, 2135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. J. Chem. Theory Comput. 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S.; Götz A. W.; Walker R. C. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. J. Chem. Theory Comput. 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- Case D. A.et al. AMBER 12; University of California: San Francisco, CA, 2012. [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar]
- Horn H. W.; Swope W. C.; Pitera J. W.; Madura J. D.; Dick T. J.; Hura G. L.; Head-Gordon T. J. Chem. Phys. 2004, 120, 9665–78. [DOI] [PubMed] [Google Scholar]
- Crowley M. F.; Williamson M. J.; Walker R. C. Int. J. Quantum Chem. 2009, 109, 3767–3772. [Google Scholar]
- Loncharich R. J.; Brooks B. R.; Pastor R. W. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Miyamoto S.; Kollman P. A. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- Darden T.; York D.; Pedersen L. J. Chem. Phys. 1993, 98, 10090–10092. [Google Scholar]
- Eastman P.; et al. J. Chem. Theory Comput. 2013, 9, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman P.; Pande V. S. Comput. Sci. Eng. 2010, 12, 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D.; de Oliveira C. A.; McCammon J. A. J. Chem. Phys. 2007, 127, 155102. [DOI] [PubMed] [Google Scholar]
- Lindert S.; Kekenes-Huskey P. M.; Huber G.; Pierce L.; McCammon J. A. J. Phys. Chem. B 2012, 116, 8449–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar D.; Harder E.; Damm W.; Friesner R.; Sherman W. J. Chem. Theory Comput. 2012, 8, 2553–2558. [DOI] [PubMed] [Google Scholar]
- Mayne C. G.; Saam J.; Schulten K.; Tajkhorshid E.; Gumbart J. C. J. Comput. Chem. 2013, 34, 2757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D.; Guidoni L.; Carloni P.; Röthlisberger U. Biophys. J. 2010, 98, L47–L49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. Ø.; Jogini V.; Eastwood M. P.; Shaw D. E. J. Gen. Physiol. 2013, 141, 619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.