

Abstract
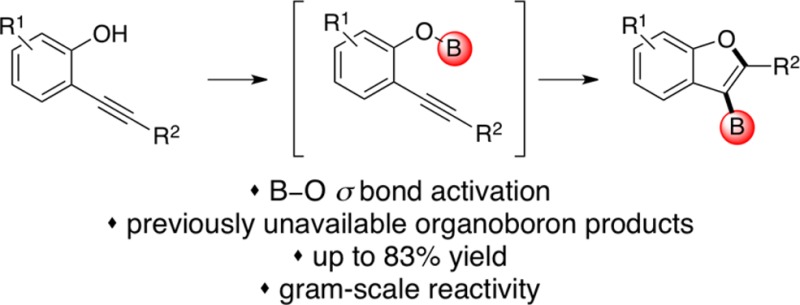
For nearly 70 years, the addition of boron–X σ bonds to carbon–carbon multiple bonds has been employed in the preparation of organoboron reagents. However, the significantly higher strength of boron–oxygen bonds has thus far precluded their activation for addition, preventing a direct route to access a potentially valuable class of oxygen-containing organoboron reagents for divergent synthesis. We herein report the realization of an alkoxyboration reaction, the addition of boron–oxygen σ bonds to alkynes. Functionalized O-heterocyclic boronic acid derivatives are produced using this transformation, which is mild and exhibits broad functional group compatibility. Our results demonstrate activation of this boron–O σ bond using a gold catalysis strategy that is fundamentally different from that used previously for other boron addition reactions.
Introduction
Boronic acids and their derivatives are versatile reagents in modern organic synthesis, and the hydroboration reaction is a well-established method for generating these building blocks through the addition of B–H bonds across C–C multiple bonds.1 First described by Hurd2 in 1948 and later developed in detail by Brown,3 this reaction has inspired many catalyzed variants.4,5 Recently, several compelling examples of related B–X bond addition reactivity have been reported for X = C,6,7 Si,5,8,9 Sn,5,10 S,11 B,5,12 Cl,13 Br,14 and I14 (Figure 1a). Many of these transformations proceed through the oxidative addition of a catalytic transition metal such Ni(0), Pd(0), or Pt(0) into the B–X σ bond.
Figure 1.

(a) Previous work developing addition of B–X bonds across alkynes. (b) This work demonstrating the first addition of B–O bonds across alkynes.
Despite this progress, the corresponding activation of B–O bonds and addition to C–C multiple bonds—alkoxyboration—has remained elusive for 65 years.15,16 This striking dearth of B–O bond activation reactivity may be due to the extremely high strength of the B–O bond, ∼136 kcal/mol, compared to less than 105 kcal/mol for all others entries in the series.17 This high stability may render the B–O bond unreactive toward oxidative addition, thus preventing the successful application of Ni, Pd, or Pt catalysis6−11 in an alkoxyboration reaction. Organoboron reagents are the building blocks of choice for medicinal chemistry and drug discovery.18 Given that ethers are found in many diverse classes of natural products19 and in nearly 25% of the top-grossing pharmaceuticals in the United States for 2012,20 the development of such a transformation allows for the preparation of oxygen-containing building blocks useful in drug discovery and materials science.20,21
Results and Discussion
Herein we report the realization of an alkoxyboration reaction of alkynes (Figure 1b), through which new O- heterocyclic organoboronate coupling partners are available for downstream functionalization. The high functional group tolerance of this reaction enables downstream divergent synthesis of functionalized benzofurans—the ability to access multiple benzofurans from one bench stable precursor. In contrast, current methods for synthesizing benzofurans often rely on harsh conditions that limit compatibility with functional groups desirable for divergent synthesis.22
We envisioned that the desired alkoxyboration reactivity could be promoted through an activation pathway employing a bifunctional Lewis acidic/Lewis basic catalyst, which could simultaneously activate both the alkyne and the B–O σ bond partners. We anticipated that this unique strategy could allow for the anti addition of B–O bonds across alkynes by circumventing the previous problematic strategy of oxidative cleavage of the B–O bond.
Our optimized one-pot procedure begins with 2-alkynyl phenols (1), which are converted into the requisite boric ester intermediate 2 using the readily available reagent B-chlorocatecholborane (Figure 2). Treatment of this intermediate with the commercially available Lewis acidic gold(I) precatalyst IPrAuCl and NaTFA affords alkoxyboration product 3 in good to excellent conversion as determined by the ERETIC method.23 Interestingly, our examination of alternative π-Lewis acidic transition metal catalysts revealed no other active catalysts aside from Au(I).24 For synthetic ease, the catechol boronic ester alkoxyboration product 3 was converted into either the organotrifluoroborate25 or N-methyliminodiacetic acid (MIDA) boronate26 derivative, 4, both of which are air stable indefinitely.
Figure 2.

Functionalized benzofuran boronic acid derivatives available through the alkoxyboration reaction. Values represent isolated yields of organotrifluoroborate or MIDA boronate products 4. Values in parentheses represent 1H NMR yields of the corresponding catechol boronic ester 3 versus an external mesitylene standard using the ERETIC method.
Organotrifluoroborate 4a is readily isolated in high yield using a chromatography-free purification, making this derivatization method particularly amenable to applying the alkoxyboration reaction on preparative scale. The corresponding MIDA derivative (4b) provides an option for purification by silica gel chromatography, but this comes at the cost of slightly diminished yield. Single-crystal X-ray diffraction analysis of 4b allowed for the unambiguous identification of the alkoxyboration product (Figure 3).
Figure 3.
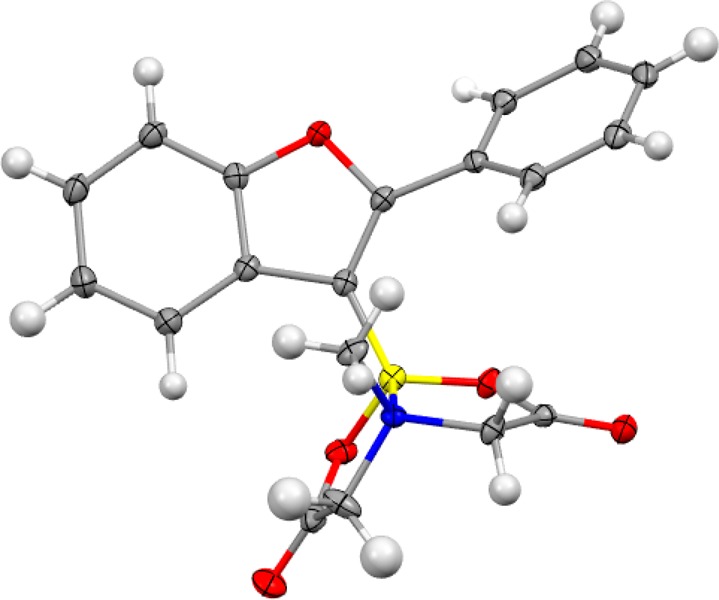
X-ray structure of 4b with the thermal ellipsoids set at the 50% probability level (B, yellow; C, gray; H, white; O, red; N, blue).
The alkoxyboration reaction is tolerant of a variety of functional groups suitable for downstream reactivity. Aryl bromide 4c, silyl-protected alcohol 4d, terminal alkyne 4f, amide 4g, esters 4h and 4i, and the functionally dense iodonitrile 4j are compatible with the reaction conditions. Many of these alkoxyboration reactions proceed smoothly at 50 °C, although the reactions generating 4d, 4g, 4h, and 4j required heating to 90 °C in order to affect full conversion. We attribute the relatively slow formation of 4d to the high steric encumbrance from the silyl ether at the 2-position of the benzofuran. The cyclization of substrates containing Lewis basic nitrogen atoms (forming 4g, 4h, and 4j) was likely retarded by reversible N–B coordination that was observed by 11B nuclear magnetic resonance (NMR) spectroscopy. For all substrates, the mass balance was largely attributable to protonolysis of the product C–B bond.
Notably, many of these products contain functional groups incompatible with commonly employed methods of benzofuran synthesis, including via other borylation techniques (Figure 4). In one frequently used borylation technique, an aryl lithium intermediate is trapped by a boron electrophile (Method 1); thus, electrophiles such as carbonyl or nitrile groups and enolizable protons are not generally tolerated due to the highly nucleophilic and basic nature of the requisite organolithium intermediate.27 Aryl halides may also suffer from undesired lithium/halogen exchange. The Miyaura borylation is a more mild alternative that is compatible with electrophilic functional groups (Method 2), but aryl halides are borylated through this Pd(0)-catalyzed reaction28 and are therefore not spectator functional groups under these conditions. Finally, the Ir-catalyzed C–H activation/borylation reaction29 is an effective means of accessing aryl boronic acid derivatives (Method 3), but this reaction is regioselective for either 2- or 7-borylation; 3-borylated benzofurans such as those available through the alkoxyboration reaction cannot be synthesized regioselectively through C–H activation/borylation.30
Figure 4.

Benzofuran boronic acid derivatives inaccessible using conventional borylation methods but newly accessible using the alkoxyboration reaction.
We set out to demonstrate the utility in divergent synthesis of the alkoxyboration products enabled through this synthesis in subsequent divergent functionalization steps (Scheme 1). Rh-catalyzed conjugate addition of 4i into methyl vinyl ketone using the method developed by Batey31 provides β-benzofuranyl ketone 6 in moderate yield. Subjection of the same benzofuran trifluoroborate to Suzuki-Miyaura coupling conditions described by Molander and Biolatto32 afforded afford 3-arylated benzofuran 8 with concomitant methanolysis of the ethyl ester. Finally, addition of 4i to an iminium ion was used to prepare aminated benzofuran 10. Thus, a single bench-stable alkoxyboration product can be functionalized in a variety of ways, which is important in diversity-oriented syntheses to develop compound catalogs for drug discovery.18
Scheme 1. Versatility of an Alkoxyboration Product in Diversity-Oriented Synthesis.

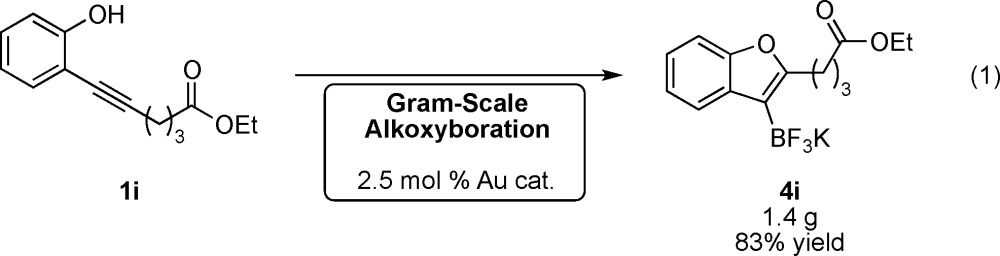
We next explored the scalability of the alkoxyboration reaction. Ester-containing phenol 1i was successfully converted to more than 1 g of organotrifluoroborate 4i on a 5 mmol scale with 2.5% gold catalyst (eq 1). Full conversion of starting material was affected even with this lower Au catalyst loading. This convenient scalability demonstrates that quantities of O-heterocyclic boronic acid derivatives sufficient for multistep synthesis may be prepared using the alkoxyboration method.
 |
1 |
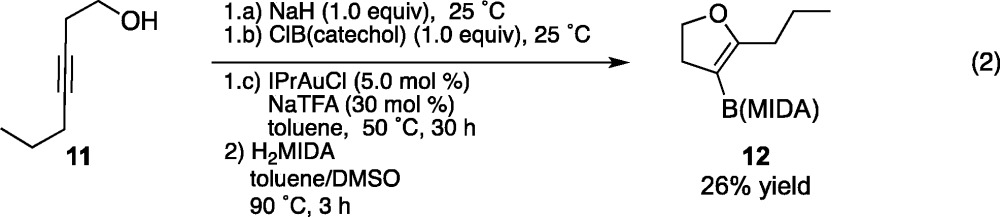
Having demonstrated the utility of this transformation in generating members of the benzofuran class of O-heteroaryl boronic acid derivatives, we explored its application to the synthesis of a nonaromatic oxygen-containing heterocycle (eq 2). Simple and commercially available homopropargyl alcohol 11 was subjected to standard alkoxyboration reaction conditions to prepare dihydrofuran product 12. A large number of unidentifiable trace coproducts were detected in this reaction, possibly consistent with intermolecular reactivity. This substrate demonstrates the potential for generality in the alkoxyboration reaction: The reaction features low labor “setup cost” by employing simple, commercially available starting materials to generate highly value-added O-heterocyclic organoboronate compounds in one synthetic step, and the cyclization proceeds without requiring the gain of product aromaticity or the need for a fused ring system that enforces a conformational bias toward cyclization.
 |
2 |
A number of Lewis-acidic metal catalysts have been developed for the addition of oxygen-electrophile bonds across alkynes, albeit not with boron and thus not generating these building blocks for downstream functionaliztion.33 We propose the catalytic cycle shown in Scheme 2 featuring bifunctional Lewis acidic/Lewis basic substrate activation. The bifunctional catalyst IPrAuTFA can be generated in situ from IPrAuCl and NaTFA. Reaction of the Lewis basic trifluoroacetate moiety with electrophilic boric ester 2a gives nucleophilic borate 14. The resulting Lewis acidic Au(I) cation may then bind to the alkyne (15), increasing its electrophilicity. Nucleophilic attack on the alkyne–Au π complex by the phenol B–O bond would provide two neutral intermediates: boron electrophile 16 and organogold nucleophile 17, which could recombine to regenerate 13 with concomitant formation of the observed alkoxyboration product 3a. Thus, the IPrAu+ moiety of the catalyst activates the alkyne for nucleophilic attack, and the TFA counterion allows for reversible tuning from a boron electrophile to a nucleophilic borate adduct. This reaction manifold is fundamentally unique from the metal-catalyzed addition of B–C, B–Si, B–Sn, and B–S σ bonds, which often proceed through oxidative addition of a low-valent metal catalyst into the B–X bond. We believe that the new activation strategy employed in the alkoxyboration reaction could be extended to other types of B–X bonds in order to provide additional reactivity complementary to preexisting methods. Notably, this approach also suggests a new generalizable mechanism for Au catalyst turnover by trapping with electrophilic boron to generate other previously inaccessible organoboron building blocks.
Scheme 2. Mechanistic Hypothesis Featuring the Bifunctional Lewis Acidic/Lewis Basic Catalyst IPrAuTFA.

Conclusions
This alkoxyboration reaction proceeds through an unprecedented activation of the strong B–O σ bond. This fundamentally new activation is showcased in a mild, scalable technique for the preparation of O-heterocyclic boronic acid derivatives and downstream-functionalized benzofurans. The reaction provides a simple new bond disconnection for constructing these motifs with different regioselectivity and broader functional group compatibility than existing methods. This compatibility yields highly functionalized bench-stable building blocks for divergent synthesis that are not directly accessible using alternative methods. The carbophilic Lewis acid activation mechanism for B–X addition suggests its broader application to other B–X addition reactions and the ability to synthesize previously inaccessible organoboron building blocks via this new strategy for turning over gold and other carbophilic metal catalysts.
Experimental Section
General Methods
All chemicals were used as received from commercial sources unless otherwise noted. Sodium trifluoroacetate was dried at 130 °C at 10 mTorr for 18 h before use. Toluene and dichloromethane were purified by passage through an alumina column under argon pressure on a push-still solvent system. Anhydrous dimethylsulfoxide was obtained by stirring over activity I alumina 18 h under N2 atmosphere, decanting the liquid, and distilling the liquid at 10 Torr over CaH2. Acetone was dried by distillation over anhydrous CaSO4 under N2 atmosphere. Toluene-d8 was dried over CaH2, degassed using three freeze–pump–thaw cycles, and vacuum transferred prior to use. All manipulations were conducted in a glovebox under nitrogen atmosphere or using standard Schlenk techniques unless otherwise specified. Analytical thin layer chromatography (TLC) was performed using Merck F250 plates. Plates were visualized under UV irradiation (254 nm) and/or using a basic aqueous solution of potassium permanganate. Flash chromatography was conducted using a Teledyne Isco Combiflash Rf 200 Automated Flash Chromatography System, and Teledyne Isco Redisep 35–70 μm silica gel. All proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on a Bruker DRX-400 spectrometer, Bruker DRX-500 spectrometer outfitted with a cryoprobe, or a Bruker AVANCE-600 spectrometer. All boron nuclear magnetic resonance (11B NMR) spectra were recorded on a Bruker AVANCE-600 spectrometer. All fluorine nuclear magnetic resonance (19F NMR) spectra were recorded on a Bruker DRX-400. All chemical shifts (δ) are reported in parts per million (ppm) downfield of tetramethylsilane, and referenced to the residual protiated solvent peak (δ = 7.26 ppm for CDCl3, δ = 2.50 ppm for DMSO-d6, or δ = 1.94 ppm for CD3CN in 1H NMR spectroscopy experiments; δ = 77.16 ppm for CDCl3, δ = 39.52 ppm for DMSO-d6, or δ = 1.34 ppm for CD3CN in 13C NMR spectroscopy experiments). 11B and 19F NMR spectroscopy experiments are referenced to the absolute frequency of 0 ppm in the 1H dimension according to the Xi scale. Low- and high-resolution mass spectrometry data were obtained at the University of California, Irvine.
General Considerations for Alkoxyboration Reactions
All alkoxyboration reactions were conducted in a N2-filled glovebox due to the high moisture sensitivity of the boric ester intermediate 2. All glassware and reagents must be rigorously dry for optimal yield. The reaction progress was monitored by removing a small aliquot of the reaction mixture from the glovebox and diluting it in 1:1 EtOAc:water. This results in rapid hydrolysis of boric ester intermediate 2 back to the phenol starting material 1. Thus, co-spotting the reaction mixture versus phenol 1 provides a convenient method for determining whether or not intermediate 2 has been fully consumed. The addition of PPh3 to quench the Au catalyst34 between the alkoxyboration step and the formation of the organotrifluoroborate or MIDA boronate was essential. Full synthetic protocols for the alkoxyboration reaction are available in the Supporting Information.
Benzofuran Trifluoroborate 4a
A solution of phenol 1a (97.0 mg, 0.500 mmol, 1.00 equiv) in 1.0 mL of toluene was added to a flame-dried 10-mL Schlenk tube. To this stirring solution was added dropwise at 25 °C a suspension of NaH (69 wt % purity, 17.4 mg, 0.500 mmol, 1.00 equiv) in 0.5 mL of toluene over 2 min. A suspension of NaTFA (20 mg, 0.15 mmol, 30 mol %) in 0.5 mL of toluene was added next, and the resulting suspension was stirred for 15 min to affect full deprotonation. To the resulting stirring sodium phenoxide suspension was added at 25 °C a solution of B-chlorocatecholborane (77.0 mg, 0.500 mmol, 1.00 equiv) in toluene (1.0 mL), using additional toluene as a rinse to ensure full transfer (2 × 0.5 mL portions). The resulting suspension was stirred vigorously for 30 min to allow for full conversion to boric ester intermediate 2a. Next, a suspension of IPrAuCl (16 mg, 0.025 mmol, 5.0 mol %) in toluene (0.5 mL) was added to the reaction mixture at 25 °C, using additional toluene as a rinse to aid in full transfer (1 × 0.5 mL portion). The reaction mixture was then sealed with a ground glass stopper and a PTFE sealing ring and placed in a preheated 50 °C copper shot heating bath. The final concentration of the reaction mixture was 0.1 M in 1a. After 24 h, analysis by TLC (20% EtOAc/hexanes) indicated full consumption of boric ester intermediate 2a. The reaction mixture was cooled to 25 °C, and a solution of PPh3 (13 mg, 0.050 mmol, 10 mol %) in toluene (0.5 mL) was added. The resulting suspension was stirred for 22 h at 25 °C in order to quench IPrAuTFA before proceeding. The quenched reaction mixture was removed from the glovebox and filtered through a fiberglass filter to remove the suspended solids. The filter was then rinsed with toluene (3 × 3 mL), and the combined filtrates were concentrated in vacuo to a pale yellow powder, which was suspended in acetone (4.5 mL) and added to a stirring solution of KHF2 (140 mg, 1.8 mmol, 3.5 equiv) in water (1.5 mL). The resulting mixture was stirred at 25 °C for 30 min, and then concentrated in vacuo to remove the solvents. To this residue was added 2 mL of Et2O and the solution was subsequently concentrated at ca. 10 mTorr for 30 min in order to remove residual acetone. The resulting pale yellow solid residue was washed with Et2O (4 × 2 mL) and extracted with acetone (4 × 2 mL). The combined acetone extracts were concentrated in vacuo, and the resulting residue was subjected to an additional washing/extraction cycle to yield 4a as a white powder (113 mg, 75% yield) after removing volatiles at 25 °C and ca. 10 mTorr for 18 h. 1H NMR (DMSO-d6, 600 MHz): δ 8.10 (d, J = 7.4 Hz, 2H), 7.88 (d, J = 7.7 Hz, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.38 (t, J = 7.6 Hz, 2H), 7.26 (t, J = 7.3 Hz, 1H), 7.13 (td, J = 6.5 Hz, 1.2 Hz, 1H), 7.08 (td, J = 7.4, 0.9 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz): δ 154.7 (q, JC–F = 4.6 Hz), 153.8, 135.4, 133.3, 127.9, 126.8, 126.7 (q, JC–F = 2.3 Hz), 124.1 (q, JC–F = 2.8 Hz), 122.7, 109.6. [Note: As with many organotrifluoroborates, the ipso carbon was not detected, presumably due to broadening through coupling to the 11B nucleus. The quaternary carbon at the benzofuran 2 position was also not detected.] 11B NMR (DMSO-d6, 193 MHz): δ 3.2 (br s). 19F NMR (DMSO-d6, 376 MHz): δ −131.9 (br s). HRMS (ESI-): Calculated for C14H9BF3O ([M – K]−), 261.0701; found, 261.0706.
MIDA Boronate 4b
A solution of phenol 1a (97.0 mg, 0.500 mmol, 1.00 equiv) in 1.0 mL of toluene was added to a flame-dried 10-mL Schlenk tube. To this stirring solution was added dropwise at 25 °C a suspension of NaH (92 wt % purity, 13.0 mg, 0.500 mmol, 1.00 equiv) in 0.5 mL of toluene over 2 min. A suspension of NaTFA (20 mg, 0.15 mmol, 30 mol %) in 0.5 mL of toluene was added next, and the resulting suspension was stirred for 15 min to affect full deprotonation. To the resulting stirring sodium phenoxide suspension was added at 25 °C a solution of B-chlorocatecholborane (77.0 mg, 0.500 mmol, 1.00 equiv) in toluene (1.0 mL), using additional toluene as a rinse to ensure full transfer (2 × 0.5 mL portions). The resulting suspension was stirred vigorously for 30 min to allow for full conversion to boric ester intermediate 2a. Next, a suspension of IPrAuCl (16 mg, 0.025 mmol, 5.0 mol %) in toluene (0.5 mL) was added to the reaction mixture at 25 °C, using additional toluene as a rinse to aid in full transfer (1 × 0.5 mL portion). The reaction mixture was then sealed with a ground glass stopper and a PTFE sealing ring and placed in a preheated 50 °C copper shot heating bath. The final concentration of the reaction mixture was 0.1 M in 1a. After 24 h, analysis by TLC (20% EtOAc/hexanes) indicated full consumption of boric ester intermediate 2a. The reaction mixture was cooled to 25 °C, and a solution of PPh3 (13 mg, 0.050 mmol, 10 mol %) in toluene (0.5 mL) was added. The resulting suspension was stirred for 16 h at 25 °C in order to quench IPrAuTFA before proceeding. Anhydrous DMSO (2.0 mL) and H2MIDA (81 mg, 0.55 mmol, 1.1 equiv) were added to the quenched alkoxyboration reaction mixture, and the resulting suspension was stirred at 90 °C for 2 h. The reaction mixture was then cooled to 25 °C and removed from the glovebox. Toluene was removed in vacuo at 25 °C and ca. 10 Torr, then DMSO was removed by Kugelrohr distillation at ca. 10 mTorr. The resulting semisolid residue was adsorbed onto Celite from a MeCN suspension and purified by silica gel chromatography using an elution gradient from 100% Et2O to 100% MeCN. Removal of volatiles at 25 °C and ca. 10 mTorr for 18 h afforded MIDA boronate 4b as a white powder (101 mg, 58% yield). Crystals suitable for X-ray diffraction analysis were prepared by slow diffusion of Et2O into a saturated solution of 4b in Et2O/acetone at 25 °C over 3 days. TLC (10% MeCN/Et2O): Rf = 0.39. 1H NMR (CD3CN, 600 MHz): δ 7.72 (dd, J = 7.8 Hz, 0.8 Hz, 1H), 7.67–7.65 (m, 2H), 7.55 (d, J = 9.7 Hz, 1H), 7.47–7.44 (m, 3H), 7.35–7.31 (m, 1H), 7.29–7.26 (m, 1H), 3.97 (d, J = 17.1 Hz, 2H), 3.65 (d, J = 17.1 Hz, 2H), 2.56 (s, 3H). 13C NMR (CD3CN, 125 MHz): δ 169.0, 156.0, 133.6, 133.0, 130.6, 130.2, 129.3, 125.2, 123.9, 123.5, 111.8, 63.0, 48.2. [Note: no signals were observed for the quaternary C–B ipso carbon or the quaternary carbon at the benzofuran 2 position.] 11B NMR (CD3CN, 193 MHz): δ 11.3 (br s). HRMS (ESI+): Calculated for C17H19BBrNO5 ([M + Na]+), 372.1023; found, 372.1016.
Acknowledgments
This work was supported by a grant from the NIH (1R01GM098512-01) and by an NSF Graduate Fellowship to J.J.H. (NSF-201015893). We thank Prof. David L. van Vranken (University of California–Irvine) for helpful conversation. We also thank Dr. Joseph W. Ziller and Ms. Jordan F. Corbey for X-ray diffraction analysis, and Dr. Phillip R. Dennison for NMR spectroscopy assistance.
Supporting Information Available
Full experimental procedures and characterization data, including CIF data for 4b. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Provisional patent applications (no. 61/836,391 and no. 61/906,040) have been filed by the University of California.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hall D. G.Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine, and Materials, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Hurd D. T. J. Am. Chem. Soc. 1948, 70, 2053–2055. [Google Scholar]
- Brown H. C. Tetrahedron 1961, 12, 117–138. [Google Scholar]
- a Männig D.; Nöth H. Angew. Chem., Int. Ed. Engl. 1985, 24, 878–879. [Google Scholar]; b Carrol A.-M.; O’Sullivan T. P.; Guiry P. J. Adv. Synth. Catal. 2005, 347, 609–631. [Google Scholar]
- Miyaura N.Hydroboration, Diboration, and Stannylboration. In Catalytic Heterofunctionalization; Togni A., Grützmacher H., Eds.; Wiley-VCH: Weinheim, 2001; pp 1–46. [Google Scholar]
- a Suginome M.; Yamamoto A.; Murakami M. J. Am. Chem. Soc. 2003, 125, 6358–6359. [DOI] [PubMed] [Google Scholar]; b Suginome M.; Shirakura M.; Yamamoto A. J. Am. Chem. Soc. 2009, 128, 14438–14439. [DOI] [PubMed] [Google Scholar]
- Suginome M. Chem. Rec. 2010, 10, 348–358. [DOI] [PubMed] [Google Scholar]
- Suginome M.; Nakamura H.; Ito Y. Chem. Commun. 1996, 2777–2778. [Google Scholar]
- Suginome M.; Nakamura H.; Ito Y. Angew. Chem., Int. Ed. Engl. 1997, 36, 2516–2518. [Google Scholar]
- Onozawa S.; Hatanaka Y.; Sakakura T.; Shimada S.; Tanaka M. Organometallics 1996, 15, 5450–5452. [Google Scholar]
- Ishiyama T.; Nishijima K.; Miyaura N.; Suzuki A. J. Am. Chem. Soc. 1993, 115, 7219–7225. [Google Scholar]
- a Ishiyama T.; Matsuda N.; Miyaura N.; Suzuki A. J. Am. Chem. Soc. 1993, 115, 11018–11019. [Google Scholar]; b Marder T. B.; Norman N. C. Top. Catal. 1998, 5, 63–73. [Google Scholar]
- Lappert M. F.; Prokai B. J. Orgmet. Chem. 1964, 1, 384–400. [Google Scholar]
- Hara S.; Dojo H.; Takinami S.; Suzuki A. Tetrahedron Lett. 1983, 24, 731–734. [Google Scholar]
- Cragg R. H.; Lappert M. F.; Tilley B. P. J. Chem. Soc. 1964, 2108–2115. [Google Scholar]
- Matsumi N.; Chujo Y. Macromolecules 1998, 31, 3802–3806. [Google Scholar]
- Sanderson R. T.Polar Covalence; Academic Press: New York, 1983. [Google Scholar]
- Burke M. D.; Berger E. M.; Schreiber S. L. J. Am. Chem. Soc. 2004, 126, 14095–14104. [DOI] [PubMed] [Google Scholar]
- Domínguez de María P.; van Gemert R. W.; Straathof A. J. J.; Hanefeld U. Nat. Prod. Rep. 2010, 27, 370–392. [DOI] [PubMed] [Google Scholar]
- Drugs.com, U.S. Pharmaceutical Sales – 2012 (http://www.drugs.com/stats/top100/2012/sales).
- Anderson S.; Taylor P. N.; Verschoor G. L. B. Chem.—Eur. J. 2004, 10, 518–527. [DOI] [PubMed] [Google Scholar]
- Joule J. A.; Mills K.. Heterocyclic Chemistry, 5th ed.; Wiley: Chichester: United Kingdom, 2010. [Google Scholar]
- Akoka S.; Barantin L.; Trierweiler M. Anal. Chem. 1999, 71, 2554–2557. [DOI] [PubMed] [Google Scholar]
- Full experimental details are available in the Supporting Information.
- Molander G. A.; Ellis N. Acc. Chem. Res. 2007, 40, 275–286. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2008, 130, 14084–14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki A.; Moriwaki Y.; Yoshida J. Chem. Commun. 2012, 48, 11211–11213. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; Murata M.; Miyaura N. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar]
- a Cho J. Y.; Tse M. K.; Holmes D.; Maleczka R. E. Jr.; Smith M. R. III. Science 2002, 295, 305–308. [DOI] [PubMed] [Google Scholar]; b Ishiyama T.; Takagi J.; Ishida K.; Miyaura N. J. Am. Chem. Soc. 2002, 124, 390–391. [DOI] [PubMed] [Google Scholar]
- Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- Batey R. A.; Thadani A. N.; Smil D. V. Org. Lett. 1999, 1, 1683–1686. [Google Scholar]
- Molander G. A.; Biolatto B. J. Org. Chem. 2003, 68, 4302–4314. [DOI] [PubMed] [Google Scholar]
- For select related examples of Lewis acid-catalyzed reactions, see:; a Nakamura I.; Mizushima Y.; Yamamoto Y. J. Am. Chem. Soc. 2005, 127, 15022–15023. [DOI] [PubMed] [Google Scholar]; b Fürstner A.; Davis P. W. J. Am. Chem. Soc. 2005, 127, 15024–15025. [DOI] [PubMed] [Google Scholar]; c Nakamura I.; Sato T.; Yamamoto I. J. Am. Chem. Soc. 2006, 45, 4473–4475. [DOI] [PubMed] [Google Scholar]; d Nakamura I.; Sato T.; Yamamoto Y. Angew. Chem., Int. Ed. 2006, 45, 4473–4475. [DOI] [PubMed] [Google Scholar]; e Körner C.; Starkov P.; Sheppard T. D. J. Am. Chem. Soc. 2010, 132, 5968–5969. [DOI] [PubMed] [Google Scholar]; f Malona J. A.; Cariou K.; Spencer W. T. III; Frontier A. J. J. Org. Chem. 2012, 77, 1891–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Blanco Jaimes M. C.; Weingand V.; Rominger F.; Hashmi A. S. K. Chem.—Eur. J. 2013, 19, 12504–12511. [DOI] [PubMed] [Google Scholar]; h Marder T. B.; Chan D. M.-T.; Fultz W. C.; Calabrese J. C.; Milstein D. J. Chem. Soc, Chem. Commun. 1987, 1885–1887. [Google Scholar]
- Shi Y.; Roth K. E.; Ramgren S. D.; Blum S. A. J. Am. Chem. Soc. 2009, 131, 18022–18023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.