

Abstract
Studies are being conducted on the applicability of genomic data to improve the accuracy of the selection process in livestock, and genome-wide association studies (GWAS) provide valuable information to enhance the understanding on the genetics of complex traits. The aim of this study was to identify genomic regions and genes that play roles in birth weight (BW), weaning weight adjusted for 210 days of age (WW), and long-yearling weight adjusted for 420 days of age (LYW) in Canchim cattle. GWAS were performed by means of the Generalized Quasi-Likelihood Score (GQLS) method using genotypes from the BovineHD BeadChip and estimated breeding values for BW, WW, and LYW. Data consisted of 285 animals from the Canchim breed and 114 from the MA genetic group (derived from crossings between Charolais sires and ½ Canchim + ½ Zebu dams). After applying a false discovery rate correction at a 10% significance level, a total of 4, 12, and 10 SNPs were significantly associated with BW, WW, and LYW, respectively. These SNPs were surveyed to their corresponding genes or to surrounding genes within a distance of 250 kb. The genes DPP6 (dipeptidyl-peptidase 6) and CLEC3B (C-type lectin domain family 3 member B) were highlighted, considering its functions on the development of the brain and skeletal system, respectively. The GQLS method identified regions on chromosome associated with birth weight, weaning weight, and long-yearling weight in Canchim and MA animals. New candidate regions for body weight traits were detected and some of them have interesting biological functions, of which most have not been previously reported. The observation of QTL reports for body weight traits, covering areas surrounding the genes (SNPs) herein identified provides more evidence for these associations. Future studies targeting these areas could provide further knowledge to uncover the genetic architecture underlying growth traits in Canchim cattle.
Introduction
Growth traits are traditionally included in selection criteria in beef cattle breeding programs, due to their association with meat production, and therefore are of great economic importance for both breeders and the industry [1]. The most common type of growth trait used in the selection process is the body weight measurement, which can be taken from birth and throughout an animal’s life. These traits are used not only for evaluation of growth and development, but also for decision making about reproduction, nutritional, and prophylactic management. Body weight usually presents heritability and genetic correlation coefficients from medium to high magnitude [2]–[4].
Technological advances allow the use of massive genotype information through single nucleotide polymorphism (SNP) panels for animal breeding. For cattle, there are several SNP panel densities commercially available from Illumina [5] and Affymetrix [6], varying from a few thousand to more than 3 million SNPs [7], which are applicable for genome-wide association and genomic selection studies. The concept of genomic selection (GS) was first presented by Meuwissen et al. [8], as an alternative to predict more accurate breeding values through genetic markers (i.e. genomic breeding values), and increase the rate of genetic gain by reducing generation intervals. Genome-wide association studies (GWAS) were designed to identify regions that could clarify the inheritance of complex traits [9]. If candidate regions are identified it could be useful for livestock improvement through GS by focusing on relevant genomic regions [10], [11].
For growth traits in crossed beef cattle, important genomic associations for birth weight, weaning weight, and yearling weight were reported by Snelling et al. [12]. These authors also found the same significant SNPs for different growth traits, indicating that a pleiotropic effect exists and genes that are responsible for body weight performance at earlier ages are also acting later on. Furthermore, many of these SNPs observed by Snelling et al. [12] were located in quantitative trait loci (QTL) previously reported for birth weight [13], [14], pre- and post-weaning body weight gain [15], yearling weight [13], stillborn calves [16], and calving difficulty [17]. The presence of QTL for birth weight and calving difficulty support that calf birth weight should be monitored in order to avoid problems with dystocia [18].
Many breeds specialized in meat production are reared in Brazil, such as Canchim, which is derived from crosses between Charolais and Zebu animals. Quantitative research has previously been conducted in this breed to evaluate genetic parameters for growth, reproductive performance, and meat quality traits. However, the specific regions or genes responsible for the genetic variation of these traits are still unknown. Recently, Mokry et al. [19] studied backfat thickness in Canchim cattle and found genomic associations that could aid in GS. Backfat thickness is also a trait of economic importance and has recently gained attention from producers, who wish to increase fat deposition in animals raised on pastures. Thus, the aim of this study was to identify, through GWAS, genomic regions and genes that play roles on birth weight, weaning weight, and long-yearling weight in Canchim cattle.
Materials and Methods
Ethics Statement
This study was performed with the approval of the Embrapa Southeast Livestock Ethical Committee of Animal Use (CEUA-CPPSE) under protocol number 02/2009.
Animals and Data
The BovineHD Beadchip SNP panel from Illumina was used for genotyping 194 males and 205 females, of which 285 were Canchim animals, and 114 were from the “MA” genetic group. The Canchim breed was developed in Brazil through mating schemes between Charolais and Zebu breeds, which generated animals with 5/8 (62.5%) Charolais and 3/8 (37.5%) Zebu fractions [20]. Different schemes were developed to ensure genetic variability and to expand the genetic base of the breed [21]. In particular, the “MA” genetic group produces individuals with an approximate proportion of 65.6% Charolais and 34.4% Zebu, and is very popular among producers. MA animals are the product of crossings between group “A” individuals (derived from crossings between Canchim and Zebu) with Charolais animals [19], [20], [22].
The genotyped individuals were the offspring of 49 sires and 355 dams, born between 1999 and 2005, and originated from seven farms in the states of São Paulo and Goiás in Brazil. In the state of São Paulo, farms were located in the municipality of Águas de Santa Bárbara (22°52′51″S - 49°14′20″W), Angatuba (23°29′24″S - 48°24′46″W), Capão Bonito (24°00′21″S - 48°20′56″W), Sandovalina (22°27′21″S - 51°45′46″W), São Carlos (22°01′04″S - 47°53′27″W), and São Miguel Arcanjo (23°52′40″S - 47°59′49″W). In the Goiás state, animals were from the Jussara municipality (15°51′54″S - 50°52′04″W).
No specific permissions were required for these locations/states and the field studies did not involve endangered or protected species. All farms participate in the Canchim breeding program, in which the database is maintained by Embrapa Beef Cattle through the responsibility of the researchers Dr. Roberto Augusto de Almeida Torres Júnior and Dr. Luiz Otávio Campos da Silva. The genomic data used in this study can be available upon request from Dr. Luciana Correia de Almeida Regitano (Embrapa Southeast Livestock. Address: Rodovia Washington Luiz, km 234, São Carlos, São Paulo, 13560-970, Brazil, Telephone: 55+16 34115600).
Data on estimated breeding values (EBVs) for birth weight (BW), weaning weight adjusted for 210 days of age (WW), and long-yearling weight adjusted for 420 days of age (LYW) were provided by the National Association of Canchim Breeders (ABCCAN) and by the Embrapa-Geneplus Beef Cattle Breeding Program. The EBVs for each trait were obtained by means of multi-trait analysis, which was done using the REMLF90 software [23] under an animal model that included the additive genetic, maternal genetic (only for BW and WW), and residual random effects; contemporary group (CG) as a fixed effect for all the traits, and age of the dam at calving as a covariate. The effects considered in the formation of CGs were sex, birth year and birth season (January to March; April to September; and October to December), farm of birth, genetic groups, and feeding regimen. The relationship matrix of the genotyped animals consisted of a total of 4,095 individuals. The average inbreeding coefficient was 0.02.
Genotype Quality Control
The SNPs with a genotype calling score lower than 15% were treated as missing genotypes, as recommended by the Infinium platform [24]. Genotype quality control was applied to exclude SNPs with significant (P<10−5) Hardy-Weinberg Equilibrium deviation; heterozygous excess (>15%); minor allele frequency (<5%); and call rate (<90%). Animals with call rate lower than 90% were also excluded and only autosomal SNPs with known genome position, according to the UMD_3.1 bovine assembly map [25], were used for the GWAS.
Genome-wide Association Study
GWAS were carried out by means of the Generalized Quasi-Likelihood Score method (GQLS), developed by Feng et al. [26] and implemented in the SLEUTH software by Dr. Mehdi Sargolzaei. In this method, a logistic regression was used to associate the EBVs (treated as a covariate) with genotypes (treated as a response variable). Analyses were done for one SNP at a time, in which 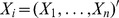 represents the EBVs (
represents the EBVs (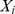 ) for the ith animal; and
) for the ith animal; and 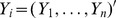 represents the genotypes (
represents the genotypes (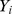 ), considering
), considering 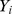 = ½* (number of alleles for the ith animal’s genotype). As the genotypes were coded as “0”, “1”, and “2”; their respective proportions would be 0, ½, and 1. The expected SNP allele frequency is represented by
= ½* (number of alleles for the ith animal’s genotype). As the genotypes were coded as “0”, “1”, and “2”; their respective proportions would be 0, ½, and 1. The expected SNP allele frequency is represented by 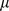 , in which
, in which 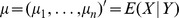 , thus
, thus 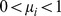 .
.
To associate 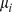 with
with 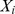 , the following logistic regression is defined as:
, the following logistic regression is defined as:
in which β0 is the constant term and β1 the angular coefficient.
The hypothesis to verify the association of the SNP with the EBV assumes:
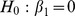 , null hypothesis (the SNP is not associated with the EBV);
, null hypothesis (the SNP is not associated with the EBV);
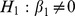 , alternative hypothesis (the SNP is associated with the EBV).
, alternative hypothesis (the SNP is associated with the EBV).
Considering the null hypothesis, 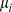 can be interpreted as
can be interpreted as 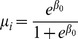 , for all
, for all 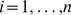 . The mean vector of
. The mean vector of 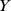 does not depend on
does not depend on 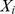 , which becomes
, which becomes 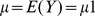 , in which “1” is a vector. The solution for the “quasi-likelihood score” equation results in an estimate of
, in which “1” is a vector. The solution for the “quasi-likelihood score” equation results in an estimate of 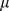 . The equation is represented by
. The equation is represented by 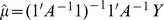 , in which
, in which 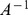 is the inverse of the relationship matrix of all individuals.
is the inverse of the relationship matrix of all individuals.
To estimate the “generalized quasi-likelihood score”, the 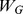 statistics can be calculated by:
statistics can be calculated by:
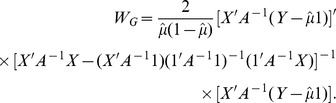 |
According to Heide [27], under a null hypothesis, 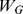 follows a Chi-squared distribution with one degree of freedom, resulting in p-values for each SNP. The advantages of the GQLS method are that it allows an unspecified distribution of EBVs, the method is suitable for studies with either a quantitative trait or a binary trait, and proposes the correction for the population stratification problem by considering the relationship matrix (
follows a Chi-squared distribution with one degree of freedom, resulting in p-values for each SNP. The advantages of the GQLS method are that it allows an unspecified distribution of EBVs, the method is suitable for studies with either a quantitative trait or a binary trait, and proposes the correction for the population stratification problem by considering the relationship matrix (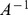 ) for calculating WG. However, the effect of the SNP in this methodology is not able to be estimated. Thus, single regression analyses for each significant SNP were carried out, as described in the following section.
) for calculating WG. However, the effect of the SNP in this methodology is not able to be estimated. Thus, single regression analyses for each significant SNP were carried out, as described in the following section.
Allele Substitution Effects
Single regression analyses were carried out to verify the genetic additive effects of each significant SNP, previously identified in the GQLS analyses. The following model was applied:
in which 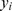 is the EBV for the trait of interest; µ is the mean value of the EBV (constant variable);
is the EBV for the trait of interest; µ is the mean value of the EBV (constant variable);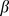 is the linear regression coefficient (allele substitution effect);
is the linear regression coefficient (allele substitution effect); 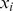 is the number of copies of a given allele (0, 1, or 2); and
is the number of copies of a given allele (0, 1, or 2); and 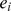 is the residual effect.
is the residual effect.
Correction for Multiple Testing
The false discovery rate (FDR) was used for multiple testing correction [28] in order to verify significant SNPs. A maximum threshold of 10% FDR for each chromosome (chromosome-wise) was considered. The p-values of each SNP were sorted in ascending order and the following formula applied:
in which q is the desired level of significance, m is the total number of SNPs, and 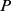 is the p-value of the
is the p-value of the 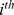 SNP. The SNPs were considered as significant when
SNP. The SNPs were considered as significant when 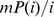 resulted in a value lower than
resulted in a value lower than 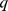 .
.
Gene Mapping and In-silico Functional Analyses
The significant SNPs were surveyed to their corresponding genes or to surrounding genes within a distance of 250 kb, using the genome databanks National Center for Biotechnology Information (NCBI) [29] and Ensembl Genome Browser [30]. Functional analysis of the mapped genes was performed by means of the UniProt website [31] to verify functional information of the genes.
When no information was available for the Bos taurus genes, annotations from human, rat or mouse orthologs were used to proceed with the in-silico functional analyses. AnimalQTLdb [32] was accessed to verify previous QTL reported for growth traits in the surrounding regions of significant SNPs.
Results
The descriptive statistics of estimated breeding values for BW, WW, and LYW are presented in Table 1. As our individuals were a sample from a larger population (of which the breeding values were estimated), it was expected that the mean EBV for each trait was different than zero.
Table 1. Descriptive statistics for birth weight (BW), weaning weight (WW), and long-yearling weight (LYW) in animals with genotype information.
| Trait | Animals | Mean | Standard-Deviation | Minimum | Maximum |
| BWEBV (kg) | 397 | 0.20 | 1.34 | −4.32 | 6.15 |
| WWEBV (kg) | 397 | 1.24 | 5.67 | −14.87 | 23.12 |
| LYWEBV (kg) | 397 | 0.95 | 8.98 | −24.38 | 29.24 |
Estimated breeding values (EBV).
A total of 786,799 SNPs were originally present for each animal, and after the quality control, a total of 672,778 SNPs remained for the GWAS. The number of SNPs evaluated in each Bos taurus autosome (BTA) is presented in Table 2. On average, 9.44% of the SNPs were excluded after the genotype quality control. The BTA22 and BTA13 presented the lowest and highest quantity of excluded SNPs, respectively.
Table 2. Number of SNPs evaluated for each Bos taurus autosome (BTA), BTA length in megabase pair (Mbp), and number of chromosome-wise significant SNPs* for birth weight (BW), weaning weight (WW), and long-yearling weight (LYW), respectively.
| BTA | BTA length (Mbp) | Number of SNPs evaluated | BW* | WW* | LYW* |
| 1 | 158.31 | 42,331 | 0 | 0 | 0 |
| 2 | 137.01 | 36,802 | 0 | 0 | 0 |
| 3 | 121.39 | 32,464 | 0 | 0 | 0 |
| 4 | 120.63 | 32,187 | 1 | 1 | 0 |
| 5 | 121.18 | 31,541 | 0 | 0 | 0 |
| 6 | 119.42 | 32,799 | 0 | 2 | 0 |
| 7 | 112.60 | 30,247 | 0 | 0 | 2 |
| 8 | 113.35 | 30,611 | 0 | 0 | 0 |
| 9 | 105.67 | 28,856 | 3 | 0 | 0 |
| 10 | 104.28 | 28,134 | 0 | 0 | 0 |
| 11 | 107.27 | 29,252 | 0 | 9 | 0 |
| 12 | 91.12 | 23,880 | 0 | 0 | 0 |
| 13 | 84.21 | 21,206 | 0 | 0 | 0 |
| 14 | 84.03 | 22,441 | 0 | 0 | 0 |
| 15 | 85.23 | 22,408 | 0 | 0 | 0 |
| 16 | 81.69 | 22,284 | 0 | 0 | 0 |
| 17 | 75.15 | 20,390 | 0 | 0 | 0 |
| 18 | 65.98 | 17,803 | 0 | 0 | 0 |
| 19 | 63.96 | 17,246 | 0 | 0 | 0 |
| 20 | 71.95 | 19,697 | 0 | 0 | 0 |
| 21 | 71.57 | 19,360 | 0 | 0 | 0 |
| 22 | 61.29 | 16,807 | 0 | 0 | 2 |
| 23 | 52.46 | 13,848 | 0 | 0 | 0 |
| 24 | 62.54 | 17,182 | 0 | 0 | 0 |
| 25 | 42.82 | 11,795 | 0 | 0 | 1 |
| 26 | 51.64 | 13,957 | 0 | 0 | 0 |
| 27 | 45.40 | 11,938 | 0 | 0 | 5 |
| 28 | 46.24 | 12,032 | 0 | 0 | 0 |
| 29 | 51.18 | 13,280 | 0 | 0 | 0 |
| Total | 2509.57 | 672,778 | 4 | 12 | 10 |
*Total of significant SNPs after multiple testing correction (FDR = 10%).
The results from the GQLS and single regression analyses, and SNP and gene identification are presented in Table 3. A total of 4, 12, and 10, SNPs were significantly associated (on a chromosome-wise level) with BW, WW, and LYW, respectively. A total of 18 SNPs were located in gene intron regions, whereas seven were located in intergenic regions, and only one was located in an upstream region. Figure 1 shows the Manhattan plots for chromosome-wise significant regions for BW, WW, and LYW, respectively. Linkage disequilibrium between significant SNPs in each chromosome (BTA) were calculated using the r2 coefficient [33], resulting in average values of 0.11 (BTA6, for WW), 0.80 (BTA7, for LYW), 0.91 (BTA9, for BW), 1.00 (BTA11, for WW), 1.00 (BTA22, for LYW), and 1.00 (BTA27, for LYW), respectively.
Table 3. Significantly associated SNPs for birth weight (BW), weaning weight (WW), and long-yearling weight (LYW) obtained by the Generalized Quasi-Likelihood Score method (GQLS).
| Trait | SNP Reference | BTA | Position (bp) | Alleles | Genes | Region | MAF | p-value | FDR | Allele substitution effect* |
| BW | rs43421095 | 4 | 117,400,491 | A,C | DPP6 | Intron | 0.2957 | 1.39E-06 | 5% | −0.5929 |
| BW | rs135754703 | 9 | 55,075,535 | A,C | LOC783932, MANEA | Intergenic | 0.4552 | 8.22E-06 | 10% | −0.41612 |
| BW | rs136146400 | 9 | 55,078,557 | A,G | LOC783932, MANEA | Intergenic | 0.4563 | 7.23E-06 | 10% | −0.41976 |
| BW | rs109313268 | 9 | 55,103,057 | T,C | LOC783932, MANEA | Intergenic | 0.4962 | 3.98E-06 | 10% | −0.44946 |
| WW | rs43421095 | 4 | 117,400,491 | A,C | DPP6 | Intron | 0.2957 | 1.91E-07 | 1% | −2.84898 |
| WW | rs135156506 | 6 | 35,008,291 | T,C | FARSB | Upstream | 0.1582 | 7.32E-07 | 5% | −2.17934 |
| WW | rs135591504 | 6 | 41,978,318 | T,C | KCNIP4 | Intron | 0.2273 | 2.74E-06 | 5% | 1.55340 |
| WW | rs136337296 | 11 | 103,096,174 | T,C | GTF3C5 | Intron | 0.365 | 1.81E-05 | 10% | 1.82227 |
| WW | rs109348820 | 11 | 103,154,983 | T,C | RALGDS | Intron | 0.3916 | 2.02E-05 | 10% | 1.76133 |
| WW | rs133132366 | 11 | 103,157,389 | T,C | RALGDS | Intron | 0.4077 | 1.28E-05 | 10% | 1.81074 |
| WW | rs134657108 | 11 | 103,162,503 | A,G | RALGDS | Intron | 0.4177 | 1.26E-05 | 10% | 1.90613 |
| WW | rs136054783 | 11 | 103,167,055 | A,G | RALGDS | Intron | 0.4066 | 8.40E-06 | 10% | 1.79279 |
| WW | rs136961684 | 11 | 103,170,500 | A,G | RALGDS | Intron | 0.4076 | 1.08E-05 | 10% | −1.80009 |
| WW | rs109945520 | 11 | 103,171,584 | G,T | RALGDS | Intron | 0.4047 | 1.17E-05 | 10% | 1.80350 |
| WW | rs109524492 | 11 | 103,172,572 | C,T | RALGDS | Intron | 0.4099 | 5.74E-06 | 10% | 1.63200 |
| WW | rs110048168 | 11 | 103,174,303 | A,C | RALGDS | Intron | 0.3886 | 3.05E-06 | 10% | −1.66682 |
| LYW | rs29011435 | 7 | 28,515,652 | T,C | MARCH3, LMNB1, PHAX, ALDH7A1, C7H5orf48, GRAMD3, MIR2458, LOC100848523 | Intergenic | 0.374 | 4.62E-06 | 10% | 3.59660 |
| LYW | rs134201365 | 7 | 28,522,539 | T,G | MARCH3, LMNB1, PHAX, ALDH7A1, C7H5orf48, GRAMD3, MIR2458, LOC100848523 | Intergenic | 0.375 | 4.62E-06 | 10% | −3.59660 |
| LYW | rs109581958 | 22 | 54,624,190 | T,C | LARS2, LOC101907967, TMEM158, LOC101908013, CDCP1, LOC614114, LOC101908094, LOC101901958, ZDHHC3, EXOSC7, LOC100847326, CLEC3B | Intergenic | 0.2335 | 1.09E-05 | 10% | −3.61775 |
| LYW | rs110246286 | 22 | 54,625,467 | T,C | LARS2, LOC101907967, TMEM158, LOC101908013, CDCP1, LOC614114, LOC101908094, LOC101901958, ZDHHC3, EXOSC7, LOC100847326, CLEC3B | Intergenic | 0.2348 | 1.09E-05 | 10% | −3.61775 |
| LYW | rs109242147 | 25 | 15,697,543 | A,G | XYLT1 | Intron | 0.1192 | 1.26E-06 | 5% | 3.24823 |
| LYW | rs109646351 | 27 | 2,614,991 | A,G | LOC101904868 | Intron | 0.4456 | 3.05E-05 | 10% | −3.91546 |
| LYW | rs109822265 | 27 | 2,619,242 | A,C | LOC101904868 | Intron | 0.4454 | 3.05E-05 | 10% | 3.91546 |
| LYW | rs110603636 | 27 | 2,620,088 | A,C | LOC101904868 | Intron | 0.4456 | 3.05E-05 | 10% | 3.91546 |
| LYW | rs110994026 | 27 | 2,620,961 | T,C | LOC101904868 | Intron | 0.4494 | 3.46E-05 | 10% | −3.85715 |
| LYW | rs134791735 | 27 | 2,623,000 | T,G | LOC101904868 | Intron | 0.4443 | 3.05E-05 | 10% | 3.91546 |
Single locus regression was carried out to estimate allele substitution effects for significant SNPs.
*Coefficient of regression (P<0.001); FDR = False discovery rate; MAF = Minor allele frequency; bp = base pairs; BTA = Bos taurus autosome.
Figure 1. Manhattan plots of p-values for birth weight (BW), weaning weight (WW), and long-yearling weight (LYW).

Significance levels were determined by false discovery rate (FDR) correction at levels of 1% (red line), 5% (blue line), and 10% (black line). Positions were presented in megabase pair (Mbp). Associations were observed in autosome (BTA) 4, 6, 7, 9, 11, 22, 25, and 27.
Discussion
Birth Weight (BW)
The SNP rs43421095 was significantly associated with BW and is located in the DPP6 (dipeptidyl-peptidase 6) gene (Table 3). The DPP6 gene is involved in proteolysis and nervous system development of mouse [34]. This SNP is located in the same region of a QTL associated with yearling weight and calving ease for Angus cattle [35]. Schrooten et al. [17] observed a QTL associated with gestation length in dairy cattle located between 102.10 and 124.80 cM.
On BTA9, the SNPs rs135754703, rs136146400, and rs109313268 are located close to MANEA (endo-alpha mannosidase) and the LOC783932 (small ubiquitin-related modifier 1-like) genes. The MANEA gene product is related to the glycoprotein endo-alpha-1,2-mannosidase activity, which acts in metabolism of protein pathways [36]. For the LOC783932 gene, no information regarding its function was observed in the consulted literature. In the same region, Alexander et al. [37] found a QTL associated with post-weaning average daily gain located between 48.73 and 80.26 cM. Snelling et al. [12] observed five significant SNPs (FDR≤10%), located between 112,474,006 and 114,565,961 bp on BTA4; and three SNPs, located between 54,832,703 and 57,515,862 bp on BTA9, associated with birth weight. One of these SNPs found by these authors (rs108988191) is also located close to the MANEA and LOC783932 genes.
For Canchim cattle, Machado et al. [22] found a QTL on BTA5 associated with BW, which contained the microsatellite markers ILSTS066, TEXAN15, and BMS1248 and the microsatellite on the IGF1 gene. Andrade et al. [20] verified association of BW with microsatellite markers in the IGF1 gene, also located on BTA5. The reason why we didn’t find associations for BW on chromosome 5 could be due to the differences in experimental designs, and because the previously cited authors studied only animals from the Embrapa Southeast Livestock herd, while in our study animals from other farms and other Brazilian states were used. The genes here identified as associated with BW seem to act on an animal’s body development, with special emphasis on the DPP6 gene, considering its function in the nervous system. Our discovery also provides more evidences for the genetic relationship of BW with gestation length and calving ease. In addition, our findings corroborate with quantitative genetic studies, which present genetic correlations of moderate to high magnitude between BW and reproductive traits [38], [39].
Weaning Weight (WW)
On BTA 4, a pleiotropic effect for rs43421095 SNP was found between BW and WW, which corroborates with the results obtained by Snelling et al. [12]. These authors found pleiotropic effects for birth weight, weaning weight, and yearling weight; which means that the genes that are acting on body weight performance at earlier ages are also acting later on. On BTA6, the rs135156506 SNP (Table 3) is located in the upstream region of the FARSB gene (phenylalanyl-tRNA synthetase, beta subunit pseudogene); which plays a role in aminoacyl-tRNA biosynthesis pathway. For WW, McClure et al. [35] found a QTL on BTA6 between 8.05 and 43.93 cM that contains the rs135156506 SNP. The KCNIP4 gene (Kv channel-interacting protein 4) has a role in the calcium ion binding, and in potassium and voltage-gated ion channel activity [40]. A QTL associated with yearling weight was found by McClure et al. [35] between 46.86 and 53.72 cM.
The RALGDS gene product (ral guanine nucleotide dissociation stimulator), located on BTA11, is related to the guanyl-nucleotide exchange factor activity and participates in the regulation of small GTPase mediated signal transduction [41], [42]. No biological function for the GTF3C5 gene (general transcription factor 3C polypeptide 5) was found in the literature. Snelling et al. [12] observed three significant SNPs for WW located between 113,506,092 and 114,302,482 bp, on BTA4; five SNPs located between 33,441,527 and 37,584,088 bp, on BTA6; and nine SNPs located between 41,178,449 and 42,609,559 bp, on BTA6. These SNPs were located in similar regions of the SNPs that were significant in the present study. Regarding the Canchim breed, Andrade et al. [20] found associations of WW with microsatellite markers in the IGF1 gene on BTA5.
Long-Yearling Weight (LYW)
For LYW, pleiotropic phenomenon was not observed with BW and WW, which means that different genes are acting in earlier and older ages. Significant SNPs close to the MARCH3 (membrane-associated ring finger (C3HC4) 3), LMNB1 (lamin B1), PHAX (phosphorylated adaptor for RNA export), ALDH7A1 (Aldehyde dehydrogenase family 7 member A1), C7H5orf48 (chromosome 7 open reading frame, human C5orf48), GRAMD3 (GRAM domain containing 3), and LOC100848523 (60S ribosomal protein L26-like) genes and micro RNA MIR2458 (microRNA mir-2458) were observed. According to Fukuda et al. [43], the MARCH3 gene participates in the processes of endocytosis and protein ubiquitination. The LMNB1 gene contributes in the maintenance of structural integrity of a complex or assembly within or outside a cell [44]. The PHAX gene acts in protein transport and in snRNA export from the nucleus to the cytoplasm [45]. The ALDH7A1 gene has a role in the glycine betaine biosynthetic pathway [46]. Information regarding the functions of C7H5orf48, LOC100848523, GRAMD3, and MIR2458 was not found in the consulted literature. Snelling et al. [12] observed a SNP (rs109544319) significantly associated with yearling weight in the C7H5orf48 gene.
In the region where these significant SNPs were located, no QTL for growth traits has been previously found. However, Sherman et al. [47] found a QTL on BTA7 associated with residual feed intake located between 11.70 and 48.50 cM in Angus, Charolais, and hybrid bulls. Maltecca et al. [48] and Druet et al. [49] verified QTL associated with gestation length and spermatic motility located between 16.70 and 39.50 cM and between 25 and 71 cM, respectively.
SNPs associated with LYW, on BTA22, were located close to LARS2 (leucyl-tRNA synthetase 2, mitochondrial), TMEM158 (transmembrane protein 158), CDCP1 (CUB domain containing protein 1), LOC101907967 (uncharacterized LOC101907967), LOC614114 (cytochrome c oxidase subunit VIb polypeptide 1 (ubiquitous) pseudogene), LOC101908013 (cAMP-regulated phosphoprotein, 19kDa pseudogene), ZDHHC3 (zinc finger, DHHC-type containing 3), EXOSC7 (exosome component 7), LOC101908094 (ribosomal protein L29 pseudogene), LOC101901958 (uncharacterized LOC101901958), and CLEC3B (C-type lectin domain family 3 member B) genes. The functions for CDCP1, TMEM158, LOC101907967, LOC614114, LOC101908013, LOC101908094, and LOC101901958 genes were not found in the consulted literature.
The LARS2 gene is involved in aminoacyl-tRNA editing leucyl-tRNA aminoacylation, and in translational fidelity [50]. The ZDHHC3 gene product participates in transferase activity (transferring acyl groups) and in interaction with zinc ion [51]. The EXOSC7 gene participates in positive regulation of cell growth and rRNA processing [52]. The CLEC3B gene participates in calcium ion binding. This gene encodes a protein that is important in bone mineralization, cellular response to transforming growth factor beta stimulus, positive regulation of plasminogen activation, and skeletal system development [25], [53]. QTL associated with yearling weight was found by McClure et al. [35], located between 64.08 and 82.93 cM.
The SNP rs109242147 is located in the intron region of the XYLT1 (xylosyltransferase I) gene, which participates in the cellular response to heat and in the glycosaminoglycan biosynthetic pathway [54]. McClure et al. [35] observed QTL for yearling weight located between 2.24 and 14.44 cM. On BTA27, five SNPs were significantly associated with LYW and are located in the LOC101904868 gene (CUB and sushi domain-containing protein 1-like). However, no biological functions or QTL were reported for this gene in the consulted literature.
Conclusions
The Generalized Quasi-Likelihood Score method identified regions on chromosome associated with birth weight, weaning weight, and long-yearling weight in Canchim and MA animals. New candidate regions for body weight traits were detected and some of them have interesting biological functions, of which most have not been previously reported. The observation of QTL reports for body weight traits, covering areas surrounding the genes (SNPs) herein identified provides more evidence for these associations. Future studies targeting these areas could provide further knowledge to uncover the genetic architecture underlying growth traits in Canchim cattle.
Acknowledgments
The authors would like to acknowledge the Brazilian Agricultural Research Corporation (Embrapa), the Canchim Breed Association, and Andrea Gondo (from Embrapa Beef Cattle) for providing the data used in this study. We thank Dr. David Z. Mokry for English revisions and corrections. The authors would also like to thank the Universidade Estadual Paulista (UNESP) - Faculdade de Ciências Agrárias e Veterinárias, and the University of Guelph for providing computational support for the conducted analyses.
Funding Statement
MEB would like to thank the “National Council of Technological and Scientific Development” (CNPq) (grantee n. 142053/2010-4) and the “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior” (Capes) (grantee n. 5285-11-9). DAG was supported by a Post-Doctoral fellowship at the University of Guelph. DPM, MVGBS, LCAR and MMA were supported by a fellowship from CNPq. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Barwick SA, Henzell AL (2005) Development successes and issues for the future in deriving and applying selection indexes for beef breeding. Aust J Exp Agric 45: 923–933. [Google Scholar]
- 2. Baldi F, Albuquerque LG, Alencar MM (2010) Random regression models on Legendre polynomials to estimate genetic parameters for weights from birth to adult age in Canchim cattle. J Anim Breed Genet 127: 289–299. [DOI] [PubMed] [Google Scholar]
- 3. Buzanskas ME, Grossi DA, Baldi F, Barrozo D, Silva LOC, et al. (2010) Genetic associations between stayability and reproductive and growth traits in Canchim beef cattle. Livest Sci 132: 107–112. [Google Scholar]
- 4. Gaviolli VRN, Buzanskas ME, Cruz VAR, Savegnago RP, Munari DP, et al. (2012) Genetic associations between weight at maturity and maturation rate with ages and weights at first and second calving in Canchim beef cattle. J Appl Genet 53: 331–335. [DOI] [PubMed] [Google Scholar]
- 5.Illumina (2013). Available: http://www.illumina.com/applications/agriculture/plant-animal-genotyping.ilmn.Accessed 2013 Nov 1.
- 6.Affymetrix (2013). Available: http://www.affymetrix.com/estore/catalog/prod440002/AFFY/Axiom%252526%252523174%25253B-Genome%252526%25252345%25253BWide-BOS-1-Bovine-Array#1_1.Accessed 2013 Nov 3.
- 7. Khatkar MS, Moser G, Hayes BJ, Raadsma HW (2012) Strategies and utility of imputed SNP genotypes for genomic analysis in dairy cattle. BMC Genomics 13: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maher B (2008) The case of the missing heritability. Nature 456: 18–21. [DOI] [PubMed] [Google Scholar]
- 10. Zhang Z, Ding X, Liu J, Zhang Q, Koning DJ (2011) Accuracy of genomic prediction using low-density marker panels. J Dairy Sci 94: 3642–3650. [DOI] [PubMed] [Google Scholar]
- 11. Moser G, Khatkar MS, Hayes BJ, Raadsma HW (2010) Accuracy of direct genomic values in Holstein bulls and cows using subsets of SNP markers. Genet Sel Evol 42: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Snelling WM, Allan MF, Keele JW, Kuehn LA, McDaneld T, et al. (2010) Genome-wide association study of growth in crossbred beef cattle. J Anim Sci 88: 837–848. [DOI] [PubMed] [Google Scholar]
- 13. Casas E, Shackelford SD, Keele JW, Stone RT, Kappes SM, et al. (2000) Quantitative trait loci affecting growth and carcass composition of cattle segregating alternate forms of myostatin. J Anim Sci 78: 560–569. [DOI] [PubMed] [Google Scholar]
- 14. Gutiérrez-Gil B, Williams JL, Homer D, Burton D, Haley CS, et al. (2009) Search for quantitative trait loci affecting growth and carcass traits in a cross population of beef and dairy cattle. J Anim Sci 87: 24–36. [DOI] [PubMed] [Google Scholar]
- 15. Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, et al. (2004) Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J Anim Sci 82: 3405–3414. [DOI] [PubMed] [Google Scholar]
- 16. Kühn C, Bennewitz J, Reinsch N, Xu N, Thomsen H, et al. (2003) Quantitative trait loci mapping of functional traits in the German Holstein cattle population. J Dairy Sci 86: 360–368. [DOI] [PubMed] [Google Scholar]
- 17. Schrooten C, Bovenhuis H, Coppieters W, Van Arendonk JAM (2000) Whole genome scan to detect quantitative trait loci for conformation and functional traits in dairy cattle. J Dairy Sci 83: 795–806. [DOI] [PubMed] [Google Scholar]
- 18. Mee JF (2008) Prevalence and risk factors for dystocia in dairy cattle: a review. Vet J 176: 93–101. [DOI] [PubMed] [Google Scholar]
- 19. Mokry FB, Higa RH, Mudadu MA, Lima AO, Meirelles SLC, et al. (2013) Genome-wide association study for backfat thickness in Canchim beef cattle using Random Forest approach. BMC Genet 14: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrade PC, Grossi DA, Paz CCP, Alencar MM, Regitano LCA, et al. (2008) Association of an insulin-like growth factor 1 gene microsatellite with phenotypic variation and estimated breeding values of growth traits in Canchim cattle. Anim Genet 39: 480–485. [DOI] [PubMed] [Google Scholar]
- 21.Barbosa PF (2000) O Canchim na Embrapa Pecuária Sudeste. Available: http://www.alice.cnptia.embrapa.br/bitstream/doc/44905/1/PROCIPFB2000.00028.pdf. Accessed 2013 Jan 10.
- 22. Machado MBB, Alencar MM, Pereira AP, Oliveira HN, Casas E, et al. (2003) QTL affecting body weight in a candidate region of cattle chromosome 5. Genet Mol Biol 26: 259–265. [Google Scholar]
- 23. Misztal I (2008) Reliable computing in estimation of variance components. J Anim Breed Genet 125: 363–370. [DOI] [PubMed] [Google Scholar]
- 24.Illumina (2013) Technical Note - Infinium Genotyping Data Analysis. Available: http://res.illumina.com/documents/products/technotes/technote_infinium_genotyping_data_analysis.pdf. Accessed 2013 Nov 1.
- 25. Zimin A V, Delcher AL, Florea L, Kelley DR, Schatz MC, et al. (2009) A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol 10: r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng Z, Wong WWL, Gao X, Schenkel F (2011) Generalized genetic association study with samples of related individuals. Ann Appl Stat 5: 2109–2130. [Google Scholar]
- 27.Heyde CC (1997) Quasi-Likelihood and its application: A general approach to optimal parameter estimation. Canberra: Springer. 235p.
- 28. Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B 57: 289–300. [Google Scholar]
- 29.National Center for Biotechnology Information (2013). Available: http://www.ncbi.nlm.nih.gov/snp.Accessed 2013 Oct 1.
- 30.Ensembl Genome Browser (2013). Available: http://www.ensembl.org/index.html.Accessed 2013 Oct 1.
- 31.UniProt (2013). Available: http://www.uniprot.org/.Accessed 2013 Oct 1.
- 32.AnimalQTLdb (2013). Available: http://www.animalgenome.org/cgi-bin/QTLdb/index.Accessed 2013 Oct 1.
- 33. Hill WGG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38: 226–231. [DOI] [PubMed] [Google Scholar]
- 34. Wada K, Yokotani N, Hunter C, Doi K, Wenthold RJ, et al. (1992) Differential expression of two distinct forms of mRNA encoding members of a dipeptidyl aminopeptidase family. Proc Natl Acad Sci U S A 89: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, et al. (2010) A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet 41: 597–607. [DOI] [PubMed] [Google Scholar]
- 36. Kukushkin N V, Easthope IS, Alonzi DS, Butters TD (2012) Restricted processing of glycans by endomannosidase in mammalian cells. Glycobiology 22: 1282–1288. [DOI] [PubMed] [Google Scholar]
- 37. Alexander LJ, Geary TW, Snelling WM, Macneil MD (2007) Quantitative trait loci with additive effects on growth and carcass traits in a Wagyu-Limousin F2 population. Anim Genet 38: 413–416. [DOI] [PubMed] [Google Scholar]
- 38. Mucari TB, Alencar MM, Barbosa PF, Barbosa RT (2011) Análise genética do período de gestação em animais de um rebanho Canchim: estimação de parâmetros genéticos e escolha entre modelos animais alternativos. Rev Bras Zootec 40: 1211–1216. [Google Scholar]
- 39. Crews Jr DH (2006) Age of dam and sex of calf adjustments and genetic parameters for gestation length in Charolais cattle. J Anim Sci 84: 25–31. [DOI] [PubMed] [Google Scholar]
- 40. An WF, Bowlby MR, Betty M, Cao J, Ling HP, et al. (2000) Modulation of A-type potassium channels by a family of calcium sensors. Nature 403: 553–556. [DOI] [PubMed] [Google Scholar]
- 41. Miller MJ, Prigent S, Kupprman E, Rioux L, Park S-H, et al. (1997) RalGDS Functions in Ras- and cAMP-mediated Growth Stimulation. J Biol Chem 272: 5600–5605. [DOI] [PubMed] [Google Scholar]
- 42. Herrmann C, Horn G, Spaargaren M, Wittinghofer A (1996) Differential interaction of the ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J Biol Chem 271: 6794–6800. [DOI] [PubMed] [Google Scholar]
- 43. Fukuda H, Nakamura N, Hirose S (2006) MARCH-III Is a novel component of endosomes with properties similar to those of MARCH-II. J Biochem 139: 137–145. [DOI] [PubMed] [Google Scholar]
- 44. Lin F, Worman HJ (1995) Structural Organization of the Human Gene (LMNB1) Encoding Nuclear Lamin B1. Genomics 27: 230–236. [DOI] [PubMed] [Google Scholar]
- 45. Ohno M, Segref A, Bachi A, Wilm M, Mattaj IW (2000) PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell 101: 187–198. [DOI] [PubMed] [Google Scholar]
- 46. Brocker C, Lassen N, Estey T, Pappa A, Cantore M, et al. (2010) Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J Biol Chem 285: 18452–18463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sherman EL, Nkrumah JD, Li C, Bartusiak R, Murdoch B, et al. (2009) Fine mapping quantitative trait loci for feed intake and feed efficiency in beef cattle. J Anim Sci 87: 37–45. [DOI] [PubMed] [Google Scholar]
- 48. Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A (2009) Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein x Jersey crossbred population. Anim Genet 40: 27–34. [DOI] [PubMed] [Google Scholar]
- 49. Druet T, Fritz S, Sellem E, Basso B, Gérard O, et al. (2009) Estimation of genetic parameters and genome scan for 15 semen characteristics traits of Holstein bulls. J Anim Breed Genet 126: 269–277. [DOI] [PubMed] [Google Scholar]
- 50. ’t Hart LM, Hansen T, Rietveld I, Dekker JM, Nijpels G, et al. (2005) Evidence that the Mitochondrial Leucyl tRNA Synthetase (LARS2) Gene Represents a Novel Type 2 Diabetes Susceptibility Gene. Diabetes 54: 1892–1895. [DOI] [PubMed] [Google Scholar]
- 51. Putilina T, Wong P, Gentleman S (1999) The DHHC domain: a new highly conserved cysteine-rich motif. Mol Cell Biochem 195: 219–226. [DOI] [PubMed] [Google Scholar]
- 52. Houseley J, Tollervey D (2009) The many pathways of RNA degradation. Cell 136: 763–776. [DOI] [PubMed] [Google Scholar]
- 53. Wewer UM, Ibaraki K, Schjørring P, Durkin ME, Young MF, et al. (1994) A potential role for tetranectin in mineralization during osteogenesis. J Cell Biol 127: 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stoolmiller AC, Horwitz AL, Dorfman A (1972) Biosynthesis of the Chondroitin Sulfate Proteoglycan. J Biol Chem 247: 3525–3532. [PubMed] [Google Scholar]