

Abstract
Background
Increasingly, evidence from studies in both animal models and patients suggests that cardiovascular dysfunction is important in HD. Previous studies measuring function of the left ventricle (LV) in the R6/2 model have found a clear cardiac abnormality, albeit with preserved LV systolic function. It was hypothesized that an impairment of RV function might play a role in this condition via mechanisms of ventricular interdependence.
Objective
To investigate RV function in the R6/2 mouse model of Huntington’s disease (HD)
Methods
Cardiac cine-magnetic resonance imaging (MRI) was used to determine functional parameters in R6/2 mice. In a first experiment, these parameters were derived longitudinally to determine deterioration of cardiac function with disease progression. A second experiment compared the response to a stress test (using dobutamine) of wildtype and early-symptomatic R6/2 mice.
Results
There was progressive deterioration of RV systolic function with age in R6/2 mice. Furthermore, beta-adrenergic stimulation with dobutamine revealed RV dysfunction in R6/2 mice before any overt symptoms of the disease were apparent.
Conclusions
This work adds to accumulating evidence of cardiovascular dysfunction in R6/2 mice, describing for the first time the involvement of the right ventricle. Cardiovascular dysfunction should be considered, both when treatment strategies are being designed, and when searching for biomarkers for HD.
Keywords: MRI, right ventricle, cognitive function, heart failure, Huntington’s Disease
Introduction
Huntington’s disease (HD) is a hereditary disorder caused by a CAG repeat expansion in the HTT gene. During the course of the disease, progressive neurodegeneration in the basal ganglia and cerebral cortex occurs, which causes motor, cognitive and psychological problems. Neurological symptoms are the most prominent and devastating consequences of the disease and understandably, over the past two decades, research has focused mostly on the central nervous system dysfunction and pathology. However, the brain is not the only organ involved in HD. The gene is ubiquitously expressed and the protein is found in most peripheral tissues, including skeletal muscle, liver and heart [1]. As a consequence, in addition to neurological symptoms, HD leads to a number of systemic complications, such as metabolic dysfunction including cachexia [2, 3].
There is a growing awareness of cardiac dysfunction in HD. Heart failure (HF) is a major cause of death in HD patients [4-6], and HD patients are 15 times more likely to suffer from heart disease than age-matched controls [7]. It is well established that HD patients suffer from impairments in the autonomic nervous system [8] and circadian control [9-12]. These abnormalities have been found in pre-symptomatic mutation carriers (PHD) and in early-symptomatic patients, in the form of sleep disturbances and an impaired cardiovascular response (e.g. diastolic arterial blood pressure and heart rate) to cognitive challenge [11] or postural changes [10]. It has been suggested that these symptoms are related to loss of dopamine receptors, microglia activation and neural inclusions in the hypothalamus of PHD individuals [9], although it is possible that the cardiovascular system is directly involved.
Circadian changes in daily activity patterns have been observed in several mouse models of HD [13], including R6/2 mice [14-16], R6/1 mice [17] and BACHD mice [16]. Interestingly, it has been shown that circadian control of autonomic regulation of heart rates was disrupted in both BACHD [16] and R6/1 mice [17]. Analysis of ECG data in early stage R6/1 mice revealed different types of arrhythmia, which are thought to be caused by impairments in cardiovagal modulation [17]. Abnormalities in areas of the brain implicated in autonomic regulation were also reported. Mild effects on systemic blood pressure and on the size of myocytes in the R6/1 model have been shown, although gross functional deficits in left ventricular (LV) function were not observed [17].
While the autonomic nervous system is very likely to play an important role in HD cardiac dysfunction, some evidence suggests that HF may be a result of direct expression of huntingtin in cardiomyocytes [7]. Indeed, a recent study has shown that cardiomyocyte-restricted expression of a protein carrying a polyglutamine repeat can alone induce heart disease in wildtype (WT) mice, in a way that is dependent on the number of glutamine repeats and not on the levels of expression of the protein [18]. Expression of low levels of a peptide containing 83 glutamine repeats caused both deposition of pre-amyloid oligomers and formation of polyglutamine-containing aggresomes within the cardiomyocytes, leading to cardiomyocyte loss, progressive cardiac dilation and eventual death from HF by approximately 5–7 months [19].
In this study we were interested in investigating the role of the right ventricle (RV) in the cardiac phenotype seen in R6/2 mice. The R6/2 mouse has proved to be a useful model of HD, displaying, as it does, neurodegeneration [20, 21], behavioural abnormalities [22-27], and altered daily activity patterns [14-16]. Cardiovascular function has also been studied in R6/2 mice using both echocardiogram [28] and magnetic resonance imaging (MRI) [29]. With both approaches, progressive dysfunction with reduced left ventricular (LV) volume was found previously, though with a preserved LV systolic function [28] [29]. Additionally, changes in the shape of the LV during systole including bowing of the septum were reported [29]. To our knowledge, analysis of the RV in this mouse model has never been reported in literature. Although the RV is known to be involved in many pathological conditions, its role in disease is less investigated compared to the LV, and is a current focus for cardiovascular research [30].
Here we hypothesized that an impaired RV function, and therefore reduced pulmonary circulation, might be an important component of the circulatory problems seen in HD mice. In this study we used in vivo MRI to determine whether the RV was involved in R6/2 cardiac pathology. We characterised this over time, and also used a pharmacological stress test (dobutamine) to determine whether or not these effects could be unmasked earlier in the disease.
Methods
This study was carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986, and with the approval of the University of Cambridge Ethical Review Panel.
Animals
Mice were taken from a colony of R6/2 transgenic mice [31] established at the University of Cambridge, and maintained by backcrossing onto CBA × C57BL6N F1 female mice. Genotyping and CAG repeat length measurement were carried out by Laragen (Los Angeles, CA, USA) as described previously [32]. The R6/2 mice used in this study had a mean CAG repeat length of 226 ± 7 (mean ± s.d.). Mice were kept in home cages comprising single sex, single genotype groups. All of the mice lived in an enhanced environment (24). Clean cages were provided twice weekly with grade 8/10-corncob bedding, and finely shredded paper for nesting. The mice were maintained on a 12:12 hour light/dark (LD) cycle, at a temperature of 21–23° C and a humidity of 55 ± 10%. The mice had ad libitum access to water (using water bottles with elongated spouts) and dry laboratory food (RM3(E) rodent pellets, Special Diet Services, Witham, UK). In addition, once a day, a mash was prepared for the R6/2 mice by soaking 100 g dry food in 230 ml water until the pellets were soft and fully expanded. The mash was placed on the cage floor, improving access to food and water. This feeding regime has been shown previously to be beneficial [33].
Magnetic Resonance Imaging
Two experiments were performed: the first to evaluate the functional parameters longitudinally in transgenic mice (n=5) that were not exposed to dobutamine, the second to evaluate the difference in the effect of dobutamine on C57BL6 WT (n=6) and R6/2 (n=6) mice at a presymptomatic timepoint.
Longitudinal characterization
Five male R6/2 mice were scanned at three time points, designed to capture an initial period before severe symptoms developed (t1=7±1 weeks of age; mean ± s.d.) and two later scans reflecting different stages of pathology. In R6/2 mice, body weight increases with development and, after the onset of neurological symptoms, it falls as a consequence of disease progression [34, 35]. Disease-induced weight loss was therefore used for pathology staging. To evaluate changes in heart size independently of bodyweight, a second scan (t2=14±1 weeks of age) was performed when the bodyweight had decreased to the same point as it was at the initial scan. A third scan was performed at a later stage of the disease, when the bodyweight was 75% of the initial weight (t3=16±1 weeks of age).
Stress test
Six male R6/2 mice and six male WT controls (C57Bl6N/CBA) were scanned at 10 weeks of age. This corresponded to the last time point before the disease phenotype starts to show and body weight starts to decrease. In addition to the protocol described below, pharmacological stress was induced with an in situ intraperitoneal (i.p.) bolus injection of dobutamine (12 μg/g) to induce chronotropic, as well as inotropic and lusitropic response [36]. For 30 minutes following injection three short axis slices were acquired in order to observe the changes in LV and RV volumes caused by beta-adrenergic stimulation.
Imaging protocol
Anaesthesia was induced with 3% isoflurane in 1l/min O2 and maintained with 1-2% isoflurane in 1l/min O2. A pressure sensor for respiration rate was used to monitor anaesthesia depth and a rectal sensor was utilised to monitor the core temperature, which was maintained at 36-37° with the use of a flowing-water heating blanket. Gating of the MRI sequences was achieved prospectively with electrocardiogram (ECG). The ECG trace was visually inspected between MRI sequences to identify severe arrhythmias if present. MRI was performed at 4.7T with a Bruker BioSpec 47/40 system (Bruker Inc., Ettlingen, Germany). A birdcage coil of 12cm was used for signal excitation and a 2cm surface coil for signal reception with the animals positioned prone. After initial localization images, 4-chamber and 2-chamber views were acquired (FISP, TR/TE 7ms/2.4ms, 13-20 frames, 3.5 cm FOV, 256×256 matrix, 1 mm slice thickness, bandwidth 64kHz, flip angle 20°, NEX 2). Using these scans as a reference, short axis slices were arranged perpendicularly to both the long-axis views (FISP, TR/TE 7ms/2.4ms, 13-20 frames, 3.5 cm FOV, 256x256 matrix, 1 mm slice thickness, bandwidth 64.1kHz, flip angle 20°, NEX 2). Full LV and RV coverage was achieved with no slice gap with 8-10 slices following Weissman et al [37].
Data analysis
All of the delineations were performed by a single investigator, blind to genotype or stage of the disease. Delineation of the LV and RV at each phase of the cardiac cycle excluded the papillary muscles and trabeculations throughout. The regions from each slice were combined using Simpson’s rule to provide LV mass as well as end diastolic volume (EDV), end systolic volume (ESV), stroke volume (SV) and ejection fraction (EF) for both LV and RV using Segment v1.9 [38]. To analyse the effects of inotropic stimulation, ejection fraction from the slices acquired 30 minutes after the injection of dobutamine was compared with the same slices acquired at baseline performing a paired Student’s t-test.
In the longitudinal analysis parameters were tested for significant differences using a paired Student’s t-test. Differences between WT and R6/2 mice were tested with a two samples different variance t-test.
Results
During MRI examinations, mean heart rates were in the range of 350-450 beats/minute, and respiratory rates were between 25-65 breaths/minute. Visual inspection of the ECG signals during the MRI studies did not reveal arrhythmia in any subject.
Cardiac functional parameters and body weight at different stages of the disease in R6/2 mice are shown in Table 1. R6/2 mice develop normally, and body weight increased as expected in the first 8-10 weeks. Body weight then falls as a result of disease progression. For this reason, weight was the same at 7 weeks (t1) and 14 weeks (t2), and had declined further by 16 weeks (t3). No significant change in heart rate was observed due to disease progression. Nevertheless, we observed a significant decrease in LV mass starting from middle stage of the disease (t2). At a later stage (t3), we observed a substantial decrease in cardiac output. The LV volume measurements (Table 1) show a progressive reduction of EDV and ESV, consistent with the reduced cardiac output, but LVEF was unaffected.
Table 1. Cardiac parameters for R6/2 mice at different disease stages.
| Age (wk) | 7± 1 | 14± 1 | 16± 1 |
|---|---|---|---|
| (presymptomatic) | (symptomatic) | (late stage) | |
| mean ± s.d. | mean ± s.d. | mean ± s.d. | |
| Body weight (g) | 23 ± 1 | 23 ± 1 | 16 ± 1 ** |
| Heart rate (bpm) | 425 ± 40 | 400 ± 80 | 375 ± 40 |
| LVM (μl) | 76 ± 10 | 65 ± 7 ** | 58 ± 9 * |
| LVEDV (μl) | 42 ± 5 | 40 ± 5 | 30 ± 5 * |
| LVESV (μl) | 15 ± 1 | 11 ± 2 | 8 ± 4 * |
| LVEF (%) | 65 ± 4 | 72 ± 6 | 73 ± 7 |
| LVSV (μl) | 28 ± 3 | 29 ± 5 | 21 ± 2 * |
| LVCO (ml/min) | 12 ± 1 | 11 ± 3 | 8.0 ± 0.7 * |
| RVEDV (μl) | 40 ± 4 | 42 ± 9 | 44 ± 6 |
| RVESV (μl) | 12 ± 2 | 14 ± 6 | 22 ± 6 * |
| RVEF (%) | 71 ± 5 | 68 ± 10 | 51 ± 9 * |
| RVSV (μl) | 28 ± 4 | 29 ± 6 | 22 ± 2 * |
| RVCO (ml/min) | 12 ± 1 | 11 ± 3 | 8.2 ± 0.7 * |
p<0.05,
p<0.01.
Visual inspection of the short-axis images (Figure 1) showed a substantial impairment of RV systolic function. Remodelling of the RV was represented by a substantial increase in ESV, as confirmed by the volumetric measurements (Table 1). RVEF measurements showed significant deterioration from 71% to 51%.
Figure 1. Right ventricular function during systole in WT and R6/2 mice.

(a) Cartoon of the long axis view of the mouse heart, showing the position of the mid-ventricular short axis slice seen in (b). (b) Images from mid-ventricular short-axis slices from WT (upper panels) and R6/2 (lower panels) mice, showing an enlarged RV throughout systole in 16 weeks old R6/2 mice.
At 10 weeks of age, when R6/2 mice show no overt symptoms, LV mass and ventricle volumes were similar in WT and R6/2 mice (Table 2). Furthermore, no significant differences in LVEF or RVEF were present between WT and R6/2 before inotropic stimulation. After dobutamine administration, we observed a significant increase in the heart rate for both groups (p<0.001), although the increase was not significantly different between the two groups (20 ± 15 % in WT, 30 ± 15 % in R6/2 mice, p>0.10). Nevertheless, under dobutamine-induced stress, RV systolic function dropped dramatically in the R6/2 mice, but not in the WT mice (Figure 2). Decrease in RV function of R6/2 mice was driven by a significant increase in both RVEDV and RVESV (Table 3). LV ejection fraction significantly increased in both groups (p<0.001), but there was no significant difference in the effect of dobutamine on LVEF on mice of different genotypes.
Table 2. Cardiac parameters in WT and R6/2 mice at 10 weeks, prior to dobutamine administration.
| Baseline values (mean ± s.d.) | ||
|---|---|---|
| Genotype | WT | R6/2 |
| Age (wk) | 10 ± 2 | 10 ± 2 |
| Body weight (g) | 25 ± 5 | 25 ± 4 |
| LVM (μl) | 84 ± 15 | 79 ± 14 |
| LVEDV (μl) | 46 ± 11 | 43 ± 10 |
| LVESV (μl) | 13 ± 5 | 14 ± 5 |
| LVEF (%) | 73 ± 4 | 68 ± 5 |
| RVEDV (μl) | 42 ± 8 | 38 ± 10 |
| RVESV (μl) | 11 ± 3 | 10 ± 4 |
| RVEF (%) | 74 ± 5 | 75 ± 4 |
Figure 2. Changes in ejection fractions during dobutamine stress test.
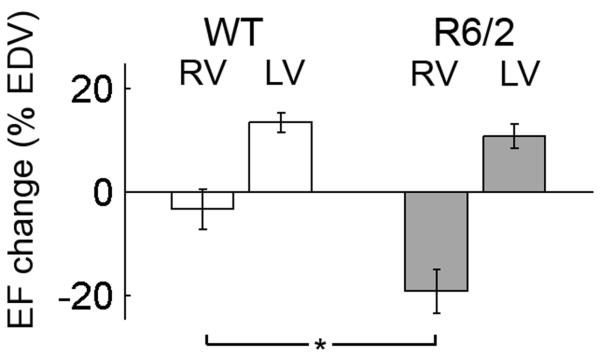
RV = right ventricle, LV = left ventricle.
Data are means ± SEM. *=p<0.05
Table 3. Percentage changes in ventricular volumes caused by dobutamine.
| % increase (mean ± s.d.) | ||
|---|---|---|
| Genotype | WT | R6/2 |
| LVEDV (μl) | −32 ± 10 | −35 ± 10 |
| LVESV (μl) | −71 ± 30 | −50 ± 15 |
| RVEDV (μl) | −5 ± 15 | 15 ± 40 |
| RVESV (μl) | 30 ± 15 | 115 ± 50 |
Discussion
Heart disease is an important contributor to physical decline and represents one of the major causes of death in HD patients [4-6]. However, the pathophysiology of heart failure in HD has been relatively unexplored during the past decades [7]. Recent studies using a cardiomyocyte-restricted model were crucial in proving that production of a polypeptide expressing a polyglutamine repeat is sufficient to cause HF [18]. Nevertheless, an in vivo model of the disease would also be of great value in understanding not only the mechanisms behind HF in HD, but also the impact of novel pharmacological strategies that might affect the circulatory system. Our data add to accumulating evidence that cardiac dysfunction is an important part of the R6/2 mouse pathological phenotype. In vivo MRI confirmed a progressive reduction in LV output with disease progression in R6/2 mice, although LVEF was preserved. This study used only a small number of animals, though the results were significant and findings were consistent with recent reports [28, 29].
Measurement of LV mass has not previously been performed in vivo in the R6/2 model. We found significant muscle loss in the myocardium at early to middle stage of the disease. We also found that muscle loss in the heart is not proportional to the reduction of bodyweight, as was previously conjectured [29]. R6/2 mice lose body weight from approximately 12 weeks of age, so body weight at 14 weeks was the same as it had been at 7 weeks. However, there was a decrease in LV mass of approximately 14% between 7 and 14 weeks time points. This disproportionate loss in LV mass indicates atrophy that is proportionally greater in the heart than the rest of the body. These data suggest that heart failure in HD is not merely a consequence of other symptoms, but may develop in parallel.
Measurements of the RV function showed for the first time a progressive deterioration in R6/2 mice, which reaches a severe stage by 16 weeks of age. Interestingly, this dysfunction can be unmasked at the initial stage of the disease using the beta-adrenergic agonist dobutamine. These results highlight a specific pathology of the RV in R6/2 mice, which might be useful as a biomarker of HD cardiac pathology in this model. The RV and the LV are highly interdependent, and in some disease states the dilation of the RV shifts the interventricular septum toward the left, changing LV geometry. These changes contribute to a low cardiac output state by decreasing LV distensibility and preload [39]. The measured impairment in RV function is therefore likely to influence the LV cardiac output seen in these mice through mechanisms of ventricular interdependence [39], thereby diminishing the systemic blood supply and contributing to physical decline. Several factors indicate that RV pathology might be synchronous with, rather than a consequence of, autonomic nervous system defects. Firstly, this pathology is present in presymptomatic animals, when there are no explicit signs of severe defects in autonomic nervous system regulation. Secondly, during inotropic stimulation the RV responds showing a dramatic decrease in systolic function, whereas heart rate and LV systolic function respond normally as previously seen in WT mice [40].
The R6/2 mouse model of HD displays LV and RV remodelling, with preserved LV function and chronotropic response, accompanied by a substantial reduction in LV mass. Interestingly, our findings resemble those seen in the mdx mouse model of muscular dystrophy [41]. The mdx mouse is a dystrophin-deficient model that has been characterised by many investigators over the past 20 years. It shows cardiomyopathy together with skeletal muscle deficiencies [42]. In mdx mice, RV remodelling begins several months before LV dysfunction. In common with the present findings in R6/2 mice, these changes can be unmasked with a dobutamine stress test before overt symptoms are present [43]. Although the phenotypes appear with different time courses in the two models, the similarities of the pathology are suggestive of a similar mechanism. As is observed in muscular dystrophy, the progressive RV dysfunction could be due to pulmonary hypertension and functional degeneration of the diaphragm muscle [44]. During dobutamine stimulation, the increased LV contractility is not matched by the RV, contributing to an elevation of RV pressure and volume [41, 45]. This stress-mediated abnormality represents an early measure of cardiovascular impairment, anticipating most of the other symptoms of the disease in the R6/2 mouse model.
During the past 70 years, research on the cardiopulmonary system has lagged behind compared to work on systemic circulation. In the past, procedures such as Fontaine surgery and cauterization of the RV lateral wall in dog models have led clinicians to consider the RV to be ‘unnecessary’ [46]. Only recently have technical developments in echocardiography and MRI led to renewed interest in the RV, seen in the broader context of the cardiopulmonary system [46]. Although there is limited information on RV function, impairment plays a significant part in a number of disease states including valvular heart disease, congenital heart disease, left HF, complications from heart surgery, idiopathic pulmonary artery hypertension, chronic pulmonary disease, acute respiratory distress and sepsis [39, 47]. Furthermore, the extent of RV dysfunction is a predictor of outcome in these diseases, and in 2006, the National Heart, Lung, and Blood Institute identified RV physiology as a priority area in cardiovascular research [30]. While respiratory function during disease progression in HD is gaining interest [48] and is being actively studied in a clinical trial (IRSCTN72770961), we have been unable to find any accounts measuring RV function, and we hope that our findings in R6/2 mice will encourage this investigation.
Imaging the RV remains a challenging task because of fibre orientation, complex geometry and contraction pattern [39]. However, MRI is an accurate tool for measuring RV function in humans [39], as well as in mice [37], since it is capable of high spatial, temporal resolution and of arranging oblique slices to form 3D stacks. In small animal systems, accurate monitoring, gating, in situ drug delivery, and slice planning still pose difficulty and require a certain degree of expertise. Different heart rates and physical size mean that MRI protocols optimised for patients need substantial modifications to be efficient in mice [49]. The ability to measure pharmacologically-induced stress in vivo with dobutamine, a drug that is commonly used in the assessment of patients with suspected coronary artery disease [50], represents an excellent tool for increasing the sensitivity of cardiac MRI exams in particular clinical conditions. As dobutamine-stress MRI measurements are suitable for use in humans, they are an appropriate platform for addressing the effects of novel cardiac drug treatments in HD mice. In particular, it will be possible to compare changes in cardiac function induced by cardioprotective agents, drugs acting on blood pressure or on the function of the diaphragm, with alterations in the longevity and neurological condition in this model. We believe that increased attention to cardiovascular dysfunction in HD patients may lead to novel treatment approaches that will be beneficial.
In conclusion, these data add to previous findings of LV pathology in the R6/2 mice by showing that RV function is also impaired. Importantly, these changes can be unmasked in young, otherwise asymptomatic animals with the beta-adrenergic agonist dobutamine. Using a serial and non-invasive method, this work successfully identified a specific dysfunction of the cardiovascular system in this model of human disease. More detailed study of the mechanisms behind these changes may help elucidate the increased mortality from heart disease seen in HD patients, and lead to new pharmacological strategies designed to improve the lives of HD sufferers.
Acknowledgements
This work was funded by Cure Huntington’s Disease Initiative (CHDI) Foundation, Inc. We thank Dr Zhiguang Zheng and Wendy Leavens for technical assistance.
List of Abbreviations
- LV
left ventricle
- RV
right ventricle
- LVM
left ventricular mass
- LVED
left ventricular end diastolic volume
- LVES
left ventricular end systolic volume
- LVSV
left ventricular stroke volume
- LVEF
left ventricular ejection fraction
- RVED
right ventricular end diastolic volume
- RVES
right ventricular end systolic volume
- RVSV
right ventricular stroke volume
- RVEF
right ventricular ejection fraction
Footnotes
Conflict of interest: The authors have no conflicts to disclose.
References
- 1.Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, et al. Formation of Polyglutamine Inclusions in Non-CNS Tissue. Human molecular genetics. 1999;8:813–22. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert GJ. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2009;73:572. doi: 10.1212/WNL.0b013e3181af0cf4. [DOI] [PubMed] [Google Scholar]
- 3.Aziz NA, van der Burg JMM, Landwehrmeyer GB, Brundin P, Stijnen T, Roos RAC. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71:1506–13. doi: 10.1212/01.wnl.0000334276.09729.0e. [DOI] [PubMed] [Google Scholar]
- 4.Chiu E, Alexander L. Causes of death in Huntington’s disease. The Medical journal of Australia. 1982;1:153. doi: 10.5694/j.1326-5377.1982.tb132224.x. [DOI] [PubMed] [Google Scholar]
- 5.Lanska DJ, Lanska MJ, Lavine L, Schoenberg BS. Conditions associated with Huntington’s disease at death. A case-control study. Archives of neurology. 1988;45:878–80. doi: 10.1001/archneur.1988.00520320068017. [DOI] [PubMed] [Google Scholar]
- 6.Lanska DJ, Lavine L, Lanska MJ, Schoenberg BS. Huntington’s disease mortality in the United States. Neurology. 1988;38:769. doi: 10.1212/wnl.38.5.769. [DOI] [PubMed] [Google Scholar]
- 7.van der Burg JMM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet neurology. 2009;8:765–74. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- 8.Andrich J, Schmitz T, Saft C, Postert T, Kraus P, Epplen JT, et al. Autonomic nervous system function in Huntington’s disease. Journal of neurology, neurosurgery, and psychiatry. 2002;72:726–31. doi: 10.1136/jnnp.72.6.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aziz NA, Anguelova GV, Marinus J, van Dijk JG, Roos RAC. Autonomic symptoms in patients and pre-manifest mutation carriers of Huntington’s disease. European journal of neurology. 2010;17:1068–74. doi: 10.1111/j.1468-1331.2010.02973.x. [DOI] [PubMed] [Google Scholar]
- 10.Bär KJ, Boettger MK, Andrich J, Epplen JT, Fischer F, Cordes J, et al. Cardiovagal modulation upon postural change is altered in Huntington’s disease. European journal of neurology. 2008;15:869–71. doi: 10.1111/j.1468-1331.2008.02173.x. [DOI] [PubMed] [Google Scholar]
- 11.Kobal J, Melik Z, Cankar K, Bajrovic FF, Meglic B, Peterlin B, et al. Autonomic dysfunction in presymptomatic and early symptomatic Huntington’s disease. Acta neurologica Scandinavica. 2010;121:392–9. doi: 10.1111/j.1600-0404.2009.01251.x. [DOI] [PubMed] [Google Scholar]
- 12.Aziz NA, Anguelova GV, Marinus J, Lammers GJ, Roos RA. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism and related disorders. 2010 Jun;16(5):345–50. doi: 10.1016/j.parkreldis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Crook ZR, Housman D. Huntington’s disease: can mice lead the way to treatment? Neuron. 2011;69:423–35. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. The Journal of neuroscience. 2005;25:157–63. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH, et al. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. The Journal of neuroscience. 2010;30:10199–204. doi: 10.1523/JNEUROSCI.1694-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, et al. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Experimental neurology. 2011;228:80–90. doi: 10.1016/j.expneurol.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiriazis H, Jennings NL, Davern P, Lambert G, Su Y, Pang T, et al. Neurocardiac dysregulation and neurogenic arrhythmias in a transgenic mouse model of Huntington’s disease. The Journal of physiology. 2012;590:5845–60. doi: 10.1113/jphysiol.2012.238113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pattison JS, Robbins J. Protein misfolding and cardiac disease: establishing cause and effect. Autophagy. 2008;4:821–3. doi: 10.4161/auto.6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation. 2008;117:2743–51. doi: 10.1161/CIRCULATIONAHA.107.750232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA. Voxel-based morphometry in the R6/2 transgenic mouse reveals differences between genotypes not seen with manual 2D morphometry. Neurobiology of disease. 2009;33:20–7. doi: 10.1016/j.nbd.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA. Use of magnetic resonance imaging for anatomical phenotyping of the R6/2 mouse model of Huntington’s disease. Neurobiology of disease. 2009;33:12–9. doi: 10.1016/j.nbd.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 22.Wood NI, Glynn D, Morton AJ. “Brain training” improves cognitive performance and survival in a transgenic mouse model of Huntington’s disease. Neurobiology of disease. 2011;42:427–37. doi: 10.1016/j.nbd.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, et al. Characterization of Progressive Motor Deficits in Mice Transgenic for the Human Huntington’s Disease Mutation. J Neurosci. 1999;19:3248–57. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciamei A, Morton AJ. Rigidity in social and emotional memory in the R6/2 mouse model of Huntington’s disease. Neurobiology of learning and memory. 2008;89:533–44. doi: 10.1016/j.nlm.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Ciamei A, Morton AJ. Progressive imbalance in the interaction between spatial and procedural memory systems in the R6/2 mouse model of Huntington’s disease. Neurobiology of learning and memory. 2009;92:417–28. doi: 10.1016/j.nlm.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. The Journal of neuroscience. 1999;19:10428–37. doi: 10.1523/JNEUROSCI.19-23-10428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wood NI, Carta V, Milde S, Skillings EA, McAllister CJ, Ang YLM, et al. Responses to environmental enrichment differ with sex and genotype in a transgenic mouse model of Huntington’s disease. PloS one. 2010;5:e9077. doi: 10.1371/journal.pone.0009077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mihm MJ, Amann DM, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR. Cardiac dysfunction in the R6/2 mouse model of Huntington’s disease. Neurobiology of disease. 2007;25:297–308. doi: 10.1016/j.nbd.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood NI, Sawiak SJ, Buonincontri G, Niu Y, Kane AD, Carpenter TA, et al. Direct Evidence of Progressive Cardiac Dysfunction in a Transgenic Mouse Model of Huntington’s Disease. Journal of Huntington’s Disease. 2012;1(1):65–72. doi: 10.3233/JHD-2012-120004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–91. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 31.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 32.Duzdevich D, Li J, Whang J, Takahashi H, Takeyasu K, Dryden DTF, et al. Unusual structures are present in DNA fragments containing super-long Huntingtin CAG repeats. PloS one. 2011;6:e17119. doi: 10.1371/journal.pone.0017119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter RJ, Hunt MJ, Morton AJ. Environmental stimulation increases survival in mice transgenic for exon 1 of the Huntington’s disease gene. Movement disorders. 2000;15:925–37. doi: 10.1002/1531-8257(200009)15:5<925::aid-mds1025>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 34.Morton AJ, Glynn D, Leavens W, Zheng Z, Faull RL, Skepper JN, et al. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiol Dis. 2009 Mar;33(3):331–41. doi: 10.1016/j.nbd.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA. Voxel-based morphometry with templates and validation in a mouse model of Huntington’s disease. Magnetic resonance imaging. 2013 Nov;31(9):1522–31. doi: 10.1016/j.mri.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyrankiewicz U, Skorka T, Jablonska M, Petkow-Dimitrow P, Chlopicki S. Characterization of the cardiac response to a low and high dose of dobutamine in the mouse model of dilated cardiomyopathy by MRI in vivo. Journal of magnetic resonance imaging : JMRI. 2013;37:669–77. doi: 10.1002/jmri.23854. [DOI] [PubMed] [Google Scholar]
- 37.Wiesmann F, Frydrychowicz A, Rautenberg J, Illinger R, Rommel E, Haase A, et al. Analysis of right ventricular function in healthy mice and a murine model of heart failure by in vivo MRI. American journal of physiology Heart and circulatory physiology. 2002;283:H1065–71. doi: 10.1152/ajpheart.00802.2001. [DOI] [PubMed] [Google Scholar]
- 38.Heiberg E, Sjögren J, Ugander M, Carlsson M, Engblom H, Arheden H. Design and validation of Segment--freely available software for cardiovascular image analysis. BMC medical imaging. 2010;10:1. doi: 10.1186/1471-2342-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117:1717–31. doi: 10.1161/CIRCULATIONAHA.107.653584. [DOI] [PubMed] [Google Scholar]
- 40.Wiesmann F, Ruff J, Engelhardt S, Hein L, Dienesch C, Leupold A, et al. Dobutamine-stress magnetic resonance microimaging in mice : acute changes of cardiac geometry and function in normal and failing murine hearts. Circulation research. 2001;88:563–9. doi: 10.1161/01.res.88.6.563. [DOI] [PubMed] [Google Scholar]
- 41.Grounds MD, Radley HG, Lynch GS, Nagaraju K, De Luca A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiology of disease. 2008;31:1–19. doi: 10.1016/j.nbd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blain A, Greally E, Laval S, Blamire A, Straub V, Macgowan GA. Beta-blockers, left and right ventricular function, and in-vivo calcium influx in muscular dystrophy cardiomyopathy. PloS one. 2013;8:e57260. doi: 10.1371/journal.pone.0057260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuckey DJ, Carr CA, Camelliti P, Tyler DJ, Davies KE, Clarke K. In vivo MRI Characterization of Progressive Cardiac Dysfunction in the mdx Mouse Model of Muscular Dystrophy. PloS one. 2012;7:e28569. doi: 10.1371/journal.pone.0028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–9. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 45.Harpole DH, Jones RH. Left ventricular function under stress before and after myocardial revascularization. American heart journal. 1992;124:273–9. doi: 10.1016/0002-8703(92)90587-l. [DOI] [PubMed] [Google Scholar]
- 46.Goetschalckx K, Rademakers F, Bogaert J. Right ventricular function by MRI. Current opinion in cardiology. 2010;25:451–5. doi: 10.1097/HCO.0b013e32833b78e6. [DOI] [PubMed] [Google Scholar]
- 47.Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: Anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008;117:1436–48. doi: 10.1161/CIRCULATIONAHA.107.653576. [DOI] [PubMed] [Google Scholar]
- 48.Jones U, Enright S, Busse M, Rosser A. A Pilot Study on Respiratory Function in People with Huntington’s Disease. J Neurol Neurosurg Psychiatry. 2010 Sep;81:A42–A43. [Google Scholar]
- 49.Buonincontri G, Methner C, Krieg T, Carpenter TA, Sawiak SJ. A fast protocol for infarct quantification in mice. Journal of magnetic resonance imaging : JMRI. 2013 doi: 10.1002/jmri.24001. [DOI] [PubMed] [Google Scholar]
- 50.van Rugge FP, van der Wall EE, de Roos A, Bruschke AV. Dobutamine stress magnetic resonance imaging for detection of coronary artery disease. Journal of the American College of Cardiology. 1993;22:431–9. doi: 10.1016/0735-1097(93)90047-5. [DOI] [PubMed] [Google Scholar]