

Abstract
In most people with type 2 diabetes, progression from obesity to diabetes is accompanied by elevated tissue exposures to a variety of lipids. Among these lipid species, ceramides and more complex sphingolipids have gained recent attention as being pathophysiologically relevant for the development of insulin resistance and impaired glycemic control. Upon excess intake of saturated fat, ceramides accumulate in insulin sensitive tissues either as a consequence of de novo synthesis or through mobilization from complex sphingolipids. Clinical studies have confirmed positive correlation between plasma and tissue levels of several ceramide species and insulin resistance. At the cellular level, it has been demonstrated that ceramides impair insulin signaling and intracellular handling of glucose and lipids with resulting deleterious effects on cellular metabolism. Hence, we are reviewing whether therapeutic interventions aiming at reducing tissue exposure to ceramides or other sphingolipids represent viable therapeutic approaches to improve glucose metabolism in people with diabetes.
Keywords: Ceramides, Glucose homeostasis
1. Introduction
In most people with type 2 diabetes, elevation of plasma glucose is the ultimate consequence of failing beta cell function occurring after extended period of waning insulin sensitivity in peripheral tissues. The underlying pathophysiological mechanism(s) leading to manifest insulin resistance are largely unknown. However, it has been suggested that circulating metabolites in people with diabetes are ultimately responsible for inducing insulin resistance. Thus, several of the circulating factors that are perturbed in people with diabetes such as key metabolites (glucose, branched chain amino acids, and lipids) have been shown to impact target tissue glucose uptake. Since the initial proposal that abnormal lipid metabolism leads to impaired insulin signaling [1], the search for underlying mechanistic explanations on cellular and tissue level have been intensified and a growing body of observational as well as mechanistic evidence has been generated demonstrating the important contribution of lipids to obesity induced insulin resistance. Deterioration of insulin sensitivity occurs after prolonged ingestions of energy-dense meals in excess of daily caloric requirements thereby introducing a lipotoxic environment in peripheral tissues [2]. In addition acute lipid infusion and consequent increase of lipid excess in non-adipose tissue like muscle is associated with impaired insulin signaling and action [3]. In in obese lipid overloaded individuals it is believed that two elements contribute to the emergence of type 2 diabetes: insulin resistance and lipo-toxicity. The liver and to some extent adipose tissue are viewed as the major contributors of these untoward metabolic perturbations because increased hepatic lipid production and export exceeds the metabolic and storage capacity of peripheral tissues leading to elevated plasma lipid levels. Beside the impaired insulin action mentioned above, sustained exposure to high lipid levels is toxic for pancreatic β-cell and normalization of plasma lipids halts progressive deterioration of β-cell failure [4].
Although the knowledge about the deleterious effects and the underlying mechanisms of neutral lipids on whole-body insulin stimulated glucose uptake, β-cell function and insulin sensitivity is growing, there is still reason to believe that other lipotoxic factors are involved. Over the past decade several investigators have been drawn to study the role of polar sphingolipids as pivotal mediators of lipotoxicity leading to insulin resistance [5,6]. Five common sphingolipids are easily detected in human plasma: ceramides, glucosylceramides, lactosylceramides, sphingomyelin, and ganglioside GM3 [7]. In particular some sphingolipids, such as ceramides have been associated with metabolic dysregulation leading to diabetes. Admittedly, the current review is rather ceramide centric as most studies have been particularly focused on the negative impact of this class of polar lipids on glycemic control.
2. Ceramide synthesis
Before considering tissue ceramide accumulation and underlying molecular mechanisms that possibly mediate negative impact of sphingolipids on insulin signaling, it is worth considering the sources of these polar lipids in the human body. Spingolipids have an important role as structural components of cellular membranes, ceramides serve also as important bioactive lipids in a variety of cellular processes like cell growth and differentiation, inflammation and apoptosis [8].
Every cell in the body has the capacity to de novo synthesize sphingolipids within the endoplasmatic reticulum. Also mitochondria seem capable of contributing to sphingolipid metabolism as many of the enzymes involved in ceramide synthesis are also localized in this organelle. Four sequential reactions lead to the formation of bioactive ceramide: the first, and rate-limiting step is the conjugation of an amino acid (typically l-serine) with the fatty acid palmitoyl-CoA catalyzed by the enzyme serine-palmitoyl transferase (SPT). The resulting product, sphinganine, is rapidly acylated by (dihydro)ceramide synthase into dihydroceramide which upon subsequently dehydrogenation catalyzed by dihydroceramide desaturase 1 (Des1) gives rise to the key intermediate ceramide. In addition to acylation of the sphinganine as a step in de novo ceramide synthesis, ceramide synthases also catalyze re-acylation of sphingosine as part of a salvage pathway (Figure 1). Interestingly, six different but closely related ceramide synthases have been identified [9], which demonstrate tissue specific expression and variable substrate selectivity thereby providing the basis for tissue specific presence of ceramides with varying acyl chain lengths. For example, ceramide synthase CerC2 is widely expressed and preferentially incorporates C20–C24 acyl residues in ceramide, whereas CerS3 is predominantly expressed in skin and incorporates very long acyl chains up to C34:0 in the resulting ceramides. Ceramide synthase CerC5 seems specifically engaged with C16 ceramide formation, whilst ceramide synthase CerC6 shows a slightly wider substrate selectivity as it is involved in C14, C16, and C18 ceramide synthesis [7]. It is important to emphasize that ceramides with different acyl chain length are generated in specific physiological and pathophysiological contexts in a tissue and cell dependent fashion which is like to constitute the basis for differentially influences of signaling pathways [8] (Figures 2–4).
Figure 1.
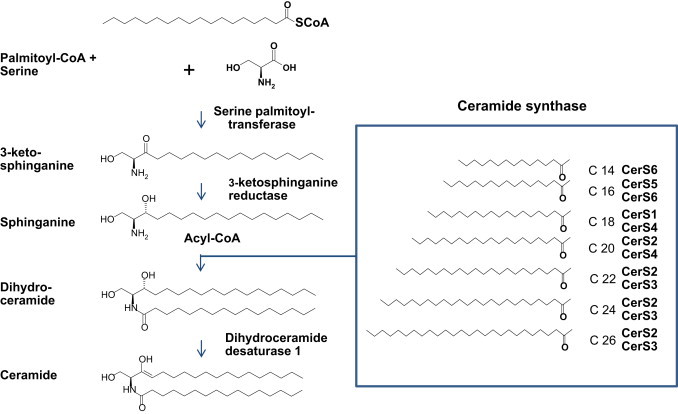
Ceramide de novo synthesis pathway. Sphingolipids are synthesized de novo in the endoplasmic reticulum. First and rate limiting step is the condensation of amino acid serine with saturated fatty acid palmitate which is catalyzed by serine palmitoyl transferase. Subsequent reduction to sphinganine is catalyzed by 3-ketosphinganine reductase. A variety of ceramide synthases (CerS1–6) catalyze the conjugation of a second acyl-CoA of variable chain length (C14-C32) leading to formation of dihydroceramide. Dihydroceramide desaturase converts the intermediaries into proper ceramides.
Figure 2.
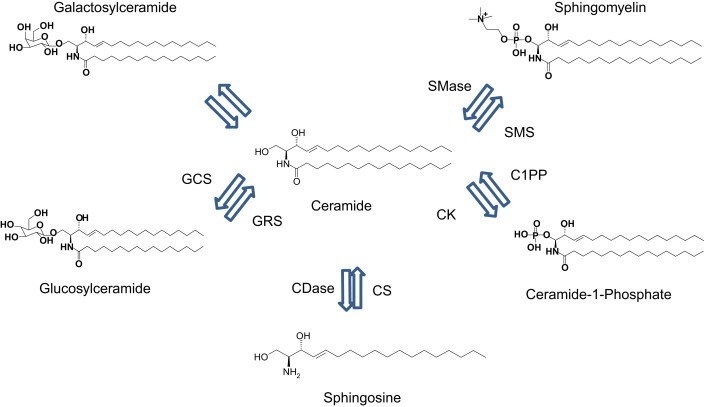
Ceramide salvage pathway. Ceramide can also be formed through several lysosomal enzymes addressing complex sphingolipids such as glycosylsphingolipid, galactosylsphingolipid, sphingomyelin, and ceramide 1-phosphate. Ceramide can be further degraded to sphingosine, which upon entry into the endoplasmic reticulum can be salvaged into ceramide. Sphingomyelinases and glycosidases act in combination with sphingolipid activator proteins. CS: ceramide synthase, CSDase: ceramidase, C1PP: Ceramide 1 phosphatase, CK: Ceramide kinase, GCS: Glycosyl ceramide synthase, GRS: glucocerebrosidase, SMS: sphingomyelin synthase, SMase: sphingomyelinase.
Figure 3.
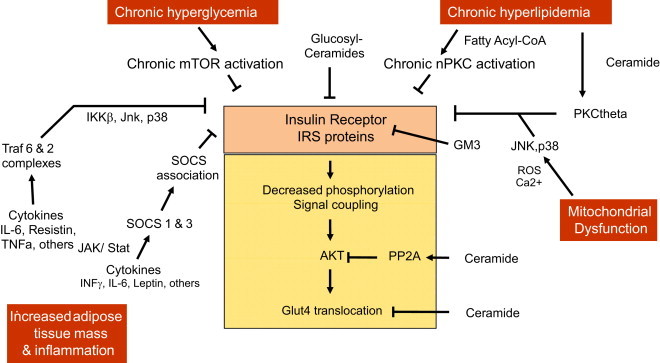
Insulin resistance occur at several levels and is mediated through several biochemical pathways. Ceramide and sphingolipids are putatively responsible for mediating part of the insulin resistance through interferences with insulin receptor function and signaling at several levels (see text for details). Ceramides activate PKCtheta, and PPA2 ultimately leading to decreased activity of Akt and hence impaired Glut4 translocation to the cell membrane. GM3 ganglioside enriched lipid domains of the cell membrane interfere with insulin receptor cell membrane caveolae which are essential for receptor signaling.
Figure 4.
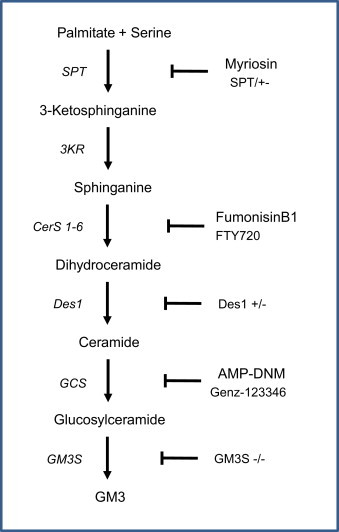
Diagram demonstrating sites of interference by various small molecule agents or genetic ablation of enzymes engaged in ceramide and sphingolipid synthesis pathway. The shown pharmacological/genetic interferences lead to improved insulin action in several preclinical animal models of diabetes. Myriocin and partial ablation of SPT1 leads to marked decrease of ceramide content in metabolically active tissues with resulting improvement of whole body insulin sensitivity. The glycosphingolipid synthesis inhibitors, ANP-DMN and Genz-123346 markedly decrease formation of several complex sphingolipids and improve whole body glucose tolerance. Similar effects are seen in GM3 synthase KO mice.
From the endoplasmatic reticulum, ceramides are transported via a specialized carrier system to the cytosolic leaflet of the trans-Golgi apparatus membrane where several metabolic fates await the molecules [10]. Thus, in the Golgi, ceramide can become glycosylated into glycosylsphingolipids, phosphorylated into ceramide 1-phosphate, or become modified with a polar head turning it into sphingomyelin [11]. Further additions of oligosaccharides and sulfate groups give rise to a broad range of very complex glycol and gangliosphingolipids [12]. These complex molecules are important components of the outer layer of the cell membrane. Ceramide is also a source of the bioactive signaling molecule sphingosine 1-phosphate (S1P) which upon secretion to the extracellular space acts as a ligand on a family of G-protein coupled receptors, S1P(1–5).
Ceramide can also be formed through the salavage pathway which involves the lysosomal degradation of complex sphingolipids into ceramide. The catabolism of the complex glucosphingolipids is predominantly located to the surface of internal membrane vesicles or at endocytosed lipoproteins. Receptor mediated endocytosis of low-density lipoprotein (LDL) delivers the glycosphingolipids to lysosomal lumen. The degradation of the simplest of the complex sphingolipids, glycosylceramide, is catalyzed through the action of glycosylceramide-beta-glucosidase (GBA1). Thus, sphingosine is generated in lysosomes from degradation of complex sphingolipids whereby it has the ability to serve as substrate for ceramide re-synthesis. Involved key enzymes for this part of the salvage pathway are sphingomyleinases and ceramide synthases. The importance of the salvage pathway is emphasized by the rare but clinically very recognizable glycospingolipidoses characterized by defects in sphingolipid degradation pathways due to mutations of specific acid hydrolases [7].
In the circulatory system, sphingolipids are transported as components of plasma lipoprotein associated particles [13]. Thus, LDL particles are the most abundantly loaded with sphingolipids including sphingomyelin and ceramide which is also transported via VLDL particles. As mentioned, all tissues are capable of de novo sphingolipid synthesis rendering locally generated sphingolipids a major source of tissue specific accumulation of ceramide. However, as ceramides synthesized in the liver are easily incorporated in LDL and VLDL particles, circulating levels of plasma ceramides are most likely of hepatic and to some extent of dietary origin. Sphingolipids are also part of the diet [14] but the daily amounts consumed are relatively modest 0.3–0.4 g/day and there is no evidence that dietary sphingolipids are essential for healthy growth or even survival [14]. Nevertheless, it is likely that individuals consuming foods particularly rich in sphingolipids will clearly exceed this level. Sphingolipids undergo considerable hydrolysis in all parts of the small and large intestine, and the resulting sphingosine is rapidly taken up intestinal epithelial cells and degraded in to fatty acids [15]. It is possible that some sphingosine escape degradation in the gut epithelium and access systemic circulation, as radiolabeled sphingoid bases given orally to rats can be found as sphingolipids in the systemic circulation [15]. Ceramides are also formed by degradation of more complex sphingolipids. Thus, sphingomyelinase is subject to activation through extracellular signaling pathways which leads to hydrolytic conversion of sphingomyelin to ceramide. A well-known activator of neutral sphingomyelinase is the proinflammatory cytokine tumor necrosis factor alpha (TNFα) [16]. This way, oxidative stress induced by TNFα enhances sphingomyelin degradation and subsequent ceramide synthesis through the salvage pathway. Other components of the innate immune system such as fatty acid activated Toll-like receptor activators can trigger ceramide synthesis thereby further enhancing the stimulatory impact of inflammatory cytokines on tissue levels of ceramide and glucosylsphingolipid [6].
Increased substrate availability seems to be the primary trigger of increased de novo ceramide synthesis. During conditions of lipid overload, mitochondrial capacity to oxidize fatty acids becomes saturated, and esterified long-chain acyl-CoA chains are channeled away from carnitine plamiotyl transferase 1 (CPT-1). By escaping this CPT-1 mediated entry to mitochondrial β-oxidation, long-chain fatty acyl-CoA are instead driven towards synthesis of triacylglycerols, and sphingolipids. Serine palmitoyltransferase activity is highly dependent on tissue levels of l-serine and palmitate acyl-CoA [17]. Thus, under circumstances characterized by excess caloric intake (in the form of palmitate and serine), ceramide levels increase in peripheral tissues substantially involved in whole body glucose homeostasis (such as skeletal muscle, liver, and adipose tissue). It has been argued that at least part of the lipotoxic effects of palmitate are due to aberrant synthesis of ceramides because of the selectivity of SPT-1 for this fatty acid [18]. This view is supported by clinical observations of human volunteers subjected to intravenous infusion of non-esterified fatty acids demonstrating a very tight correlation between concentrations and time course of rising plasma levels of free fatty acids and ceramides as well as an increased ceramide content in the muscle [19,20].
More acute regulation of ceramide synthesis is mediated via endocrine regulators of metabolism such as glucocorticoids. The acutely induced impairment of glycemic control by dexamethasone is corroborated by prior inhibition of SPT suggesting that increased ceramide levels are downstream mediators of glucocorticoids [21]. This hypothesis is further supported by observations that dexamethasone induces hepatic expression of a number of sphingolipid synthesizing enzymes such as SPT2, GCS1, and GCS6 [21]. Strangely and in contrast to ceramides, glucosylsphingolipid levels are differentially affected in a tissue dependent fashion during high fat feeding, with elevated levels of GM3 in adipose tissue and concomitantly decreased GM3 levels in skeletal muscle [22]. Actually, this recent study from the Summers laboratory has nuanced our understanding of the putatively deleterious role of glycosylceramides on glucose metabolism in peripheral insulin sensitive tissues. Apparently, glycosylceramides impair insulin action in adipocytes whilst in skeletal muscle overexpression of glycosylceramide synthase not only elevates cellular levels of glycosylceramide but also improves insulin action [22].
3. Sphingolipids and metabolic disease
A variety of diabetic animal models such as leptin signaling deficient db/db mice and Zucker Diabetic fatty (ZDF) rats display profound changes in tissue sphingolipid levels [6]. Also in non-human primates having developed diabetes due to persistent high-fat feeding, plasma levels of ceramide and dihydroceramide are elevated [23]. Interestingly, the rare sphingolipid species, C18:0 deoxysphinganine, which is a marker of increased flux through the serine palmitoyl transferase pathway was also elevated in these diabetic non-human primates suggesting that de novo sphingolipid synthesis could drive the impaired glycemic control in these animals.
In people with type 2 diabetes, insulin resistance correlates with plasma levels of ceramide [24–27], but longitudinal studies confirming causality of rising ceramide levels preceding development of insulin resistance are missing. In a recent study, examining people in various stages of type 2 diabetes, concluded that plasma levels of long-chain GM3 and ceramide are clearly positively correlated to impaired insulin sensitivity [26]. Although it is obvious that alterations of plasma ceramide and long-chain GM3 levels constitute early markers of impaired insulin action it is not possible to conclude whether these are causal mediators of insulin resistance or passive bystanders of other underlying mechanisms of dysmetabolic syndrome. However, high-fat fed mice lacking GM3 synthase have both lower plasma glucose levels and improved insulin sensitivity compared to high-fat fed wild type mice [28]. This observation suggest that complex sphingolipids are also interfering with insulin action, although the lack of metabolomic profiling of the GM3 synthase null mice makes it difficult to address which sphingolipid species in particular could mediate these untoward effects. Carefully conducted clinical studies scrutinizing the correlation between plasma and tissue levels of sphingolipids are rare and mostly of quite limited size [24,29–31]. High quality quantitative analytical methodologies have not been widely available for classification of tissue and plasma sphingolipids, which has made comparison of different reports difficult. Most clinical trials reporting plasma levels of sphingolipids have measured total ceramides accompanied by a limited subfractionation. With recent methodological advancements it has been possible to analyze specific molecular sphingolipid subspecies in a quantitative fashion and with reasonable throughput. Using high-specificity quantitative methods, it has it seems evident that plasma levels of longer chain ceramides (>16) and ganglioside GM3 (>18) are associated with impaired insulin sensitivity [26]. However, far more studies are needed to conclusively confirm which of the many sphingolipid species detectable in plasma are the most reliable surrogate markers of metabolic disease.
Further, a number of studies have found increased intramyocellular levels of ceramides in skeletal muscle biopsies from obese insulin resistant human volunteers [19,32]. However, not all clinical observations confirm a connection between muscle ceramides and insulin resistance, as improvement of skeletal muscle insulin sensitivity through endurance training seemingly have no impact on intramyocellular ceramide content in young males [33]. More recently a correlation between HOMA-IR and total ceramide as well as ceramide C16:0 in subcutaneous and epicardial adipose tissue could be established, which suggest that the bioactive lipid content in this tissue might be more predictive for insulin resistance. Furthermore increased ceramide content in this tissue might result in disturbance of its metabolism and gene expression capabilities. Interestingly, dramatic decrease in body adiposity and improvement of insulin sensitivity as obtained in obese subjects after bariatric surgery is accompanied by reduction of plasma C16:0 and C24:0 ceramides in particular [34].
4. Insulin signaling and glucose disposal
Numerous in vitro experiments have consistently demonstrated that increased exposure to ceramide and glucosphingolipids impair functional activity of several of the molecular components of the insulin signaling pathway. The primary target of ceramides in the insulin signaling pathway is protein kinase B (PKB)/Akt. Two distinct underlying molecular mechanisms appear to be involved in ceramide induced impairment of PKB/Akt activity [35]. Ceramide interferes with both a kinase and a phosphatase required for insulin mediated activation of PKB/Akt: firstly, ceramide activates cytosolic protein phosphatase-2A (PP2A) which is a serine/threonine phosphatase responsible for PKB/Akt dephosphorylation. Dephosphorylation of Thr308 and/or Ser473 in PKB/Akt leaves it inactivated whereby translocation to the plasma membrane does not occur [36]. Secondly, upon accrual of ceramide in the outer layer of the cell membrane it accumulates in the caveolin-enriched domains. In this location, ceramide interferes with PKCzeta catalyzed phosphorylation of PKB/Akt Thr34 whereby PKB/Akt sequesters in a repressed state preventing downstream insulin receptor signaling. A third albeit lesser validated mechanism via which ceramide inhibits glucose transport has to do with ceramide enriched microdomains in phosphatidylcholine containing membranes. Due to altered membrane fluidity ceramides interferes with GLUT4 translocation and fusion to the cell membrane [37]. It has also been proposed that ceramide inhibits the expression of GLUT4 in 3T3-L1 adipocytes which would synergize aforementioned effects on GLUT4 translocation [38].
For glycosphigolipids a direct interaction with the insulin receptor has been proposed [39,40]. The interaction of GM3 seems to be mediated by a specific lysine residue in the transmembrane domain of the receptor and excess levels of this ganglioside promote the dissociation of the IR from the caveolae hence hindering signal transduction. Consistent with this theory mice lacking GM3 have been reported to show enhanced phosphorylation of the IR [28].
There is a growing literature on chain length-specific cellular activities of ceramides [7]. Thus it has been demonstrated that elevation of intracellular levels of very-long chain-ceramides (>24) through increased CerS2 activity promotes proliferation whilst increase in long-chain-ceramides (C16:0 and C18:0) promotes apoptosis. Further, the ratio between very-long-chain ceramides and long-chain-ceramides influences autophagy in a variety of pathophysiological settings. Unfortunately, studies scrutinizing the importance of specific ceramide chain lengths on insulin signaling and glucose uptake are lacking. However, it has been shown that aforementioned ceramide induced activation of PP2A is particularly mediated via C16:0 ceramide suggesting that elevated Cers5 and/or CerS6 activity could be linked to impaired insulin action [41,42].
Recently, another very interesting link between ceramide and insulin action was discovered. Through a thorough series of experiments, Holland and colleagues demonstrated that adipocyte derived adiponectin imposes its beneficial effects on insulin signaling through effects on sphingolipid metabolism [43]. Apparently, both adiponectin receptors, AdipoR1 and AdipoR2 possess intrinsic ceramidase activity which upon receptor activation promotes degradation of intracellular ceramide species to sphingosine-1-phosphate (S1P). However, it cannot be excluded the adiponectin induced S1P formation is also the result of AdipoR1/AdipoR2 mediated activation of other ceramidases. Irrespective of the synthesis pathway, Holland et al. further demonstrated that adiponectin induced S1P formation is required as an important messenger between AdipoR1/AdipoR2 and AMPK activation [43]. The similar beneficial effects on insulin sensitivity are also obtained when acid ceramidase is overexpressed in free fatty acid overexposed myotubes [44].
Whereas the negative impact of ceramide upon insulin signaling affects all studied insulin receptor expressing tissues, a recent study has demonstrated that interference of glucosylceramide with insulin signaling predominantly affects adipose tissue whereas myotubes are unaffected by this sphingolipid [22]. However, the cellular mechanisms have largely been studied using cell systems, and future studies are required to translate these in vitro findings to in vivo models of insulin resistance and humans [45].
5. Sphingolipids and whole body glucose homeostasis
Given the evidence that alterations of cellular sphingolipid levels alters insulin action it has been speculated whether therapeutic interventions modifying synthesis and tissue distribution of ceramides and/or complex sphingolipids will improve whole body glucose homeostasis. Unfortunately, studies bridging in vitro observations of deleterious effects of ceramide and GM3 on insulin signaling to conclusive evidence of ceramide induced impairment of whole body glucose homeostasis are not entirely conclusive. So far, clinical phenotypes with impaired insulin action and/or overt diabetes have not been convincingly associated with genetic variations of enzymes involved in sphingolipid synthesis pathways. However, this may simply be due to the fact that people suffering from serious sphingolipidoses exert rather severe phenotypes primarily characterized by neurological complications and early fatality rendering potential metabolic disorders undetected. With the absence of human genetic data it remains to be proven that transgenic animal models carrying gain or loss of function mutations of sphingolipid synthesis enzymes have any translational validity.
Mice carrying a tissue wide deletion of GM3 synthase have improved insulin sensitivity in skeletal muscle compared to wild type animals and display a clear resistance against diet-induced impaired insulin function [28]. Apparently, the absence of GM3 synthase has no deleterious effects on vital functions. In contrast, mice lacking other sphingolipid synthesis enzymes have overt phenotypes rendering interpretation of the metabolic consequences of the absence of these specific enzymes difficult. Genetic ablation of dihydroceramide desaturase 1 (Des1) affects the animals ability to form ceramide. Animals lacking both alleles encoding Des1 are growth retarded and display increased mortality due to a complex phenotype with hepatomegaly, scaly skin, and practically no hair probably resulting a loss of core temperature and impaired liver function [21]. However, heterozygous Des1±mice are characterized by markedly lower ceramide levels in peripheral tissues with concomitant significant improvements of insulin sensitivity and insensitivity to glucocorticoid induced metabolic impairment [21].
Dependent on substrate acyl chain length one of several ceramide synthases is involved in adding a second acyl chain to sphinganine to form dihydroceramide. Genetic ablation of ceramide synthase 1, which primarily catalyzes C18:0 acylation leads to a severe cerebellar Purkinje cell loss and accompanying ataxia [46]. Complete deletion of ceramide synthase 2 leads to cerebral depletion of myelin sheet formation, cerebellar degeneration and increased prevalence of hepatocellular carcinoma [47]. Genetic ablation of ceramide synthase 6 in mice leads to behavioral abnormalities indicating a vital role of C16:0 sphingolipids in central nervous system function [48]. Another more profound phenotype is seen in mice carrying a tissue selective deletion of glucosylceramide synthase (GCS) in the epithelial cells of the intestine [49]. Newborn mice with a villin promoter driven GCS knock-out die soon after birth due to grossly impaired ability to absorb nutrients from intestine. Also adult mice carrying an inducible deletion of the GCS gene in intestinal epithelial cells die shortly after activating the deletion. The malabsorption is caused by grossly impaired ability to form intracellular vesicles trafficking lipids from the apical plasma membrane of the enterocyte to intracellular lipid depots [49]. Obviously, it is impossible to deduct which of the complex glycosphingolipids being formed down-stream of the GCS catalyzed step are required for normal intestinal absorption of lipids, but given the seemingly normal phenotype of GM3 synthase knock-out mice, it seems evident that sphingolipids formed upstream of GM3 synthase are involved.
5.1. Therapeutic interventions
Presence of elevated ceramide levels in plasma as well as tissues of importance for glucose metabolism such as liver, skeletal, muscle, and adipose tissue does not necessarily confirm causality between ceramide exposure and deterioration of insulin signaling. For example, it is well recognized that plasma levels of triglycerides correlate positively as a surrogate marker of insulin resistance whereas data demonstrating that aggressive triglyceride lowering therapy with fibrates and niacin are incapable of improving glycemic control. Although several studies have demonstrated that increased ceramide exposure deteriorate insulin signaling in vitro, it was only recently that in vivo evidence linking plasma levels of ceramides to insulin sensitivity became available. Due to their lipophilic nature, ceramides do not readily lend themselves for formulation suitable for in vivo studies. However, when using LDL particles as carriers of ceramides it has been possible to elevate plasma ceramide levels to patophysiologically relevant levels in mice with concomitant deterioration of whole body glucose handling due to increased insulin resistance [27]. Interestingly, Boon et al. demonstrated that intraveneous LDL–ceramide infusions increase ceramide content in skeletal muscle cell membranes but not in any other examined tissues [27]. Thus, it seems evident that increased plasma levels of ceramide impair glycemic control, but clear cut evidence from in vivo experimentation demonstrating that therapeutic improvement of insulin sensitivity is causally linked to lowering of plasma ceramide levels is still missing.
Till this moment, there is no publicly available information about clinical experience with specific inhibitors of sphingolipid synthesizing enzymes in diabetes patients. However, the glycosylceramide synthase (GCS) inhibitor, eliglustat, is currently undergoing late phase clinical development targeting patients with Gaucher׳s disease as the inhibition of GCS reduces the substrate load into the glycosphingolipid synthesis pathway [50,51]. Patients with Gaucher׳s disease are characterized by an inborn error of the enzyme glycosylceramide-beta-glucosidase (GBA1), which leads to accumulation of glucocerebroside within lysosomes of macrophages. Some patients with this disease show a markedly increased hepatic glucose production, elevated insulin levels and reduced insulin mediated whole body glucose uptake as demonstrated in euglycemic clamp experiments, which might be the result of the increased production of gangliosides like GM3 [12]. However following enzyme replacement therapy some patients gain body weight and develop diabetes, which might be the result of the loss of glucose-consuming macrophages. Therefore it is impossible to extrapolate effects of GCS inhibition on glycemic control from these clinical trials, and only rigorous assessment of the GCS inhibitors in adequate patient populations can tell if inhibition of glycosylceramide production will translate into clinical benefits for people with diabetes.
A wide variety of experimental molecules have been tested in cellular in vitro systems as well as in preclinical in vivo models. The atypical amino acid, myriocin, is a potent inhibitor of SPT1 [52]. Although it is likely that myriocin has other molecular targets, it has been widely used as a tool compound to inhibit SPT1 activity in a variety of animal models. Thus, myriocin treatment significantly improves glycemic control in both obese and overtly diabetic rodent models. Oral administration of myriocin is associated with moderate weight loss, but as parenteral administration of myriocin does not affect body weight it is reasonable to assume that improved glycemia is not simply the result of negative energy balance. Formal assessment of myriocin׳s chronic effects on whole body energy balance has yet to be conducted but chronic dosing of myriocin (0.3–0.5 mg/kg/day) to ZDF rats significantly improve metabolic control in parallel with dramatic reduction of skeletal muscle and plasma ceramide [53]. In addition to eliglustat, other inhibitors of GCS are available and some of these have been tested in animal models of diabetes. Small molecule compounds like fumonisin B1 and the imonosugar N-(5'-adamantine-1'-yl-methoxy)-pentyl-1-deoxynojirimycin (AMP-DNM) have been used in chronic in vivo studies to demonstrate that pharmacologically induced depletion of glycosphingolipids is associated with improved insulin sensitivity in mice [54]. In addition to improved glycemia, ob/ob mice treated with AMP-DNM also display improved hepatic lipid deposition, amelioration of cirrhosis markers, and reduced hepatic gluconeogenesis [54]. The data obtained with AMP-DNM should be interpreted with caution as this molecule also infers inhibition of the intestinal enzyme sucrose-isomaltase whereby intestinal carbohydrate absorption is likely to be hampered. However, the far more specific inhibitor of GCS, GENZ-123346, also concomitantly reduces formation of glucosphingolipids and improves insulin sensitivity and corrects hepatosteatosis [55].
Given it becomes possible to decipher which of the ceramide species exert most deleterious effects on glucose homeostasis it may be possible an interesting therapeutic approach could be to target specific CerS1–6 isoforms rather than SPT1. Such approach would potentially provide selective reduction of the unwanted ceramides whilst leaving others unaffected. Further addressing specific Ceramide synthases will affect both de novo synthesis as well as salvage pathway synthesis of ceramides.
6. Conclusion
Although clinical experience with pharmacological tools reducing tissue and plasma levels of sphingolipids is still missing, a solid body of circumstantial evidence is available to justify investment in experimental clinical studies bridging existing gaps in the translation of preclinical data to relevant clinical setting. Most importantly, we are short of thoroughly safety profiled agents that can be applied clinically to deliver graded tissue specific inhibition of enzymes involved in sphingolipid synthesis. Provided such investigational compounds become available it will become possible to link quantifiable tissue specific reductions of sphingolipids to level of improvement of insulin sensitivity in both normal individuals as well as in people with insulin resistance.
Conflict of interest
None declared.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.McGarry J.D. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992;258:766–770. doi: 10.1126/science.1439783. [DOI] [PubMed] [Google Scholar]
- 2.Unger R.H. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 3.Nowotny B., Zahiragic L., Krog D., Nowotny P.J., Herder C., Carstensen M., Yoshimura T., Szendroedi J., Phielix E., Schadewaldt P., Schloot N.C., Shulman G.I., Roden M. Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes. 2013;62:2240–2248. doi: 10.2337/db12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wajchenberg B.L. beta-cell failure in diabetes and preservation by clinical treatment. Endocrine Reviews. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- 5.Summers S.A., Nelson D.H. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing׳s syndrome. Diabetes. 2005;54:591–602. doi: 10.2337/diabetes.54.3.591. [DOI] [PubMed] [Google Scholar]
- 6.Holland W.L., Summers S.A. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocrine Reviews. 2008;29:381–402. doi: 10.1210/er.2007-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grosch S., Schiffmann S., Geisslinger G. Chain length-specific properties of ceramides. Progress in Lipid Research. 2012;51:50–62. doi: 10.1016/j.plipres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Ogretmen B., Hannun Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nature Reviews Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 9.Mizutani Y., Kihara A., Chiba H., Tojo H., Igarashi Y. 2-Hydroxy-ceramide synthesis by ceramide synthase family: enzymatic basis for the preference of FA chain length. Journal of Lipid Research. 2008;49:2356–2364. doi: 10.1194/jlr.M800158-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 11.Bartke N., Hannun Y.A. Bioactive sphingolipids: metabolism and function. Journal of Lipid Research. 2009;50(Suppl.):S91–S96. doi: 10.1194/jlr.R800080-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aertz J.M., Boot R.G., van eijk M., Groener M., Bijl N., Lombardo E., Bietrix F.M., Dekker N., Goren A.K., Ottenhoff R., Van Roomen C.P., Aten J., Serlie M.J., Langeveld M., Wennekes T., Overkleeft H.S. Glycosphingolipids and insulin resistance. In: Cowart L.A., editor. Sphingolipids and metabolic disease. Landes Bioscience and Springer Science+Business Media, LLC; 2011. pp. 99–119. [Google Scholar]
- 13.Merrill A.H., Jr., Lingrell S., Wang E., Nikolova-Karakashian M., Vales T.R., Vance D.E. Sphingolipid biosynthesis de novo by rat hepatocytes in culture. Ceramide and sphingomyelin are associated with, but not required for, very low density lipoprotein secretion. Journal of Biological Chemistry. 1995;270:13834–13841. doi: 10.1074/jbc.270.23.13834. [DOI] [PubMed] [Google Scholar]
- 14.Vesper H., Schmelz E.M., Nikolova-Karakashian M.N., Dillehay D.L., Lynch D.V., Merrill A.H., Jr. Sphingolipids in food and the emerging importance of sphingolipids to nutrition. Journal of Nutrition. 1999;129:1239–1250. doi: 10.1093/jn/129.7.1239. [DOI] [PubMed] [Google Scholar]
- 15.Schmelz E.M., Crall K.J., Larocque R., Dillehay D.L., Merrill A.H., Jr. Uptake and metabolism of sphingolipids in isolated intestinal loops of mice. Journal of Nutrition. 1994;124:702–712. doi: 10.1093/jn/124.5.702. [DOI] [PubMed] [Google Scholar]
- 16.Peraldi P., Hotamisligil G.S., Buurman W.A., White M.F., Spiegelman B.M. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. Journal of Biological Chemistry. 1996;271:13018–13022. doi: 10.1074/jbc.271.22.13018. [DOI] [PubMed] [Google Scholar]
- 17.Merrill A.H., Jr., Wang E., Mullins R.E. Kinetics of long-chain (sphingoid) base biosynthesis in intact LM cells: effects of varying the extracellular concentrations of serine and fatty acid precursors of this pathway. Biochemistry. 1988;27:340–345. doi: 10.1021/bi00401a051. [DOI] [PubMed] [Google Scholar]
- 18.Paumen M.B., Ishida Y., Muramatsu M., Yamamoto M., Honjo T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. Journal of Biological Chemistry. 1997;272:3324–3329. doi: 10.1074/jbc.272.6.3324. [DOI] [PubMed] [Google Scholar]
- 19.Straczkowski M., Kowalska I., Baranowski M., Nikolajuk A., Otziomek E., Zabielski P., Adamska A., Blachnio A., Gorski J., Gorska M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia. 2007;50:2366–2373. doi: 10.1007/s00125-007-0781-2. [DOI] [PubMed] [Google Scholar]
- 20.Watt M.J., Barnett A.C., Bruce C.R., Schenk S., Horowitz J.F., Hoy A.J. Regulation of plasma ceramide levels with fatty acid oversupply: evidence that the liver detects and secretes de novo synthesised ceramide. Diabetologia. 2012;55:2741–2746. doi: 10.1007/s00125-012-2649-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holland W.L., Brozinick J.T., Wang L.P., Hawkins E.D., Sargent K.M., Liu Y., Narra K., Hoehn K.L., Knotts T.A., Siesky A., Nelson D.H., Karathanasis S.K., Fontenot G.K., Birnbaum M.J., Summers S.A. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metabolism. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Chavez J.A., Siddique M.M., Wang S.T., Ching J., Shayman J.A., Summers S.A. Ceramides and Glucosylceramides are independent antagonists of insulin signaling. Journal of Biological Chemistry. 2013 doi: 10.1074/jbc.M113.522847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brozinick J.T., Hawkins E., Hoang Bui H., Kuo M.S., Tan B., Kievit P., Grove K. Plasma sphingolipids are biomarkers of metabolic syndrome in non-human primates maintained on a Western-style diet. International Journal of Obesity (London) 2013;37:1064–1070. doi: 10.1038/ijo.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haus J.M., Kashyap S.R., Kasumov T., Zhang R., Kelly K.R., Defronzo R.A., Kirwan J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes. 2009;58:337–343. doi: 10.2337/db08-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Mello V.D., Lankinen M., Schwab U., Kolehmainen M., Lehto S., Seppanen-Laakso T., Oresic M., Pulkkinen L., Uusitupa M., Erkkila A.T. Link between plasma ceramides, inflammation and insulin resistance: association with serum IL-6 concentration in patients with coronary heart disease. Diabetologia. 2009;52:2612–2615. doi: 10.1007/s00125-009-1482-9. [DOI] [PubMed] [Google Scholar]
- 26.Shui G., Lam S.M., Stebbins J., Kusunoki J., Duan X., Li B., Cheong W.F., Soon D., Kelly R.P., Wenk M.R. Polar lipid derangements in type 2 diabetes mellitus: potential pathological relevance of fatty acyl heterogeneity in sphingolipids. Metabolomics. 2013;9:786–799. [Google Scholar]
- 27.Boon J., Hoy A.J., Stark R., Brown R.D., Meex R.C., Henstridge D.C., Schenk S., Meikle P.J., Horowitz J.F., Kingwell B.A., Bruce C.R., Watt M.J. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes. 2013;62:401–410. doi: 10.2337/db12-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashita T., Hashiramoto A., Haluzik M., Mizukami H., Beck S., Norton A., Kono M., Tsuji S., Daniotti J.L., Werth N., Sandhoff R., Sandhoff K., Proia R.L. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3445–3449. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blachnio-Zabielska A.U., Koutsari C., Tchkonia T., Jensen M.D. Sphingolipid content of human adipose tissue: relationship to adiponectin and insulin resistance. Obesity. 2012;20:2341–2347. doi: 10.1038/oby.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heilbronn L.K., Coster A.C., Campbell L.V., Greenfield J.R., Lange K., Christopher M.J., Meikle P.J., Samocha-Bonet D. The effect of short-term overfeeding on serum lipids in healthy humans. Obesity (Silver Spring) 2013;21:E649–E659. doi: 10.1002/oby.20508. [DOI] [PubMed] [Google Scholar]
- 31.Amati F., Dubé J.J., Alvarez-Carnero E., Edreira M.M., Chomentowski P., Coen P.M., Switzer G.E., Bickel P.E., Stefanovic-Racic M., Toledo F.G.S., Goodpaster B.H. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes. 2011;60:2588–2597. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams J.M., 2nd, Pratipanawatr T., Berria R., Wang E., DeFronzo R.A., Sullards M.C., Mandarino L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 33.Helge J.W., Dobrzyn A., Saltin B., Gorski J. Exercise and training effects on ceramide metabolism in human skeletal muscle. Experimental Physiology. 2004;89:119–127. doi: 10.1113/expphysiol.2003.002605. [DOI] [PubMed] [Google Scholar]
- 34.Huang H., Kasumov T., Gatmaitan P., Heneghan H.M., Kashyap S.R., Schauer P.R., Brethauer S.A., Kirwan J.P. Gastric bypass surgery reduces plasma ceramide subspecies and improves insulin sensitivity in severely obese patients. Obesity (Silver Spring) 2011;19:2235–2240. doi: 10.1038/oby.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blouin C.M., Prado C., Takane K.K., Lasnier F., Garcia-Ocana A., Ferre P., Dugail I., Hajduch E. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes. 2010;59:600–610. doi: 10.2337/db09-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chavez J.A., Knotts T.A., Wang L.-P., Li G., Dobrowsky R.T., Florant G.L., Summers S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. Journal of Biological Chemistry. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 37.JeBailey L., Wanono O., Niu W., Roessler J., Rudich A., Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56:394–403. doi: 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- 38.Long S.D., Pekala P.H. Lipid mediators of insulin resistance: ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochemical Journal. 1996;319(Pt. 1):179–184. doi: 10.1042/bj3190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nojiri H., Stroud M., Hakomori S. A specific type of ganglioside as a modulator of insulin-dependent cell growth and insulin receptor tyrosine kinase activity. Possible association of ganglioside-induced inhibition of insulin receptor function and monocytic differentiation induction in HL-60 cells. Journal of Biological Chemistry. 1991;266:4531–4537. [PubMed] [Google Scholar]
- 40.Tagami S., Inokuchi J.i.J., Kabayama K., Yoshimura H., Kitamura F., Uemura S., Ogawa C., Ishii A., Saito M., Ohtsuka Y., Sakaue S., Igarashi Y. Ganglioside GM3 participates in the pathological conditions of insulin resistance. Journal of Biological Chemistry. 2002;277:3085–3092. doi: 10.1074/jbc.M103705200. [DOI] [PubMed] [Google Scholar]
- 41.Dobrowsky R.T., Hannun Y.A. Ceramide-activated protein phosphatase: partial purification and relationship to protein phosphatase 2A. Advances in Lipid Research. 1993;25:91–104. [PubMed] [Google Scholar]
- 42.Zeidan Y.H., Jenkins R.W., Hannun Y.A. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. Journal of Cell Biology. 2008;181:335–350. doi: 10.1083/jcb.200705060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holland W.L., Miller R.A., Wang Z.V., Sun K., Barth B.M., Bui H.H., Davis K.E., Bikman B.T., Halberg N., Rutkowski J.M., Wade M.R., Tenorio V.M., Kuo M.S., Brozinick J.T., Zhang B.B., Birnbaum M.J., Summers S.A., Scherer P.E. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nature Medicine. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chavez J.A., Holland W.L., Bar J., Sandhoff K., Summers S.A. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. Journal of Biological Chemistry. 2005;280:20148–20153. doi: 10.1074/jbc.M412769200. [DOI] [PubMed] [Google Scholar]
- 45.Samuel V.T., Shulman G.I. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao L., Spassieva S.D., Jucius T.J., Shultz L.D., Shick H.E., Macklin W.B., Hannun Y.A., Obeid L.M., Ackerman S.L. A deficiency of ceramide biosynthesis causes cerebellar purkinje cell neurodegeneration and lipofuscin accumulation. PLoS Genetics. 2011;7:e1002063. doi: 10.1371/journal.pgen.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imgrund S., Hartmann D., Farwanah H., Eckhardt M., Sandhoff R., Degen J., Gieselmann V., Sandhoff K., Willecke K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. Journal of Biological Chemistry. 2009;284:33549–33560. doi: 10.1074/jbc.M109.031971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ebel P., Vom Dorp K., Petrasch-Parwez E., Zlomuzica A., Kinugawa K., Mariani J., Minich D., Ginkel C., Welcker J., Degen J., Eckhardt M., Dere E., Dormann P., Willecke K. Inactivation of ceramide synthase 6 in mice results in an altered sphingolipid metabolism and behavioral abnormalities. Journal of Biological Chemistry. 2013;288:21433–21447. doi: 10.1074/jbc.M113.479907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jennemann R., Kaden S., Sandhoff R., Nordstrom V., Wang S., Volz M., Robine S., Amen N., Rothermel U., Wiegandt H., Grone H.J. Glycosphingolipids are essential for intestinal endocytic function. Journal of Biological Chemistry. 2012;287:32598–32616. doi: 10.1074/jbc.M112.371005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lukina E., Watman N., Arreguin E.A., Banikazemi M., Dragosky M., Iastrebner M., Rosenbaum H., Phillips M., Pastores G.M., Rosenthal D.I., Kaper M., Singh T., Puga A.C., Bonate P.L., Peterschmitt M.J. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood. 2010;116:893–899. doi: 10.1182/blood-2010-03-273151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peterschmitt M.J., Burke A., Blankstein L., Smith S.E., Puga A.C., Kramer W.G., Harris J.A., Mathews D., Bonate P.L. Safety, tolerability, and pharmacokinetics of eliglustat tartrate (Genz-112638) after single doses, multiple doses, and food in healthy volunteers. Journal of Clinical Pharmacology. 2011;51:695–705. doi: 10.1177/0091270010372387. [DOI] [PubMed] [Google Scholar]
- 52.Miyake Y., Kozutsumi Y., Nakamura S., Fujita T., Kawasaki T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochemical and Biophysical Research Communications. 1995;211:396–403. doi: 10.1006/bbrc.1995.1827. [DOI] [PubMed] [Google Scholar]
- 53.Holland W.L., Knotts T.A., Chavez J.A., Wang L.P., Hoehn K.L., Summers S.A. Lipid mediators of insulin resistance. Nutrition Reviews. 2007;65:S39–S46. doi: 10.1111/j.1753-4887.2007.tb00327.x. [DOI] [PubMed] [Google Scholar]
- 54.Bijl N., Sokolovic M., Vrins C., Langeveld M., Moerland P.D., Ottenhoff R., van Roomen C.P., Claessen N., Boot R.G., Aten J., Groen A.K., Aerts J.M., van Eijk M. Modulation of glycosphingolipid metabolism significantly improves hepatic insulin sensitivity and reverses hepatic steatosis in mice. Hepatology. 2009;50:1431–1441. doi: 10.1002/hep.23175. [DOI] [PubMed] [Google Scholar]
- 55.Zhao H., Przybylska M., Wu I.H., Zhang J., Maniatis P., Pacheco J., Piepenhagen P., Copeland D., Arbeeny C., Shayman J.A., Aerts J.M., Jiang C., Cheng S.H., Yew N.S. Inhibiting glycosphingolipid synthesis ameliorates hepatic steatosis in obese mice. Hepatology. 2009;50:85–93. doi: 10.1002/hep.22970. [DOI] [PubMed] [Google Scholar]