

Abstract
Until recently, options for therapy in metastatic melanoma were limited. The understanding of immune check-point blockade and the discovery of molecular pathways involving driver mutations like BRAF has transformed the therapeutic landscape in this disease. Ipilimumab was the first drug shown to improve survival while vemurafenib demonstrated rapid responses never seen before in melanoma. Drugs from these classes and others are now in advanced stages of development and primed to positively impact patient survival in an incremental fashion. In this review, we highlight some of the developments during this renaissance in melanoma therapy and discuss agents of promise. Clinical challenges we face include individualizing therapy for patients, overcoming resistance to molecularly targeted therapy and developing rationale combinations or sequences of drugs. A concerted bench and bedside effort in this direction will undoubtedly keep melanoma in the forefront in an era of personalized medicine.
Keywords: BRAF, immunotherapy, melanoma, MEK
INTRODUCTION
The therapeutic landscape in melanoma has undergone paradigm-defining changes in the past 3 years. Prior to 2011, there were only two drugs (dacarbazine and high-dose interleukin-2, [HD IL-2]) approved in the United States (US) for the treatment of advanced or unresectable disease. This has now tripled in 2013 with approvals of ipilimumab and vemurafenib in 2011 and dabrafenib and trametinib in 2013. For the first time in decades, the median survival for patients with metastatic melanoma has exceeded 12 months. Much of this improvement has come with greater understanding of the molecular biology in this malignancy. Despite these advances, nearly 9500 patients will succumb to melanoma in 2013 in the US, stressing the ongoing need for investigation in this field.[1]
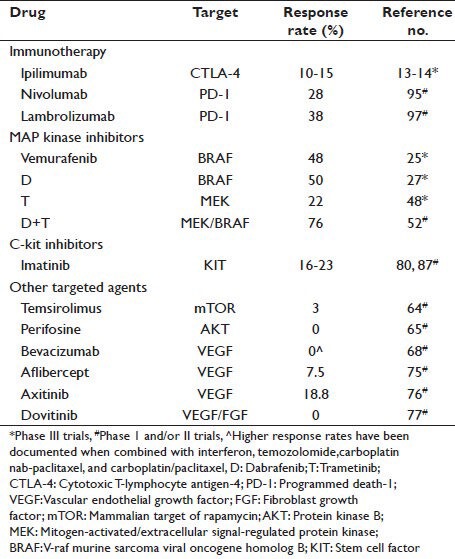
In this review, we highlight the drugs that have impacted these changes and provide our insight on their use for the individual melanoma patient. We also discuss agents of promise in clinical trials in melanoma. A summary of the agents reviewed in this report with the response rate (RR) observed in melanoma is collated in Table 1.
Table 1.
Therapeutic agents listed in review
CYTOTOXIC T-LYMPHOCYTE ANTIGEN-4
Melanoma has been characterized as one of the most immunogenic tumors stemming from initial reports of spontaneous regressions.[2] From years of clinical experience we also know that melanoma responds to immune stimulation in the form of interferon-alpha and HD IL-2.[3,4,5] Responses to HD IL-2 are around 16%, with 6% being complete responses (CR) and 60% of CRs being durable. Multiple approaches that aim at triggering immune responses have been trialed over the years, but disappointingly with little success until recently. For example, vaccines aimed at various melanoma targets, have failed to demonstrate significant clinical responses to date.[6]
The discovery of CD28 and CTLA-4's opposing effects on the response of T-cells to stimulation laid the foundation for the development of drugs targeting the CTLA-4 receptor.[7,8] The CTLA-4 receptor is a negative regulator of the immune system, which is expressed on T-cells 48 h after T-cell activation.[9] CTLA-4 binds to B7 on the antigen presenting cell with a higher affinity than CD28, thereby inhibiting the second signal required for T-cell activation and essentially “puts the brakes on the immune system”.[10] Two monoclonal antibodies targeting the CTLA-4 receptor have been developed, tremelimumab and ipilimumab and tested extensively in clinical trials. Both bind the CTLA-4 receptor allowing immune stimulation and T-cells to become activated and maintain activation. In CTLA-4 knockout mice, a massive expansion of T-cells occurs, killing mice within 3-4 weeks as a result of extensive lymphoproliferation.[11,12] Presently, only ipilimumab has received US Food and Drug Administration (FDA) approval in the treatment of advanced melanoma, based on an improvement in overall survival (OS) from two Phase III clinical trials.[13,14] This benefit however was not seen with tremelimumab when compared to dacarbazine or temozolomide in a separate trial.[15]
Ipilimumab is a fully humanized immunoglobin G1 (IgG) recombinant monoclonal antibody. When compared to the gp100 peptide vaccine in a randomized, controlled Phase III trial in previously treated advanced melanoma patients, ipilimumab improved OS by 3.6 months.[13] In a separate Phase III trial in treatment-naïve advanced melanoma patients, the addition of ipilimumab to dacarbazine significantly improved OS by 2.1 months compared with dacarbazine alone.[14] RRs by response evaluation criteria in solid tumors (RECIST) in these trials were only about 10-15%, but this does not account for stable disease with a prolonged response, progression of disease before regression and regression of target lesions in the presence of new lesions. These observations prompted the development of the immune-related response criteria, which takes a dynamic account of disease burden into consideration, including the development of new lesions, which are simply classified as progressive disease by RECIST.[16] Approximately, 30% of patients have disease control with ipilimumab, which translates to significant durable responses in many patients. Importantly, ipilimumab has improved survival by 10% at 2 and 3 years when compared with other therapies. Durability of responses and assessing the plateau at the “tail” of the survival curve with ipilimumab appears to mimic the experience with IL-2 suggesting that selected patients may be cured with this approach.
Unfortunately, the benefits of ipilimumab are not without significant toxicities, specifically immune related adverse events (iRAE's), which resulted in 14 deaths in the first Phase III clinical trial (7 of which were attributed to immune toxicity).[13] At the 3 mg/kg dose, approximately 60% of patients have an iRAE, with 10-15% grade 3/4 in nature.[13,17] The most frequently encountered iRAE's occur in the gastrointestinal tract, skin and liver. The most recent Phase III trial did not report any deaths, suggesting that the development of standardized management protocols for iRAEs and vigilance in monitoring and treating iRAEs is paramount along with good provider-patient communication.[14] This appears to mirror the learning curve of HD IL-2 administration with progressive improvement in the morbidity and mortality related to this therapy over time.[18]
BRAF
The discovery of v-Raf murine sarcoma viral oncogene homolog B (BRAF) mutations by the Sanger Institute has been the defining moment in melanoma molecular biology to date.[19] BRAF is a key member of the Rat sarcoma (RAS)mitogen-activated protein kinase (MAPK) pathway, a known regulator of cell growth and proliferation and when mutated can act as an oncogene. BRAF is highly expressed in testis, hematopoietic stems cells, neuronal tissue and melanocytes with mutations identified most frequently in papillary thyroid cancer, colorectal cancer and melanoma (BRAF mutations have also been discovered in a multitude of other cancers, but at a much lower rates). Approximately, 50% of cutaneous melanomas harbor a BRAF mutation, 97% of which are a result of a substitution of valine for glutamate at the 600 position of the amino acid sequence, the so-called V600E mutation.[20,21] This revelation re-invigorated targeted drug investigations in melanoma that were initially tempered by the failure of sorafenib, a moderate pan-inhibitor of BRAF in metastatic melanoma.[22,23] Development of specific and highly potent BRAF mutant inhibitors such as vemurafenib resulted in responses of approximately 80% in a Phase I/II trial.[24] Using the established oral dose of 960 mg twice a day from this Phase I trial, vemurafenib was tested versus dacarbazine in a Phase III randomized clinical trial in advanced treatment-naïve melanoma patients. Vemurafenib had significantly better response and survival rates leading to its approval in 2011 for use in patients harboring the BRAFV600E mutation.[25]
Dabrafenib is another orally administered potent inhibitor of mutant BRAF and showed promising results in the early Phase I/II trials, with a RR of 69% and a dose of 150 mg twice daily was established.[26] The randomized, controlled Phase III clinical trial in treatment-naïve advanced melanoma patients revealed a better RR and progression-free survival (PFS) (5.1 months vs. 2.7 months; hazard ratio [HR] 0.33, 95% confidence interval [CI], 0.18-0.51, P < 0.0001) when compared to dacarbazine.[27] In the initial report, the difference in OS was not statistically different (HR 0.61 in favor of dabrafenib, 95% CI, 0.25-1.48), but this may have been in part due to the permitted crossover to the dabrafenib arm for patients who progressed on dacarbazine. Based on these results dabrafenib became the second BRAF inhibitor to receive US FDA approval in May 2013 for BRAFV600E mutant melanoma.
Despite an initial remarkably high RR of 50% with both dabrafenib and vemurafenib, nearly all patients universally progress by 12 months. The average time of response is approximately 6 months, with disease progression that can often be rapid and lethal.[25,28] Resistance has been documented to occur via a number of MAPK dependent pathways (e.g., mitogen-activated/extracellular signal - regulated protein kinase, MEK mutations; NRAS up-regulation) and non-MAPK dependent pathways (e.g. insulin growth factor receptor, platelet derived growth factor receptor, etc.).[28,29,30,31,32,33,34,35,36,37,38] Further research will continue to define novel combinations and drugs that may be able to overcome resistance mechanisms.
Toxicity to BRAF inhibitors includes prominent photosensitivity, skin rash that is predominantly maculo papular in nature, QT interval prolongation, arthralgia and often most alarming to patients, the development of keratoacanthomas (KA).[25,39] Approximately 20% of patients will develop KAs during therapy, highlighting the importance of referral to a dermatologist for monitoring and management.[40] KA are usually treated with simple surgical excision, not necessitating a dose reduction or withholding of the BRAF inhibitor.[40]
MEK
MEK is a downstream target in the MAPK pathway and is the only known substrate for BRAF. Preclinical work has established MEK as a valid therapeutic target in the treatment of melanoma.[41] Multiple drugs targeting MEK have been developed and are currently being investigated clinically. Selumetinib is a potent, orally available, MEK 1/2 inhibitor which has been shown to possess activity in melanoma cell lines.[42] A Phase I trial showed disease stabilization in patients with melanoma.[43] A Phase II, open-label, randomized controlled trial of selumetinib versus temozolomide in treatment-naïve advanced melanoma patients showed no difference in objective RRs or PFS between the two arms.[44] Although this was a population unselected for mutations in BRAF/NRAS, five of the six partial responders to selumetinib were BRAF mutant suggesting its utility as a biomarker for further study.
Trametinib is another orally available potent MEK 1/2 inhibitor, which has shown preclinical evidence of antitumor activity in melanoma.[45] Phase I and II trial data demonstrated its activity in BRAF inhibitor-naïve BRAF mutant melanoma.[46,47] Phase I data established 2 mg once daily as the recommended dose and also revealed better responses (40%) in patients not previously treated with a BRAF inhibitor (compared to 17% in those with BRAF-mutant tumors who had prior exposure to BRAF inhibitor therapy).[46] A subsequent Phase II study in BRAF positive cutaneous melanoma with two cohorts, previously treated with or without a BRAF inhibitor, also confirmed improved outcomes in patients who were BRAF inhibitor naïve.[47] Patients who were naïve had a RR of 25% and a PFS of 4.0 months, compared with 0% and 1.8 months in the previously treated group.
METRIC was a Phase III randomized, controlled clinical trial of trametinib 2 mg once daily as compared to chemotherapy (dacarbazine or paclitaxel, investigator preference) in treatment-naïve advanced melanoma patients who were BRAF V600E or K mutant.[48] The 6-month OS (81% vs. 67%), median PFS (4.8 months vs. 1.5 months) and RR (22% vs. 8%) were all significantly improved with trametinib. The median OS had not yet been reached at the time of publication. Rash (predominantly papulopustular as opposed to maculopapular with BRAF inhibitors), diarrhea and peripheral edema were the most common adverse effects encountered. Other significant adverse effects included ventricular dysfunction and/or a decreased ejection fraction and ocular toxicity. Unlike BRAF inhibitors, squamous cell carcinoma (SCC) or KAs were not observed with trametinib. Based on these data, trametinib was approved by the US FDA in May 2013 for the treatment of BRAFV600E/K mutant melanoma advanced melanoma that had not been previously treated with a BRAF inhibitor. It is unclear where the true utility of this drug as a single agent may lie in this patient population as the RRs are less compared with BRAF inhibitors. The preferred patient population would likely be one where a BRAF inhibitor is not tolerated, but not where resistance has already developed.
COMBINING MEK AND BRAF INHIBITORS
Until date, combining orally available targeted therapies has been a challenge with significant dose-limiting toxicities and without a true clinical benefit gleaned. The majority of patients eventually develop resistance to BRAF inhibitors 6 to 7 months after initiating treatment via several mechanisms, both dependent and independent of the MAPK pathway. Pre-clinically, complete inhibition of this pathway can be attained by combining a MEK and BRAF inhibitor, which may forestall resistance to either therapy alone.[49] In addition, another benefit of MEK and BRAF combination therapy is the potential for reduced rates of SCC development, which has been associated with MAPK activation.[50,51]
To explore these hypotheses, a Phase I/II open label trial of dabrafenib and trametinib in advanced melanoma patients with BRAFV600E/K mutations was conducted in three parts.[52] Part A evaluated the pharmacokinetic interaction, part B evaluated escalating doses of both drugs and part C was the Phase 2 portion that randomized patients 1:1:1 to receive dabrafenib 150 mg twice a day alone or in combination with trametinib at a dose of 1 or 2 mg once daily respectively. The median PFS (9.4 months vs. 5.8 months; HR for progression or death = 0.39, 95% CI, 0.25-0.62, P < 0.001), RR (76% vs. 54%) and 1-year survival with no progression (41% vs. 9%) all were significantly improved with the addition of trametinib (2 mg daily) to dabrafenib compared with the latter as a single agent. The rate of cutaneous SCC was also less with the combination (for both arms with trametinib). The combination arms experienced a higher rate of pyrexia and chills, including the need for hospitalization in some patients. The use of combination MEK/BRAF inhibitor therapy is currently being explored in the Phase III setting to determine whether it will supplant the use of single agent BRAF inhibitor therapy in the appropriate patient population.
PHOSPHOINOSITIDE-3-KINASE/MAMMALIAN TARGET OF RAPAMYCIN
The PI3K-AKT-mTOR pathway has an essential role in many growth related physiologic functions and survival processes in human cells and tumorigenesis.[53] Deregulation of AKT3 has been shown to promote melanomagenesis and may occur in up to 60% of melanomas.[54] Phosphatase and tensin homolog (PTEN), a tumor suppressor gene, also has a role in the regulation of this pathway and if inactivated will result in high levels of PI3K-AKT activation. Decreases in PTEN arise in 10-43% of cutaneous melanomas.[55,56] mTOR, a key regulator of cell growth and survival, is also involved in the PI3K-AKT pathway, as mTORC1 is a direct target of AKT. Rapamycin, an mTOR analog, has been shown to decrease growth of melanoma cells.[57] Recently, combined inhibition of MEK and the PI3K-AKT-mTOR pathways was shown to be required for complete inhibition of NRAS mutant melanomas.[58]
RAS, a member of the MAPK pathway, also has direct interaction with PI3K with notions of crosstalk as evidenced by RAS's ability to increase PI3K activity. There is intriguing data that PTEN loss and constitutive activation of the PI3K-AKT pathway may play a role in resistance patterns seen with BRAF and MEK inhibitors.[29,59] Pre-clinically, it has been shown that persistent activation of the AKT pathway is associated with resistance to BRAF/MEK inhibitors.[59,60] Shorter PFS with a BRAF inhibitor has been shown to occur in patients with the loss of PTEN function. Combining BRAF/MEK inhibitors with the PI3K/mTOR inhibitor, GSK2126458, decreased cell growth in vitro among cell lines resistant to BRAF inhibition.[61]
There are a number of investigational drugs in various development stages targeting this pathway, along with two commercially available mTOR inhibitors, everolimus and temsirolimus. While the PI3K-Akt-mTOR pathway has been shown to effectively sensitize melanoma cells in vitro to alkylating agents,[62] a Phase II study combining temozolomide with everolimus failed to show significant activity.[63] Similarly, temsirolimus has also been trialed in metastatic melanoma through a Phase II trial of the California Cancer Consortium, but unfortunately with limited success.[64] It has been postulated that mTOR inhibitors alone may not work in melanoma due to a compensatory hyper-activation of AKT.
Perifosine, an alkylphosphocholine analog, has been shown to decrease levels of AKT. A Phase II study of perifosine in previously untreated patients with metastatic melanoma did not have any objective responses in the 14 evaluable patients.[65] To the best of our knowledge there is no current human clinical data available with the use of PI3K targeted drugs. Trials are now underway enrolling patients to new drugs targeting the PI3K-AKT pathway in melanoma. Until more in-depth data is obtained, the significance of the PI3K-AKT and mTOR pathways in melanoma remains undefined.
VASCULAR ENDOTHELIAL GROWTH FACTOR
Angiogenesis has been shown to an important component of tumor growth and progression in many tumor types. VEGF and fibroblast growth factor (FGF) are angiogenic factors that have been implicated in melanoma progression and growth.[66] VEGF is also up-regulated in melanoma cells when exposed to chemotherapy.[67] Bevacizumab, a monoclonal antibody that binds extracellular VEGF, has been evaluated in multiple studies for the treatment of metastatic melanoma. As a single-agent, with or without the addition of low dose interferon-alpha 2b, bevacizumab failed to produce significant responses with one patient on the combined arm demonstrating a partial response.[68] With the addition of HD interferon alpha-2b, responses have been noted as high as 24%.[69] von Moos et al. conducted a Phase II trial evaluating the efficacy of temozolomide with the addition of bevacizumab 10 mg/kg every 2 weeks.[70] 62 patients with untreated metastatic melanoma were enrolled with 1 CR and 9 PRs, for an overall RR of 16.1%.
Bevacizumab has also been combined with carboplatin and paclitaxel in two separate Phase II trials. The first was a single arm Phase II study which had 9 PRs for an overall RR of 17% with 57% having stable disease for at least 8 weeks.[71] The second, termed the BEAM study, was a randomized multicenter Phase II study of carboplatin and paclitaxel with either bevacizumab 15 mg/kg every 3 weeks or placebo in previously untreated metastatic melanoma.[72] The primary endpoint was PFS, with secondary endpoints of OS and safety. With 214 patients randomized in a 2:1 ratio to bevacizumab or placebo, the trial failed to meet its primary endpoint, with a PFS of 4.2 months in the placebo arm versus 5.6 months in the bevacizumab arm (P = 0.1414). There was a trend towards improved RR (25.55% vs. 16.4%) and OS (12.3 vs. 8.6 months), but neither was statistically significant. One trial has also been conducted evaluating the addition of bevacizumab to carboplatin and nab-paclitaxel or temozolomide in chemotherapy-naïve metastatic melanoma patients.[73] Temozolomide and bevacizumab had a RR of 23.8%, with a median PFS and OS of 3.8 and 12.3 months, respectively. The bevacizumab, carboplatin, nab-paclitaxel arm had an impressive RR of 33.3% with median PFS and OS of 6.7 and 13.9 months, respectively. In an upcoming Phase II randomized study, the combination of nab-paclitaxel plus bevacizumab will be compared to ipilimumab in advanced melanoma (NCT01879306). Bevacizumab has also been examined in combination with temsirolimus in a Phase II trial in patients with advanced melanoma.[74] In 17 patients enrolled, there were three partial responses (18%) noted and 8 patients (47%) had stable disease. Eight of 10 patients with wild-type BRAF had a response or stable disease. Aflibercept (VEGF-trap), is another VEGF targeted molecule with potent preclinical activity against melanoma and acts as a decoy VEGF receptor. In a multicenter, Phase II study in advanced chemotherapy-naïve melanoma (cutaneous or uveal origin) patients, 50% of evaluable patients were progression-free for at least 4 months with a 1-year OS of 56%.[75]
Oral VEGF targeted therapies have also been trialed in melanoma. Axitinib, a VEGF 1-3 inhibitor, was tested in a multicenter Phase II trial with 6 of 32 patients having a partial response (18.8%).[76] The median duration of response was 5.9 months with a median OS of 6.6 months. Dovitinib, a dual inhibitor of VEGF and FGF, has also been tested in advanced melanoma. A Phase I/II trial in advanced melanoma patients refractory to or relapsed after standard therapy evaluated dovitinib 200-500 mg/day.[77] 47 patients were enrolled, with no responses gleaned and a best overall response of stable disease in 12 patients. Although VEGF targeted therapy has yielded responses in melanoma, the jury is still out on its role in improving outcomes. Until such data is available, VEGF-targeted therapy should still be considered investigational.
KIT
The KIT gene encodes for the protein c-kit or CD117 and is expressed on a variety of cells including interstitial cells of Cajal, mast cells, hematopoietic progenitor cells and melanocytes. In melanoma, KIT has been shown to be an important factor in melanocyte growth.[78] When the ligand for c-kit, stem cell factor, binds to the receptor it causes dimerization and internal activation of tyrosine kinase signaling leading to cell proliferation and survival. Mutations in c-kit, resulting in constitutive activation of the pathway, are common in gastrointestinal stromal tumors (GIST). In the unselected melanoma population, c-kit mutations occur at a low rate.[79] However, in selected melanoma patients with chronic sun damaged skin, acral lentiginous or mucosal melanoma sub-types, mutations or amplification of c-kit have been described in up 25% of cases.[80]
Imatinib, a tyrosine kinase inhibitor approved for GIST and chronic myeloid leukemia is known to inhibit c-kit, abl and platelet derived growth factor. Early clinic trials utilizing imatinib in advanced melanoma were disappointing with a response in only one patient across 3 Phase II trials.[81,82,83] Importantly, the sole patient with a response had the highest c-kit expression, prompting further interest of imatinib use in patients with c-kit aberrations. Subsequent case reports noted major responses in melanoma patients with KIT mutated melanoma.[84,85,86]
Two Phase II trials utilizing imatinib in melanoma patients with c-kit aberrations have been completed. Carvajal et al. conducted a single group, open-label, Phase II trial in patients with metastatic melanoma arising from mucosal, acral or chronic sun damaged and c-kit mutations or amplification.[80] The overall durable RR was 16%, with 2 CR lasting 94 and 95 weeks and a median OS of 46.3 weeks. Guo et al. also conducted a Phase II open label single arm trial of imatinib in metastatic melanoma patients with c-kit mutations or amplification.[87] 10 of 43 patients had a partial response (23%), with 42% of patients demonstrating regression of tumors. 9 of the 10 responses had c-kit mutations in exons 11 or 13. Other oral TKI's with KIT inhibitor activity-dasatinib, nilotinib, sorafenib and sunitinib, have also been reported to induce responses in melanoma patient with KIT mutations.[88,89,90,91,92] Ongoing clinical trials utilizing drugs targeting c-kit mutations in melanoma will continue to define their role in this disease.
PROGRAMMED DEATH-1 RECEPTOR
As discussed previously, melanoma is one of the most immunogenic cancers known to mankind. There are various checkpoints which limit immune activation, thus preventing immune destruction of healthy tissues, but this unfortunately also allows cancer cells to undergo “immune escape”. PD-1 receptor, like CTLA-4, is a negative regulator of the immune system resulting in termination of the immune response. PD-L1 and PD-L2 are the ligands which bind to PD-1 and are expressed primarily by inflamed tissue and in the tumor microenvironment.[9] PD-L1 is the predominant ligand preferentially expressed by solid tumors.[93] Different from CTLA-4's interaction with B7, which occurs in lymphoid tissue during the priming phase of a T-cell response, PD-1's interaction is predominantly in the effector phase of a T-cell response thus preventing tissue damage.[9] Due to this, blockade of the PD-1 pathway has been postulated to cause fewer side effects when compared to CTLA-4 blockade. Pre-clinically, blockade of the interaction of PD-L1 with PD-1 has been shown to induce immunity and tumor regression.[94]
Nivolumab is a fully human IgG4 blocking monoclonal antibody targeting PD-1. In a Phase I dose escalation trial across several tumor types, 28% (26/94) of melanoma patients achieved a response.[95] Of these, 50% were durable, lasting a year or more. Of patients that had a tumor biopsy done, patient's tumors expressing PD-L1 had a higher objective RR (36%) than those that did not (0%). Nivolumab has also been shown to produce responses with re-induction of therapy after relapse of disease.[96] A second anti-PD-1 antibody, lambrolizumab produced a similar high RR of 38% in a Phase 1 trial in melanoma in patients (n = 135) who had previously been treated with other therapy, which included ipilimumab in 36% of patients.[97] The PFS for the entire cohort was greater than 7 months and the vast majority of patients who experienced a response had it ongoing at the time of analysis. A third antibody, BMS-936559 in the investigation in this class is a fully human IgG4 monoclonal antibody targeting PD-L1. In a Phase 1 trial reported in 2012, the RR in melanoma was 17.3% with 3 CRs and 55% of responses lasting 1 year or longer.[98]
Encouragingly, data from the Phase I trials targeting the PD-1 axis appear to suggest that iRAE's occur less often and at a lower grade when compared to CTLA-4 inhibitors. However one serious new safety signal gleaned was pneumonitis, which occurred in 3% (9 of 296) of patients and proved fatal in 3 patients (1%) treated with nivolumab. Phase II and III clinical trials are currently underway evaluating the efficacy of these three drugs in melanoma, either as single agents as well as in combination with other immunotherapy and targeted agents. Several other compounds targeting the PD-1 pathway (Pidilizumab, AMP224, MPDL3280A) are in early stage clinical development. With their demonstrated efficacy in patients refractory to other standard lines of therapy, it is very likely that this class will move to the forefront of melanoma therapy over the next decade.
OTHER PATHWAYS OF INTEREST OR IN DEVELOPMENT
Mutations in NRAS, which modulates survival and proliferation of melanoma, are present in 15-20% of cutaneous melanomas and are virtually mutually exclusive with BRAF (<1%).[38,99] To date, no drugs have been developed which inhibit mutant NRAS. MEK inhibition alone was shown to be effective in NRAS mutant melanoma cells lines.[100] The MEK inhibitors selumetinib and trametinib failed to show activity in NRAS mutant melanoma.[44,46] Recently MEK162, a MEK1/2 inhibitor, has shown some promising initial activity in NRAS mutant melanoma, with a 20% RR in a Phase II trial.[101] Overall, 63% of patients had disease control, but this was unfortunately short lived, with median PFS of 3.7 months. Pre-clinically, in vitro and in vivo, combined inhibition of MEK and PI3K/mTOR has been shown to more effectively inhibit NRAS mutant melanoma.[58] Immunotherapy also appears to be promising in this patient cohort as mutant NRAS was shown to be predictive of response to HD IL-2 in one retrospective review combining data from 2 high-volume treatment centers.[102]
GNAQ or GNA11 mutations, present in 83% of uveal melanomas cause activation of the MAPK pathway.[103] Currently, there are no drugs which directly target either mutation, but MEK inhibitors have shown some initial promising activity. Of 18 metastatic uveal melanoma patients treated with selumetinib, five had radiographic regression with two exhibiting a partial response.[104] TAK733, another MEK inhibitor, has also shown activity against uveal melanoma cell lines.[92] Ongoing work is aiming to improve treatment options for patients with metastatic uveal melanoma and GNAQ/GNA11 mutations as chemotherapy and immunotherapy trials have largely been unsuccessful in this disease.
IS IT TIME TO PERSONALIZE AN APPROACH YET?
Despite dramatic improvements in our therapeutic armamentarium, the vast majority of patients with metastatic melanoma succumb to their disease suggesting that ongoing investigations remain pivotal to further progress in this field. However, we are at a juncture where we can initiate a streamlined approach to an individual patient based on genotype in addition to the usual clinical characteristics such as disease burden and performance status. After two decades of therapeutic nihilism in melanoma, the challenge for the treating oncologist during this renaissance in melanoma therapy is to identify the most appropriate and safest approach for a patient taking into account the nuances of the new therapies now available. While it is tempting to combine available drugs given their non-overlapping mechanisms of action, this practice should only be guided by rigorously conducted trials. Combined CTLA4 and BRAF inhibition appeared to be an intuitive and exciting approach, but Phase I data revealed significant dose-limiting hepatotoxicity with the combination of ipilimumab and vemurafenib.[105]
All patients with unresectable or advanced melanoma must routinely have their tumor tissue tested for BRAFV600 mutations as initial stratification. Mutational testing for CKIT should be sought under appropriate clinical circumstances within an enriched population, e.g. patients with mucosal or acral melanomas. Immunotherapy offers the greatest potential for long-term durable responses, including cure in a minority of patients with metastatic melanoma. This curative potential is unique within the domain of solid-tumor medicine where palliation remains the norm for most advanced cancers. Hence it is our belief that patients with metastatic melanoma should be treated with immunotherapy in the first-line, including HD IL-2 for the appropriate candidate who meets rigorous cardio-pulmonary criteria to receive this drug. For patients not candidates for HD IL-2 or who progress after treatment with HD IL-2, ipilimumab as a single agent is a very appropriate choice for therapy. For patients with symptomatic or rapidly progressive disease that is BRAFV600 mutant, initiating therapy with either of the approved BRAF inhibitors is the obvious choice given their ability to induce rapid responses. Delaying a BRAF inhibitor to second-line and beyond therapy has not been shown to be detrimental to patient outcome. The role of MEK inhibitors in BRAFV600 mutant melanoma will likely become better defined once efficacy and survival results examining it in combination with BRAF inhibitors in ongoing trials are available. Finally, the rapidly evolving role of PD-1 blockade within this dynamic field will undoubtedly change paradigms of management in the very near future.[106]
The importance of clinical trials to enable us better understand the optimal treatment and sequencing in melanoma cannot be overstated. Genomic profiling and biomarker driven studies will hopefully pave the way to truly personalize therapy for patients with the eventual goal of redefining survivorship in this deadly disease where nihilism was the rule, rather than the exception. We believe we will see this change soon.
AUTHOR'S PROFILE
Anthony Jarkowski, III, Department of Pharmacy, James P. Wilmot Cancer Center, University of Rochester Medical Center, Rochester, NY 14642, USA.
Nikhil I. Khushalani, Department of Medicine, Roswell Park Cancer Institute, Buffalo, NY 14263, USA.
Footnotes
Source of Support: Grant funding from the National Comprehensive Cancer Network from general research support from Roche, Allos and Pfizer.
Conflict of Interest: None declared.
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Kalialis LV, Drzewiecki KT, Klyver H. Spontaneous regression of metastases from melanoma: Review of the literature. Melanoma Res. 2009;19:275–82. doi: 10.1097/CMR.0b013e32832eabd5. [DOI] [PubMed] [Google Scholar]
- 3.Eggermont AM, Suciu S, Testori A, Santinami M, Kruit WH, Marsden J, et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J Clin Oncol. 2012;30:3810–8. doi: 10.1200/JCO.2011.41.3799. [DOI] [PubMed] [Google Scholar]
- 4.Davar D, Tarhini AA, Kirkwood JM. Adjuvant therapy for melanoma. Cancer J. 2012;18:192–202. doi: 10.1097/PPO.0b013e31824f118b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–16. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 6.Blanchard T, Srivastava PK, Duan F. Vaccines against advanced melanoma. Clin Dermatol. 2013;31:179–90. doi: 10.1016/j.clindermatol.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Allison JP, Krummel MF. The Yin and Yang of T cell costimulation. Science. 1995;270:932–3. doi: 10.1126/science.270.5238.932. [DOI] [PubMed] [Google Scholar]
- 8.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366:2517–9. doi: 10.1056/NEJMe1205943. [DOI] [PubMed] [Google Scholar]
- 10.McArthur GA, Ribas A. Targeting oncogenic drivers and the immune system in melanoma. J Clin Oncol. 2013;31:499–506. doi: 10.1200/JCO.2012.45.5568. [DOI] [PubMed] [Google Scholar]
- 11.Tivol EA, Boyd SD, McKeon S, Borriello F, Nickerson P, Strom TB, et al. CTLA4Ig prevents lymphoproliferation and fatal multiorgan tissue destruction in CTLA-4-deficient mice. J Immunol. 1997;158:5091–4. [PubMed] [Google Scholar]
- 12.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 13.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert C, Thomas L, Bondarenko I, O’Day S, MD JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 15.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616–22. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 17.Weber JS, Dummer R, de Pril V, Lebbé C, Hodi FS MDX010-20 Investigators. Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: Detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer. 2013;119:1675–82. doi: 10.1002/cncr.27969. [DOI] [PubMed] [Google Scholar]
- 18.Kammula US, White DE, Rosenberg SA. Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer. 1998;83:797–805. [PubMed] [Google Scholar]
- 19.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 20.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty KT, McArthur G. BRAF, a target in melanoma: Implications for solid tumor drug development. Cancer. 2010;116:4902–13. doi: 10.1002/cncr.25261. [DOI] [PubMed] [Google Scholar]
- 22.Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–6. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flaherty KT, Lee SJ, Zhao F, Schuchter LM, Flaherty L, Kefford R, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol. 2013;31:373–9. doi: 10.1200/JCO.2012.42.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet. 2012;379:1893–901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 28.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–60. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–81. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71:5067–74. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem. 2012;287:28087–98. doi: 10.1074/jbc.M112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–74. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 39.Boussemart L, Routier E, Mateus C, Opletalova K, Sebille G, Kamsu-Kom N, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: A study of 42 patients. Ann Oncol. 2013;24:1691–7. doi: 10.1093/annonc/mdt015. [DOI] [PubMed] [Google Scholar]
- 40.Lacouture ME, Duvic M, Hauschild A, Prieto VG, Robert C, Schadendorf D, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314–22. doi: 10.1634/theoncologist.2012-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 43.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 46.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–9. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–9. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 49.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–30. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30:316–21. doi: 10.1200/JCO.2011.36.7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–88. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 54.Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–10. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 55.Mikhail M, Velazquez E, Shapiro R, Berman R, Pavlick A, Sorhaindo L, et al. PTEN expression in melanoma: Relationship with patient survival, Bcl-2 expression, and proliferation. Clin Cancer Res. 2005;11:5153–7. doi: 10.1158/1078-0432.CCR-05-0397. [DOI] [PubMed] [Google Scholar]
- 56.Davies MA. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012;18:142–7. doi: 10.1097/PPO.0b013e31824d448c. [DOI] [PubMed] [Google Scholar]
- 57.Buscà R, Bertolotto C, Ortonne JP, Ballotti R. Inhibition of the phosphatidylinositol 3-kinase/p70(S6)-kinase pathway induces B16 melanoma cell differentiation. J Biol Chem. 1996;271:31824–30. doi: 10.1074/jbc.271.50.31824. [DOI] [PubMed] [Google Scholar]
- 58.Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A. 2013;110:4015–20. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–47. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–20. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- 62.Sinnberg T, Lasithiotakis K, Niessner H, Schittek B, Flaherty KT, Kulms D, et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J Invest Dermatol. 2009;129:1500–15. doi: 10.1038/jid.2008.379. [DOI] [PubMed] [Google Scholar]
- 63.Dronca RS, Allred JB, Perez DG, Nevala WK, Lieser EA, Thompson M, et al. Phase II Study of Temozolomide (TMZ) and Everolimus (RAD001) Therapy for Metastatic Melanoma: A North Central Cancer Treatment Group Study, N0675. Am J Clin Oncol. 2013 Jan 24; doi: 10.1097/COC.0b013e31827b45d4. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, et al. CCI-779 in metastatic melanoma: A phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–8. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 65.Ernst DS, Eisenhauer E, Wainman N, Davis M, Lohmann R, Baetz T, et al. Phase II study of perifosine in previously untreated patients with metastatic melanoma. Invest New Drugs. 2005;23:569–75. doi: 10.1007/s10637-005-1157-4. [DOI] [PubMed] [Google Scholar]
- 66.Lázár-Molnár E, Hegyesi H, Tóth S, Falus A. Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine. 2000;12:547–54. doi: 10.1006/cyto.1999.0614. [DOI] [PubMed] [Google Scholar]
- 67.Lev DC, Ruiz M, Mills L, McGary EC, Price JE, Bar-Eli M. Dacarbazine causes transcriptional up-regulation of interleukin 8 and vascular endothelial growth factor in melanoma cells: A possible escape mechanism from chemotherapy. Mol Cancer Ther. 2003;2:753–63. [PubMed] [Google Scholar]
- 68.Varker KA, Biber JE, Kefauver C, Jensen R, Lehman A, Young D, et al. A randomized phase 2 trial of bevacizumab with or without daily low-dose interferon alfa-2b in metastatic malignant melanoma. Ann Surg Oncol. 2007;14:2367–76. doi: 10.1245/s10434-007-9389-5. [DOI] [PubMed] [Google Scholar]
- 69.Grignol VP, Olencki T, Relekar K, Taylor C, Kibler A, Kefauver C, et al. A phase 2 trial of bevacizumab and high-dose interferon alpha 2B in metastatic melanoma. J Immunother. 2011;34:509–15. doi: 10.1097/CJI.0b013e31821dcefd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.von Moos R, Seifert B, Simcock M, Goldinger SM, Gillessen S, Ochsenbein A, et al. First-line temozolomide combined with bevacizumab in metastatic melanoma: A multicentre phase II trial (SAKK 50/07) Ann Oncol. 2012;23:531–6. doi: 10.1093/annonc/mdr126. [DOI] [PubMed] [Google Scholar]
- 71.Perez DG, Suman VJ, Fitch TR, Amatruda T, 3rd, Morton RF, Jilani SZ, et al. Phase 2 trial of carboplatin, weekly paclitaxel, and biweekly bevacizumab in patients with unresectable stage IV melanoma: A North Central Cancer Treatment Group study, N047A. Cancer. 2009;115:119–27. doi: 10.1002/cncr.23987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim KB, Sosman JA, Fruehauf JP, Linette GP, Markovic SN, McDermott DF, et al. BEAM: A randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. J Clin Oncol. 2012;30:34–41. doi: 10.1200/JCO.2011.34.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kottschade LA, Suman VJ, Perez DG, McWilliams RR, Kaur JS, Amatruda TT, 3rd, et al. A randomized phase 2 study of temozolomide and bevacizumab or nab-paclitaxel, carboplatin, and bevacizumab in patients with unresectable stage IV melanoma: A North Central Cancer Treatment Group study, N0775. Cancer. 2013;119:586–92. doi: 10.1002/cncr.27760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Slingluff CL, Jr, Petroni GR, Molhoek KR, Brautigan DL, Chianese-Bullock KA, Shada AL, et al. Clinical activity and safety of combination therapy with temsirolimus and bevacizumab for advanced melanoma: A phase II trial (CTEP 7190/Mel47) Clin Cancer Res. 2013;19:3611–20. doi: 10.1158/1078-0432.CCR-12-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tarhini AA, Frankel P, Margolin KA, Christensen S, Ruel C, Shipe-Spotloe J, et al. Aflibercept (VEGF Trap) in inoperable stage III or stage iv melanoma of cutaneous or uveal origin. Clin Cancer Res. 2011;17:6574–81. doi: 10.1158/1078-0432.CCR-11-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fruehauf J, Lutzky J, McDermott D, Brown CK, Meric JB, Rosbrook B, et al. Multicenter, phase II study of axitinib, a selective second-generation inhibitor of vascular endothelial growth factor receptors 1, 2, and 3, in patients with metastatic melanoma. Clin Cancer Res. 2011;17:7462–9. doi: 10.1158/1078-0432.CCR-11-0534. [DOI] [PubMed] [Google Scholar]
- 77.Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM. Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res. 2011;17:7451–61. doi: 10.1158/1078-0432.CCR-11-1747. [DOI] [PubMed] [Google Scholar]
- 78.Wehrle-Haller B. The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res. 2003;16:287–96. doi: 10.1034/j.1600-0749.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- 79.Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821–8. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 80.Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–34. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wyman K, Atkins MB, Prieto V, Eton O, McDermott DF, Hubbard F, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy. Cancer. 2006;106:2005–11. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- 82.Ugurel S, Hildenbrand R, Zimpfer A, La Rosée P, Paschka P, Sucker A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;92:1398–405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim KB, Eton O, Davis DW, Frazier ML, McConkey DJ, Diwan AH, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008;99:734–40. doi: 10.1038/sj.bjc.6604482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hodi FS, Friedlander P, Corless CL, Heinrich MC, Mac Rae S, Kruse A, et al. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol. 2008;26:2046–51. doi: 10.1200/JCO.2007.14.0707. [DOI] [PubMed] [Google Scholar]
- 85.Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492–3. doi: 10.1111/j.1755-148X.2008.00475.x. [DOI] [PubMed] [Google Scholar]
- 86.Satzger I, Küttler U, Völker B, Schenck F, Kapp A, Gutzmer R. Anal mucosal melanoma with KIT-activating mutation and response to imatinib therapy - Case report and review of the literature. Dermatology. 2010;220:77–81. doi: 10.1159/000265558. [DOI] [PubMed] [Google Scholar]
- 87.Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904–9. doi: 10.1200/JCO.2010.33.9275. [DOI] [PubMed] [Google Scholar]
- 88.Woodman SE, Trent JC, Stemke-Hale K, Lazar AJ, Pricl S, Pavan GM, et al. Activity of dasatinib against L576P KIT mutant melanoma: Molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8:2079–85. doi: 10.1158/1535-7163.MCT-09-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kluger HM, Dudek AZ, McCann C, Ritacco J, Southard N, Jilaveanu LB, et al. A phase 2 trial of dasatinib in advanced melanoma. Cancer. 2011;117:2202–8. doi: 10.1002/cncr.25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18:1457–63. doi: 10.1158/1078-0432.CCR-11-1987. [DOI] [PubMed] [Google Scholar]
- 91.Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012;30:2008–14. doi: 10.1007/s10637-011-9763-9. [DOI] [PubMed] [Google Scholar]
- 92.Quintás-Cardama A, Lazar AJ, Woodman SE, Kim K, Ross M, Hwu P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008;5:737–40. doi: 10.1038/ncponc1251. [DOI] [PubMed] [Google Scholar]
- 93.Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010;116:1757–66. doi: 10.1002/cncr.24899. [DOI] [PubMed] [Google Scholar]
- 94.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 95.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–8. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–10. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244) Cancer Res. 2010;70:2264–73. doi: 10.1158/0008-5472.CAN-09-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–56. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 102.Joseph RW, Sullivan RJ, Harrell R, Stemke-Hale K, Panka D, Manoukian G, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35:66–72. doi: 10.1097/CJI.0b013e3182372636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carvajal RD, Ambrosini G, Wolchok JD, Chapman PD, Dockson MA, D’Angelo SP, et al. Pharmacodynamic activity of selumetinib to predict radiographic response in advanced uveal melanoma. J Clin Oncol (Meeting Abstracts) 2012 May;30(no 15_suppl):8598. [Google Scholar]
- 105.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 106.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]