

Abstract
Background
Tumor necrosis factor-alpha (TNF-α) plays a central role in the molecular pathogenesis of periodontal disease. However, the epigenetic regulation attributable to microbial and inflammatory signals at the biofilm gingival interface are poorly understood. In this study, we investigated the DNA methylation alteration within the TNFA promoter in human gingival biopsies from different stages of periodontal disease, and explored the regulatory mechanism of TNFA transcription by DNA methylation.
Methods
Gingival biopsies were harvested from 17 chronic periodontitis patients and 18 subjects with periodontal health. Another 11 subjects participated in an experimentally induced gingivitis study, and gingival biopsies were collected at the baseline, induction, and resolution phase. To confirm that TNFA promoter methylation modulated TNFA transcription we treated THP.1 cells with a DNA methyltransferase inhibitor, 5-aza-2-deoxycytidine and used a RAW 294.7 cell line transfected with a TNFA promoter-specific luciferase reporter system with or without methlyaiton,
Results
In gingival biopsies from subjects with severe chronic periodontitis two individual CpG sites within the TNFA promoter (at -163bp and -161bp) displayed increased methylation in periodontitis samples as compared to gingival health (16.1±5.1% vs. 11.0±4.6%, p=0.02, 19.8±4.1% vs. 15.4±3.6%, p=0.04, respectively). The methylation level at -163bp was inversely associated with the transcription level of TNFA (p=0.018). However, no significant difference in the TNFA promoter methylation pattern was observed in samples biopsied during the induction or resolution phase of experimentally induced gingivitis, which represented a reversible periodontal lesion. THP.1 cells treated with 5-aza-2-deoxycytidine demonstrated a time-dependent increase in TNFA messenger level. We also found that the luciferase activity decreased 2.6 fold in a construct containing an in vitro methylated TNFA promoter as compared to the unmethylated insert (p=0.03).
Conclusion
Although the biopsy samples represented a mixed cell population, the change in promoter methylation status in chronic periodontal disease suggested that DNA methylation may be an important regulatory mechanism in controlling TNFA transcriptional expression in disease.
Introduction
Periodontal disease as a chronic infection has often been described as an abnormal inflammatory response to the commensal bacteria within the biofilm. 1, 2, 3 Tumor necrosis factor –alpha (TNF-α), a primary inflammatory cytokine produced largely by macrophages/monocytes and periodontal resident cells as the early innate immune response to the bacterial challenge, can sustain or amplify inflammation by inducing a cascade of secondary inflammatory mediators such as cyclooxygenase-2, and matrix metalloproteinases (MMPs) that are responsible for extracellular matrix degradation and bone resorption.4 Abnormal levels of TNF-α have been closely associated with aggressive or actively progressing periodontitis. Duarte et al. 5 found that the gingival crevicular fluid (GCF) level of TNF-α in patients with generalized aggressive periodontitis was significantly elevated as compared to patients exhibiting gingival health or chronic periodontitis. In subjects with severe periodontitis, at their terminal (hopeless) dentition stage, the TNFA mRNA level was higher in the isolated gingival monocytes as compared to the level seen in cells harvested from chronic periodontitis subjects in a non-terminal stage.6 Although TNF-α has been shown to increase in periodontal tissues that exhibit active inflammation or progression, its level is not always elevated in the chronic and quiescent periodontal lesion. Offenbacher and colleagues 7 were unable to detect a difference in the GCF TNF-α level from periodontitis patients with deep probing depths and low bleeding scores as compared to subjects with gingival health. Similar findings have been also reported by other investigators. 5 For example, GCF TNF-α levels do not change during the induction and resolution phase of experimentally-induced gingivitis, 8 whereas others reported increases in GCF TNF-α in naturally occurring gingivitis relative to health,9, 10. Undoubtedly, during biofilm overgrowth, tissue TNF-α expression increases with activation of the innate immune response. However, since TNF-α is secreted very early and transiently in the inflammatory response and free TNF-α becomes tightly bound to tissue receptors, the failure to detect any changes in GCF is not surprising. In addition, persistent high tissue levels of TNF-α as a consequence of chronic infection would biologically lead to tissue necrosis and apoptosis. It is possible that in chronic infection, with inflammation, the host would potentially subdue the expression of TNF-α by different mechanisms. Local attenuation of TNF-α would potentially avert massive tissue destruction within the periodontium and potentially minimize systemic exposure to TNF-α, thereby protecting the host.
Although much is known about the control of TNF-α production achieved through signaling networks and genetic variations, the potential role of epigenetic regulation of TNF-α expression in periodontal diseases remains largely unknown. As compared to other transient epigenetic regulatory mechanisms such as post-translational modifications of histone molecules, and small non-coding (micro) RNA modulation, DNA methylation is heritable and the least reversible epigenetic modification mark. It has been generally accepted that the degree of DNA methylation, which almost always occurs in cytosines within CpG dinucleotide context, present in gene promoter regions is usually inversely related to the transcriptional level of those genes, 11,12 although exceptions have also been reported. 13 Recently, we have described an increased methylation (hypermethylation) of CpG rich region within the PTGS2 promoter region in human gingival biopsies with chronic periodontitis, which is associated with a subdued PTGS2mRNA level. 14 Bobetsis and co-workers15 using a mouse model demonstrated that maternal infection with a periodontal pathogen, Campylobacter rectus (C. rectus), can induce a hypermethylation profile within the P0 promoter region of IGF2 in placental tissues. These data suggest that inflammation and/or pathogenic oral bacteria may induce epigenetic changes in the promoter regions of specific genes to modify gene expression.
In the gingival sulcus, the chronic exposure to the omnipresent oral microorganisms resident in the biofilm and the inflammatory state of periodontal diseases may alter the gene expression profile of the local gingival tissue by inducing epigenetic changes. Therefore, we conducted experiments to test the hypothesis that in chronic periodontitis there would be a hypermethylation profile within the TNFA promoter region that would serve to suppress the expression of this gene. Additional experiments were performed in vitro to confirm that TNFA promoter methylation modulates TNFA mRNA expression.
Materials & Methods
Participants and tissue specimens
A total of forty-six participants provided informed consent and were recruited into two clinical studies wherein all components were approved by the Institutional Review Board (IRB) of the University of North Carolina at Chapel Hill (UNC-CH). In the first study, gingival samples were obtained from thirty-five participants, aged between 19 and 63 years. This cross-sectional study included 17 gingival biopsies harvested from different patients with chronic periodontitis and 18 control samples from subjects with periodontal health. All participants reported no use of either antibiotics or non-steroidal anti-inflammatory drugs (NSAIDs) within 1 month before enrollment. All participants reported having no medical treatment for systemic diseases 3 months prior to the gingival biopsy or periodontal surgery. Periodontal clinical measurements, including probing depth (PD), clinical attachment loss (CAL), and bleeding on probing (BOP) were obtained prior to the biopsies. Biopsies harvested from periodontitis patients were obtained from sites exhibiting at least 5mm of probing depth and radiographic evidence of alveolar bone loss. The control biopsies were collected from different participants who were periodontally healthy or had mild gingivitis at non-study sites. Those biopsy samples, which were harvested from either volunteers or participants having undergone crown-lengthening procedures, exhibited PD measurement of 4mm or less, no BOP, and no evidence of radiographic alveolar bone loss.
In the second clinical study, gingivitis was experimentally induced in 11 participants using an established protocol. 16 Gingival biopsies were collected at the baseline, the peak of gingivitis (21 days), and again at resolution phase using previously published methods. 8 Participants with BOP scores of 10% and probing depth <5 mm were re-established to periodontal health through daily oral hygiene and dental prophylaxes. One biopsy per participant representing baseline gingival health was harvested at one interproximal site one week after the completion of prophylaxis. The participants were then instructed to wear two stents during routine toothbrushing and not to floss the stent-covered teeth. The plaque in the stent-covered teeth induced gingivitis in a 3-week period. At day 21, another biopsy from each participant was collected at an interproximal site of gingiva in one of the stent-covered sextants (induction phase). Then, the participants were instructed to resume dental hygiene measures on the stent-covered teeth. After 4 weeks of resuming dental hygiene practice the third piece of biopsy representing resolved gingivitis (resolution phase) was collected at a different interproximal site from the second biopsy site, but was within the stent coverage.
The surgical procedure and specimen dissection has been described elsewhere 14. Upon removal, all biopsied gingival tissues were divided into two pieces. One piece, used for DNA methylation analysis, was kept in the −80°C freezer immediately, while the other half, used for real-time polymerase chain reaction (RT-PCR), was incubated with RNA-later (Applied Biosystems/Ambion, Austin, TX, USA) overnight at 4°C, and then transferred to −80°C for storage.
Cell cultures
THP-1 cells, a human monocytic cell line, originally purchased from American Type Culture Collection (ATCC# TIB-202, Manassas, VA, USA) were obtained from Tissue Culture Facility at UNC-CH. The cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, California, USA) supplemented with 10% fetal bovine serum (FBS) (Mediatech, Manassas, VA), 5x10-5M 2-mercaptoethanol (Sigma, St. Louis, MO, USA), and 1% penicillin/streptomycin (Invitrogen). RAW264.7 cells, a murine monocytic cell line, were generously given by Dr. Patrick Flood at the School of Dentistry, UNC-CH. Cells were maintained in DMEM medium (Lonza, Basel, Switzerland) supplemented with 10% FBS, and 1% penicillin/streptomycin, 2mM L-glutamine (Cat.# 17-605E, Lonza). All cells were grown in a humidified incubator with 5% CO2 at 37°C.
C. rectus strain 314 was grown under an anaerobic condition at 37 °C on Enriched Tryptic Soy agar (Cat.# AS-546, Anaerobe systems, Morgan Hill, CA, USA) in an anaerobic chamber. C. rectus were collected at the late logarithmic phase and resuspended in the antibiotic-free culture medium to an optical density of 1.00 (at 600 nm).
DNA isolation and sodium bisulfite conversion
Genomic DNA was extracted from the collected gingival biopsies using a DNeasy Mini kit (QIAgen, Valencia, CA, USA) according to manufacturer's manual. Genomic DNA was treated with sodium bisulfite by published procedures. 17 Briefly, 500ng∼1μg genomic DNA in 45μl of water was denatured at 42°C for 20 minutes in freshly prepared 3M sodium hydroxide solution. Then DNA was incubated with saturated sodium bisulfite (Cat.# 243937, Sigma) solution containing 10mM of hydroquinone (Cat.# H9003, Sigma), with a final pH at 5.0, at 55°C overnight. Converted DNA was purified using a Wizard DNA Clean-up Column (Promega, Madison, WI, USA) and desulfonated by incubation with the 3M sodium hydroxide solution at 37°C for 20 minutes. Sodium bisulfite treated DNA was finally precipitated with ethanol, and then resolved in 25ul of 1mM Tris-Cl (pH 8).
Bisulfite-specific PCR and pyrosequencing
The primers and annealing temperatures used in the bisulfite-specific PCR are listed in Table 1. Four PCR amplicons, which were amplified by a HotStar Taq kit (QIAgen), encompass a total of 10 CpG sites within the sequence upstream of TNFA transcription start. Quantification of the percentage of methylated cytosine at each CpG site was determined by pyrosequencing as mentioned previously. 18 Corresponding sequencer specific to each amplicon can be also found in Table 1. An artificially methylated genomic DNA (Cat.#S7821, Millipore, Billerica, MA, USA) was used in the bisulfite conversion, bisulfite-specific PCR and pyrosequencing with the primers and sequencers mentioned above as a technical control.
Table 1. Oligonucleotides used for bisulfite specific PCR and pyrosequencing.
| CpG site (*) Included | Details | Sequences | Annealing T (°C) |
|---|---|---|---|
| -244, -238 | Forward | 5′-TAGGTTTTGAGGGGTATGGG -3′ | 62 |
| Reverse | 5′-[Biotin]TCAAAAATACCCCTCACACTCC -3′ | ||
| Sequencer | 5′-GTTAGTGGTTTAGAAGATTT-3′ | ||
| -169, -163, -161 |
Forward | 5′-[Biotin]GAGTGTGAGGGGTATTTTTGATG -3′ | 63 |
| Reverse | 5′-GCAACCATAATAAACCCTACACCTTC-3′ | ||
| Sequencer | 5′-AAACCCTACACCTTCTATCT -3′ | ||
| -146, -119 | Forward | 5′-GAGGGGTATTTTTGATGTTTGTGT -3′ | 61 |
| Reverse | 5′-[Biotin] CAACCAACCAAAAACTTCCTTAAT-3′ | ||
| Sequencer | 5′-TTTAGAGATGGAGAAGAAA-3′ | ||
| -72, -49, -38 | Forward | 5′-GAGGGGTATTTTTGATGTTTGTGT-3′ | 63 |
| Reverse | 5′-[Biotin]CCAACAACTACCTTTATATATCCC -3′ | ||
| Sequencer | 5′-TTATGGGTTTTTTTATTAAG-3′ |
CpG sites indicate nucleotide position in relation to transcription start (TSS).
“−” or “+”, indicates upstream or downstream of TSS, respectively.
RNA Isolation and quantitative real-time PCR
Total RNA was isolated from gingival tissues and cells with an RNeasy Mini Kit (Qiagen). cDNA from 500ng of isolated RNA was synthesized by an Omniscript Kit (Qiagen) and random decamer primers (Cat. # 5722E, Ambion, Austin, TX, USA). Real-time PCR of the messenger level of TNFA was determined by a 20x assay on demand gene expression assay mix (Cat. # hs_99999043_m1, Applied Biosystems) in a 7000 Sequence Detection System (ABI Prism, Applied Biosystems). Ribosome 18s RNA (part# 4308329, Applied Biosystems) was employed as an internal control.
5-azacytidin treatment of cells
Dissolved 5-Aza-2′-deoxycytidine (5-Aza-2dC, Cat.# A3656, Sigma), an inhibitor of DNA methylation, was added to THP-1 cells to achieve a final concentration of 5μM. Cells were washed and re-treated with freshly prepared 5-Aza-2dC every the other day.
Cloning of TNFA promoter, transfection and luciferase reporter assay
A fragment ranging from -291bp and +44bp relative to the transcription start of TNFA, which includes 11 CpG dinucleotides, was generated by PCR using primers: 5′-TCCGGTACCCCTCCAGGGTCCTACACACA-3 (forward); 5′- TCCAAGCTT TAGCTGGTCCTCTGCTGTCC-3′ (reverse). Two restrictive endonuclease recognition sites KpnI and HindIII were underlined, respectively. Digested PCR product was ligated using a T4 DNA ligase to a pGL-3 luciferase reporter vector (Cat.#E1751, Promega) that was also pre-digested with KpnI and HindIII. After transformation of E. Coli DH5α with the construct (pGL3-PTNFA291), the inserted promoter fragment was confirmed by sequencing.
Approximately 1.8×105 RAW 294.7 cells per well on a 48-well plate were transfected with either 120ng pGL3-PTNFA291 or 220ng modified pGL3-PTNFA291 (methylated or mock-methylated) with a Lipofectamin™ 2000 reagent (Invitrogen). The luciferase reporter constructs were always co-transfected with a Renilla luciferase vector (pRL-TK vector, Cat.# E2241, Promega) to account for transfection efficiency. After 18 hours post transfection, the cells were challenged with E. coli LPS (500ng/μl) for another 16 hours.
Cells were harvested and the luciferase activities were measured by the Dual Luciferase Reporter Assay System (Cat.# 1910, Promega) following the manufacturer's instruction using a Lumat LB9507 luminometer (Berthold, Oak Ridge, TN, USA).
In vitro methylation
In vitro methylation of cloned TNFA promoter fragment was performed according to a protocol developed by Dell et al. 19 Briefly, 80μg of pGL3-PTNFA291 were digested by HindIII and KpnI. The recovered insert was then incubated with 24 units of M. SssI, a CpG methyltransferase from New England Biolabs (Cat.# M0226S, Ipswich, MA, USA), in the presence of S-adenosylmethionine (SAM), which is a methyl- donor, with a final concentration of 160μM. In parallel, a mock methylation was also performed without the supplement of SAM. The completion of the in vitro methylation was confirmed by BstUI (Cat.#R0518S, New England Biolabs) digestion. After purification, modified insert was directly religated with the digested pGL3 vector by the T4 DNA ligase to achieve either pGL3-PTNFA291Met (methylated promoter insert) or pGL3-PTNFA291Mo (mock-methylated promoter insert).
Statistical analysis
Analysis of variance (ANOVA) was applied for the statistical analyses of both clinical measurements and the realtime-PCR data of experimentally induced gingivitis. Fisher's exact test was used to test gender difference among participants. Hotelling's two-sample T2 test was applied to compare the overall methylation difference between chronic periodontitis and control samples, and Hotelling's one-sample T2 test was used for within subject comparisons among different phases of gingivitis samples. We employed Holm (step-down Bonferroni) technique to address the multiple comparisons consisting of individual tests for methylation percentage at each of the 10 CpG sites. All analyses of methylation data used the arcsin transformation to achieve constant variance. Student t-tests were employed to test the difference of other quantitative data. Linear regression analysis was applied to test for the significance of slope to evaluate the association between the methylation level of specific CpG site and TNFA transcription from the gingival biopsies. The significance level was set to 0.05.
Results
Participants
The demographic information and periodontal indices of these study participants are listed in the Table 2. No significant differences in gender and age were found among participants with either chronic periodontitis or with periodontal health. As expected, sites with chronic periodontitis exhibited higher readings for both probing depth and attachment loss when compared to the sites with periodontal health.
Table 2. Demographic information of the participants and clinical parameters in the biopsied gingival sites.
| Demographic/ Clinical parameters |
Periodontal Health (n=17) |
Periodontitis (n=18) |
Experimentally Induced Gingivitis (n=11) |
|---|---|---|---|
| Mean age (years) | 40.9± 13.5 | 48.7±8.7 | 36.8±9.7 |
| Gender | |||
| Males/Females | 5/12 | 11/7 | 5/6 |
| Probing Depth (Mean±SD, mm) |
1.9±0.9 | 5.7±1.1** | 2.4±0.3 |
| Clinic Attachment Level (Mean± SD, mm) |
0.9±0.6 | 4.1±1.0** | 1.1±0.7 |
| Alveolar bone loss | No | Yes | No |
indicates p<0.001 as compared with periodontal health
Promoter methylation level and transcription of TNFA in chronic periodontitis samples
Because DNA methylation affects the architecture of chromatin structure and the assembly of transcription machinery around the transcription start site (TSS), we analyzed the methylation status of 10 CpG dinucleotides that are just present upstream of TNFA TSS (Fig. 1). These 10 CpG sites are interspersed among several transcription factor binding sequences, such as NF-κB, AP1, Sp1 and CRE, which have been described within this region.
Figure 1.
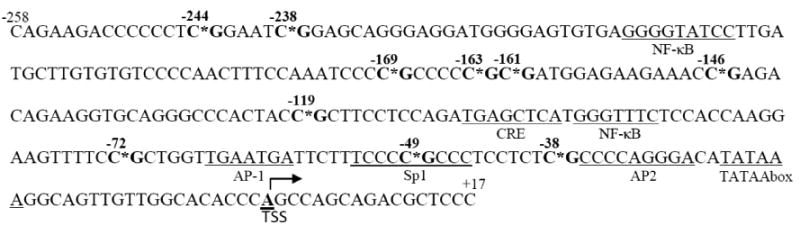
Genomic sequence of TNFA promoter fragment. Sequence ranging from -258bp to +17bp relative to TSS before bisulfite modification is presented. This promoter fragment just upstream of TSS contains ten CpG dinucleotides that are marked with “*” and in bold. Sites for potential transcriptional factor binding and TATA box are underlined.
The overall difference of percentage methylation in all 10 CpG sites between the chronic periodontitis and periodontal health samples was marginally significant (p=0.05) as determined by the two-sample Hotelling T2-test. However, site-specific comparisons of methylation level revealed that the percentage methylation at CpG site -163 and -161bp was significantly higher in the periodontitis samples than the same sites from biopsies with gingival health (16.1±5.1% vs. 11.0±4.6%, p=0.02, 19.8±4.1% vs. 15.4±3.6%, p=0.04, respectively, Fig. 2A). The messenger levels of TNFA showed a non-significant reduction in chronic periodontitis biopsies in comparison to samples of periodontal health. (p=0.08, Fig. 2B). We then examined the association between the mRNA expression and the methylation level of the two key sites -163 and -161 in Figure 2C. A significant inverse association between promoter methylation level at -163bp and its mRNA expression is present (r=0.16, p=0.018, Fig. 2C). No such a significant association between mRNA expression of TNFA and promoter methylation at -161bp can be detected (data not shown).
Figure 2.
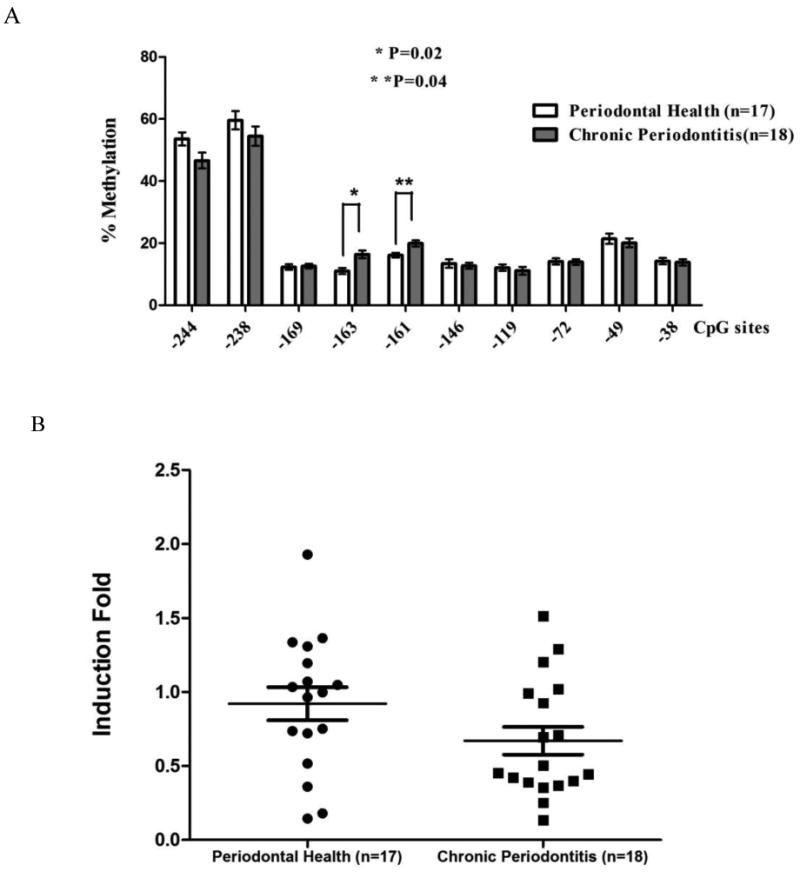
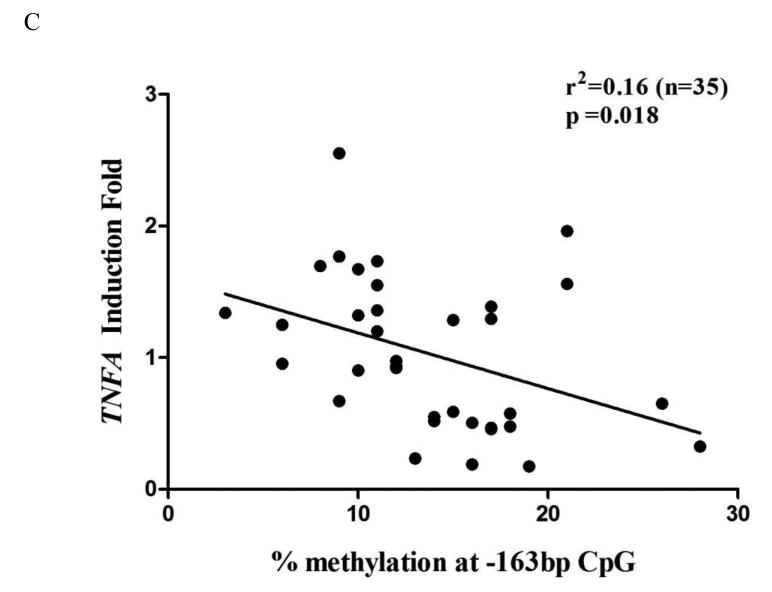
Methylation level of TNFA promoter region and transcriptional level of TNFA in clinical gingival biopsies collected from sites either with chronic periodontitis or periodontal health. A) The percentage methylation of each individual CpG dinucleotide from chronic periodontitis tissues is compared with gingival tissues with periodontal health. “*” indicates significantly higher methylation level at site -163bp in chronic periodontitis samples (p=0.02); “**” indicates significantly higher methylation level at -161bp in chronic periodontitis samples (p=0.04). B) TNFA transcriptional expression from periodontitis biopsies is compared with gingival biopsies with periodontal health. The transcription of TNFA is not significantly different from the samples with periodontal health (p=0.08). C) The messenger level of TNFA of individual sample from both biopsy groups is plotted against its methylation level at site -163bp. Regression analysis indicates that the transcriptional level of TNFA is significantly and inversely related to the methylation level at -163bp (r=0.16, p=0.018).
Promoter methylation level and transcription of TNFA in experimentally induced gingivitis samples
No significant difference was found at any of the CpG sites among those gingival biopsies collected in different phases of experimentally-induced gingivitis from the same participants by a Hotelling one-sample T2 test (p=0.49, Fig. 3A) . It seemed that there was a trend for the transcription level of TNFA to increase in the induction phase of gingivitis, as compared to the baseline level, and its transcription decreased in the resolution phase. However, those changes were not significantly different (p=0.51, Fig. 3B).
Figure 3.
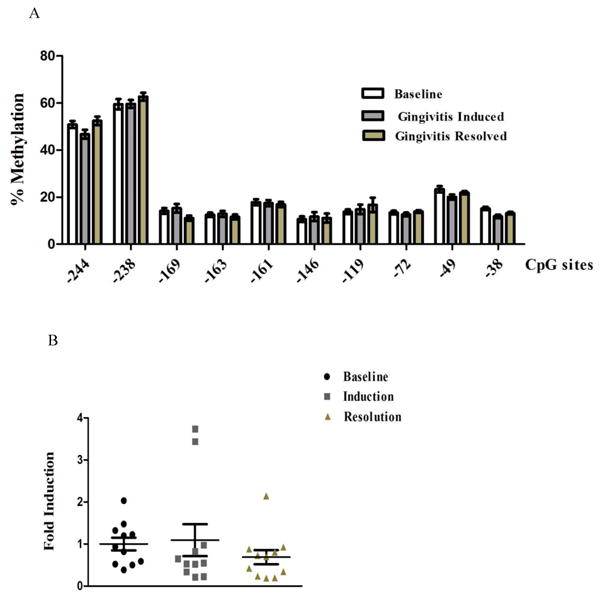
Methylation level of TNFA promoter and transcriptional level of TNFA in biopsies collected from different stages of experimentally induced gingivitis. A) The percentage methylation of each individual CpG dinucleotide from experimentally induced gingivitis is compared with gingival tissues with gingivitis resolution. No overall statistical difference is detected among the samples collected from different stages of gingivitis (p=0.49). B) No significant change of messenger level of TNFA among the samples collected from baseline, induction, and resolution phase of experimentally-induced gingivitis (p=0.53).
Effects of DNA methylation inhibitor (5azacytidine) on TNFA transcriptional expression in monocytic cells
To understand whether DNA methylation modulates TNFA transcriptional expression we explored the effect of a DNA methylation inhibitor on TNFA gene expression using a monocytic cell line, THP.1. A methylation inhibitor, 5-azacytidine, which blocks global DNA methylation during replication, was used to pretreat cells prior to stimulation and maintained during bacterial challenge. Treatment with 5-azacytidine increased TNFA expression in a time dependent manner as compared to vehicle control (mock) (Fig. 4A). After 24 hours of treatment, messenger level of TNFA was induced 1.6-fold as compared to mock treated cells (p=0.03), while the induction level was increased to 3-fold after 96 hours treatment (p=0.003, Fig. 4A) in unstimulated cells. Pretreatment also increased the TNFA transcription upon stimulation by live C.rectus314 as compared to mock challenged cells (p=0.006, Fig. 4B). Pyrosequencing analysis of the promoter region following 5-azacytidine treatment confirmed a loss of methylation across the promoter region of interest (data not shown).
Figure 4.
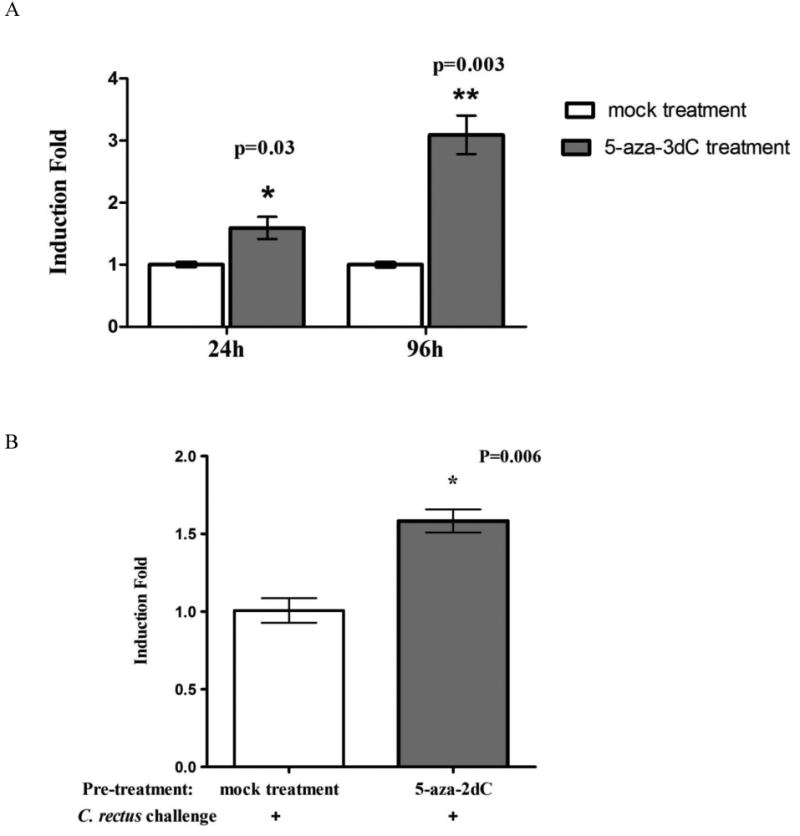
Transcription of TNFA in THP-1 cells treated with 5-aza-2dC. A) Cells treated with 5-aza-2dC exhibited an increase of TNFA transcription in a time-dependent manner in comparison to mock treated cells (“*” indicates p=0.03 and “**” indicates p=0.003). B) THP-1 cells pretreated with 5-aza-2dC are more responsive to C. rectus challenge by increasing TNFA transcription (p=0.006, as indicated by “*”).
The direct effects of TNFA promoter methylation on promoter activation
To further investigate the direct effect of the TNFA promoter methylation on its transcription, we measured the luciferase activity in RAW297.4 cells transfected with the reporter construct pGL3-PTNFA291 containing all the CpG sites upstream of TNFA that were analyzed. The activity of this luciferase reporter increased drastically upon LPS induction (Fig. 5A). We then generated hypermethylated TNFA promoter (pGL3-PTNFA291Met) and mock methylated constructs (pGL3-PTNFA291Mo) prior to transfection by an in vitro enzymatic methylation technique. The methylation efficiency of the modified TNFA promoter sequence was confirmed by electrophoresis analysis by digesting modified TNFA promoter fragment with a methylation sensitive restrictive endonuclease, BstUI. The resistance to BstUI digestion as reflected by the integral band in agarose gel indicated successful modification (Fig. 5B). After transfection of the reporter constructs with either pGL3-PTNFA291Mo or pGL3-PTNFA291Met and induction by LPS, we observed that the promoter activity of pGL3-PTNFA291Met showed a 2.6 fold reduction in expression as compared to the mock modified construct (p=0.03, Fig. 5C).
Figure 5.
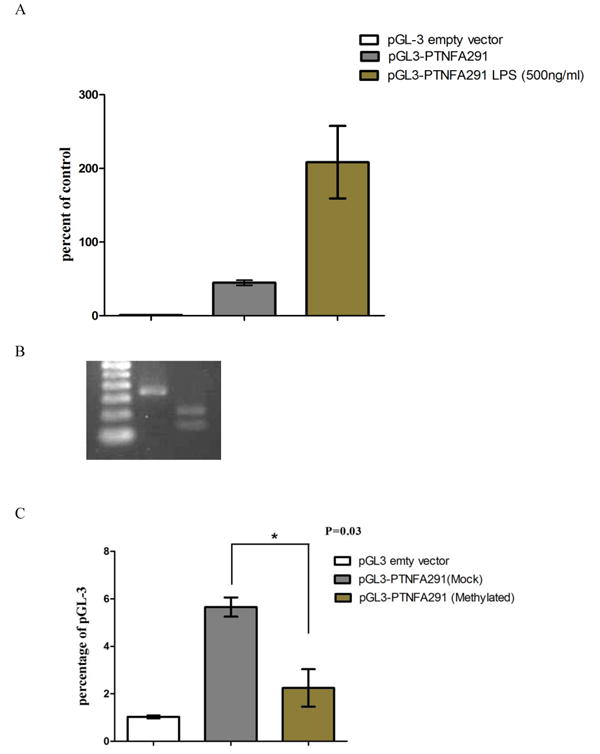
Activity of luciferase reporter construct containing either unmodified or modified TNFA promoter fragment ranging from -291bp to +44bp relative to TSS. A) Cells transfected with pGL-3PTNFA291 construct showed increased luciferase activity upon induction by LPS as compared to uninduced construct. Lucfirase activity is relative to the empty vector. B) Cells transfected with in vitro methylated pGL-3PTNFA291Met showed a significantly decreased luciferase activity as compared to cells transfected with a mock methylated pGL-3PTNFA291Mo upon LPS induction (*p=0.03).
Discussion
As the most stable epigenetic modification, DNA methylation plays an important role in the regulation of various inflammatory cytokines in chronic inflammatory conditions like rheumatoid arthritis, lupus, atherosclerosis, diabetes and periodontitis. 18, 20-26 In this report we demonstrated that the methylation status of TNFA promoter was not modified at the peak of gingivitis induction or during resolution, but is hypermethylated in chronic periodontitis. To gain a mechanistic insight into how the methylation status of the TNFA promoter region affects the transcription of this gene, we first treated THP.1 cells with a DNA methylation inhibitor. Treating resting cells with 5aza-2dC induced a time dependent increase in TNFA mRNA synthesis, which is related to the loss of methylation in CpG sites upstream of TNFA TSS. This may indicate that the alteration of DNA methylation is one of the regulatory mechanisms that govern TNFA transcription. However, a key limitation in this experimental approach was that 5aza-2dC induces global demethylation that has an effect across the entire genome. Thus, the continuous treatment with this DNA methylation inhibitor may not be specifically linked to TNFA promoter transcriptional activation, as it may be a secondary consequence of global demethylation. To address this caveat, we constructed a luciferase reporter containing the sequence ranging from -291∼+34bp of TNFA that includes the CpG sites just upstream of TNFA TSS. This region of the promoter was selected, in part, because it contained CpG sites that were discovered to be differentially methylated in human periodontitis tissues. The luciferase reporter assay demonstrated that the cloned sequence displays promoter activity, as inducible by LPS stimulation. By comparing the luciferase activities from the methylation-modified reporter constructs with mock methylation-modified promoter region of TNFA in transfected cells, we proved that TNFA transcription is directly dependent upon the methylation status of the TNFA promoter region.
This information provides insight into the observed promoter methylation changes seen among clinical gingival biopsies. Epigenetic modification within this region would be expected to affect the transcription of TNFA. In chronically inflamed gingival biopsies the methylation pattern of TNFA is significantly different from samples of gingival health with higher methylation level present at both -163 and -161bp. However, no significant change has been observed at different phases of experimentally induced gingivitis comparing baseline, induction and resolution. The hypermethylation pattern seen within the TNFA promoter in chronic periodontitis likely reflects stable epigenetic modification induced by the chronic exposure of periodontal tissue to a pathogenic environment present in the biofilm. We do not know whether changes in methylation are induced by bacteria alone or whether inflammatory responses are needed. The non-significant trend for inhibition of TNFα mRNA in chronic periodontitis samples, as compared to gingival health, is consistent with the hypermethylation of CpG sites in the TNFA promoter. These findings remind us of our previous PTGS2 epigenetic study that demonstrated a similar suppressed transcriptional and increased promoter methylation pattern using gingival biopsies from chronic periodontitis subjects.14 Interestingly, the regulation of PGE2 production by TNF-α has been elucidated in periodontal diseases.4, 27 Therefore, inflammatory molecules within the same regulatory network appear to be epigenetically regulated in a concerted manner. This epigenetic regulation may favor a dampening mechanism that protects the host from the unconstrained and prolonged environmental insults. Such an epigenetic mechanism could potentially serve to create an adaptive homeostasis to minimize the insult caused by bacteria in a manner that is analogous to the gastrointestinal tract. Takahashi et al. 28, 29 have reported, for example, that the gut microbiome induces epigenetic modification of the intestinal epithelium of the gut mucosa to dampen the inflammatory response.
The alteration of promoter methylation patterns at specific CpG sites has been also reported for many physiopathological conditions. For example, Camion et al 30 found that the methylation levels at -169bp and -119bp within TNFA promoter region were associated with successful weight loss in adult obese males. In the current study, we observed that the methylation level at site -163bp is correlated with the transcription of TNFA. This promoter region contains transcriptional binding elements for key transcriptional factors, such as NF-κB, Sp1, Ap1, Ap2, etc., suggesting that methylation alterations may modulate binding by these transcriptional factors. Additional experiments to induce hypermethylation in specific individual CpG sites within the promoter region will be required to resolve the unbalanced effect of methylation at “key” CpG sites on TNFA transcription. 31, 32
In this study, one major factor that limits our interpretation is the mixed cell types present within the in gingival biopsies. The heterogeneity of cell populations present in the gingival biopsies doesn't allow us to identify the level of methylation alteration within TNFA promoter in each individual cell type. Although we demonstrated a significant overall change in tissue methylation, this potentially could be attributable to a change in cell composition However, the fact that there no changes in TNFA methylation during the induction and resolution of gingivitis- a process that leads to dramatic changes in cellular composition of the tissues, suggest perhaps that the observed changes in methylation status in chronic periodontitis relate to the methylation status of the resident cells, which comprise the chronic lesion. Laser microdissection or culture of primary cells isolated from gingival tissue may provide a useful tool in fine-tuning the analysis of cell type-specific methylation changes in periodontal inflammation.
In conclusion, DNA methylation may be an important regulatory mechanism in controlling TNFA transcriptional expression in chronic periodontitis, a non-reversible form of periodontal disease. The methylation profile present in the promoter region of TNFA is mechanistically related to its transcription level.
References
- 1.Offenbacher S, Barros SP, Beck JD. Rethinking periodontal inflammation. J Periodontol. 2008;79(8 Suppl):1577–1584. doi: 10.1902/jop.2008.080220. [DOI] [PubMed] [Google Scholar]
- 2.Graves D. Cytokines that promote periodontal tissue destruction. J Periodontol. 2008;79(8 Suppl):1585–1591. doi: 10.1902/jop.2008.080183. [DOI] [PubMed] [Google Scholar]
- 3.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89(12):1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 4.Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74(3):391–401. doi: 10.1902/jop.2003.74.3.391. [DOI] [PubMed] [Google Scholar]
- 5.Duarte PM, da Rocha M, Sampaio E, et al. Serum levels of cytokines in subjects with generalized chronic and aggressive periodontitis before and after non-surgical periodontal therapy: a pilot study. J Periodontol. 2010;81(7):1056–1063. doi: 10.1902/jop.2010.090732. [DOI] [PubMed] [Google Scholar]
- 6.Salvi GE, Brown CE, Fujihashi K, et al. Inflammatory mediators of the terminal dentition in adult and early onset periodontitis. J Periodontal Res. 1998;33(4):212–225. doi: 10.1111/j.1600-0765.1998.tb02193.x. [DOI] [PubMed] [Google Scholar]
- 7.Offenbacher S, Barros SP, Singer RE, Moss K, Williams RC, Beck JD. Periodontal disease at the biofilm-gingival interface. J Periodontol. 2007;78(10):1911–1925. doi: 10.1902/jop.2007.060465. [DOI] [PubMed] [Google Scholar]
- 8.Offenbacher S, Barros S, Mendoza L, et al. Changes in gingival crevicular fluid inflammatory mediator levels during the induction and resolution of experimental gingivitis in humans. J Clin Periodontol. 2010;37(4):324–333. doi: 10.1111/j.1600-051X.2010.01543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garlet GP, Cardoso CR, Campanelli AP, Martins W, Jr, Silva JS. Expression of suppressors of cytokine signaling in diseased periodontal tissues: a stop signal for disease progression? J Periodontal Res. 2006;41(6):580–584. doi: 10.1111/j.1600-0765.2006.00908.x. [DOI] [PubMed] [Google Scholar]
- 10.Rossomando EF, Kennedy JE, Hadjimichael J. Tumour necrosis factor alpha in gingival crevicular fluid as a possible indicator of periodontal disease in humans. Arch Oral Biol. 1990;35(6):431–434. doi: 10.1016/0003-9969(90)90205-o. [DOI] [PubMed] [Google Scholar]
- 11.Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99(5):451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 12.Shuto T, Furuta T, Oba M, et al. Promoter hypomethylation of Toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J. 2006;20(6):782–784. doi: 10.1096/fj.05-4934fje. [DOI] [PubMed] [Google Scholar]
- 13.Kelavkar UP, Harya NS, Hutzley J, et al. DNA methylation paradigm shift: 15-lipoxygenase-1 upregulation in prostatic intraepithelial neoplasia and prostate cancer by atypical promoter hypermethylation. Prostaglandins Other Lipid Mediat. 2007;82(1-4):185–197. doi: 10.1016/j.prostaglandins.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Barros SP, Niculescu MD, Moretti AJ, Preisser JS, Offenbacher S. Alteration of PTGS2 promoter methylation in chronic periodontitis. J Dent Res. 2010;89(2):133–137. doi: 10.1177/0022034509356512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobetsis YA, Barros SP, Lin DM, et al. Bacterial infection promotes DNA hypermethylation. J Dent Res. 2007;86(2):169–174. doi: 10.1177/154405910708600212. [DOI] [PubMed] [Google Scholar]
- 16.Loe H, Theilade E, Jensen SB. Experimental Gingivitis in Man. J Periodontol. 1965;36:177–187. doi: 10.1902/jop.1965.36.3.177. [DOI] [PubMed] [Google Scholar]
- 17.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29(13):E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S, Crivello A, Offenbacher S, Moretti A, Paquette DW, Barros SP. Interferon-gamma promoter hypomethylation and increased expression in chronic periodontitis. J Clin Periodontol. 2010;37(11):953–961. doi: 10.1111/j.1600-051X.2010.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dell G, Charalambous M, Ward A. In vitro methylation of specific regions in recombinant DNA constructs by excision and religation. Methods Mol Biol. 2001;181:251–258. doi: 10.1385/1-59259-211-2:251. [DOI] [PubMed] [Google Scholar]
- 20.Ishida K, Kobayashi T, Ito S, et al. Interleukin-6 Gene Promoter Methylation in Rheumatoid Arthritis and Chronic Periodontitis. J Periodontol. 2011 doi: 10.1902/jop.2011.110356. [DOI] [PubMed] [Google Scholar]
- 21.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58(12):2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation. J Cell Mol Med. 2010;14(6A):1225–1240. doi: 10.1111/j.1582-4934.2010.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu ZH, Chen LL, Deng XL, et al. Methylation Status of CpG Sites in the MCP-1 Promoter is Correlated to Serum MCP-1 in type 2 Diabetes. J Endocrinol Invest. 2011 doi: 10.3275/7981. [DOI] [PubMed] [Google Scholar]
- 24.Sekigawa I, Okada M, Ogasawara H, Kaneko H, Hishikawa T, Hashimoto H. DNA methylation in systemic lupus erythematosus. Lupus. 2003;12(2):79–85. doi: 10.1191/0961203303lu321oa. [DOI] [PubMed] [Google Scholar]
- 25.Viana MB, Cardoso FP, Diniz MG, et al. Methylation pattern of IFN-gamma and IL-10 genes in periodontal tissues. Immunobiology. 2011;216(8):936–941. doi: 10.1016/j.imbio.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 26.De Oliveira NF, Andia DC, Planello AC, et al. TLR2 and TLR4 gene promoter methylation status during chronic periodontitis. J Clin Periodontol. 2011;38(11):975–983. doi: 10.1111/j.1600-051X.2011.01765.x. [DOI] [PubMed] [Google Scholar]
- 27.Taylor JJ. Cytokine regulation of immune responses to porphyromonas gingivalis. Periodontol. 2010;200054:160–194. doi: 10.1111/j.1600-0757.2009.00344.x. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi K, Sugi Y, Hosono A, Kaminogawa S. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J Immunol. 2009;183(10):6522–6529. doi: 10.4049/jimmunol.0901271. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi K, Sugi Y, Nakano K, et al. Epigenetic control of the host gene by commensal bacteria in large intestinal epithelial cells. J Biol Chem. 2011;286(41):35755–35762. doi: 10.1074/jbc.M111.271007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campion J, Milagro FI, Goyenechea E, Martinez JA. TNF-alpha promoter methylation as a predictive biomarker for weight-loss response. Obesity (Silver Spring) 2009;17(6):1293–1297. doi: 10.1038/oby.2008.679. [DOI] [PubMed] [Google Scholar]
- 31.Murayama A, Sakura K, Nakama M, et al. A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. EMBO J. 2006;25(5):1081–1092. doi: 10.1038/sj.emboj.7601012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305(5688):1289–1292. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]