

Abstract
Neural stem cells (NSCs) were recently revealed to exist in the hypothalamus of adult mice. Here, following our observation showing that a partial loss of hypothalamic NSCs caused weight gain and glucose intolerance, we studied if NSCs-based cell therapy could be developed to control these disorders. While hypothalamus-implanted NSCs failed to survive in mice with obesity, NF-κB inhibition induced survival and neurogenesis of these cells, leading to effects in counteracting obesity and glucose intolerance. To generate an alternative cell source, we revealed that iPS-derived NSCs were converted into htNSCs by neuropeptide treatment. Of note, obesity condition potentiated the transfer of carotid artery-injected NSCs into the hypothalamus. These iPS-derived cells when engineered with NF-κB inhibition were also effective in reducing obesity and glucose intolerance, and neurogenesis towards POMCergic and GABAergic lineages was accountable. In conclusion, building NSCs in the hypothalamus represents a strategy for controlling obesity and glucose disorders.
Keywords: Neural stem cells, iPS, Hypothalamus, NF-κB, Neuropeptide, Obesity, Glucose tolerance
1. Introduction
Feeding, body weight and glucose balance are regulated by the mediobasal hypothalamus (MBH) in the central nervous system (CNS), and this regulation is mediated importantly by melanocortin signals from the arcuate nucleus (ARC), in particular anorexigenic proopiomelanocortin (POMC) neurons and orexigenic agouti-related peptide (AGRP) neurons [1–6]. These two neuronal types are sensitively and dynamically affected by systemic hormones such as leptin and insulin which fluctuate according to feeding and fat mass conditions, and this process is physiologically critical for maintaining body weight homeostasis [1–7]. However, when chronically challenged under high-fat diet (HFD) condition, these neurons reduce the responsiveness to leptin and insulin, and these changes contribute to the mechanism of HFD-induced obesity and type-2 diabetes (T2D) [1–7]. In addition to these signaling changes, research during recent years has begun to show that hypothalamic neural organization and neuronal numbers are altered under chronic HFD feeding [8–10]. For example, it was revealed that prolonged HFD feeding leads to a fractional loss (~10%) in POMC neurons in the hypothalamus [10–12]. Intriguingly, Pomc gene expression is known to be divergently regulated [13,14], and was shown to be present in neural precursors which give rise to various neuronal types [15]. In relation to this background, we have been interested in understanding if impaired hypothalamic neurogenesis might play a role in the development of HFD-induced obesity. In agreement with this idea, several evidences were recently documented to indicate that the hypothalamus of adult rodents has neurogenesis [16–21]. We showed that the MBH contain adult hypothalamic NSCs (htNSCs), and these cells appear to be involved in the hypothalamic control of metabolic physiology [11]. Herein, we extended to study if htNSCs could be developed to treat obesity and related glucose disorders, and if so, what technological approaches could be introduced to promote the practical employment of this technique.
2. Results
2.1. Partial loss of htNSCs leads to weight gain and glucose intolerance
We recently showed that dietary obesity is associated with impaired survival and neurogenesis of htNSCs [11]. In this context, we studied if htNSCs could be developed to treat or prevent against obesity and related metabolic disorders. We first tested if a loss of endogenous htNSCs in mice could be sufficient to lead to these metabolic disorders. To do so, we generated a mouse model with adult-onset ablation of dividing Sox2-positive htNSCs in the MBH. Briefly, adult C57BL/6 mice received MBH injection of lentiviruses expressing Sox2 promoter-driven Herpes simplex virus type-1 thymidine kinase (Hsv1-TK), a kinase that works to convert a nontoxic nucleoside analog, ganciclovir (GCV), into the toxic product which acts as a chain terminator during DNA replication and therefore kills dividing Sox2-positive cells [22–25]. Injection of matched control lentiviruses was performed in control mice for comparison. All these mice were then chronically treated with GCV to induce Hsv1-TK/GCV-mediated ablation of Sox2-positive htNSCs, and as assessed in Figure S1A and B, 3-month GCV treatment resulted in a partial loss of htNSCs in the ARC. Using this model, we studied if such a loss of htNSCs could affect the population of POMC cells in the hypothalamus, since POMC neurons were recently shown to undergo constant although slow-speed turnover [10,11]. Data revealed that there was ~10% reduction in POMC cells over a 4-month follow-up period in our model (Figure S1C). Through physiological profiling, we observed that these mice displayed a collection of metabolic disorders, including glucose intolerance, hyperinsulinemia, increased food intake and moderate weight gain (Figure S2). Thus, these data indicate that adult htNSCs play a significant role in body weight and glucose control, supporting us to study if htNSCs therapy could be developed to control related diseases.
2.2. htNSCs fail to survive under HFD-altered hypothalamic microenvironment
Subsequently, we investigated if exogenous htNSCs could be implanted into the MBH of mice to control HFD-induced obesity and related glucose disorders. To do so, we employed a line of htNSCs which we recently established [11]; in order to track htNSCs-induced neurogenesis, GFP was introduced into this cell line via lentiviral infection (Figure 1A and B), similarly as we performed previously [11], and we also verified the stem cell identify of these cells using immunostaining for a NSC marker (Figure 1B). For hypothalamic implantation, we targeted the MBH for cell injection shown in Figure 1C, given that the MBH is the hypothalamic region that is enriched with htNSCs. However, when these cells were injected in mice which underwent the development of HFD-induced obesity, this trial was unsuccessful, because implanted htNSCs survived poorly in the hypothalamic microenvironment of these HFD-fed mice. As shown in Figure 1D and E, the survival rate of implanted htNSCs decreased at Day 7, and then became undetectable at Days 15–30 post-injection. In contrast to HFD-fed mice, much better survival rate of these cells was seen when we implanted them into the MBH of chow-fed normal mice (Figure 1E). This data further supported that the hypothalamic microenvironment is altered under the condition of chronic HFD feeding to a level which was hostile for the survival of htNSCs. Thus, it is necessary to engineer htNSCs to increase the hypothalamic survival of these cells, in order to evaluate the potential of htNSCs-based cell therapy in treating overnutrition-related diseases such as obesity and T2D.
Figure 1.
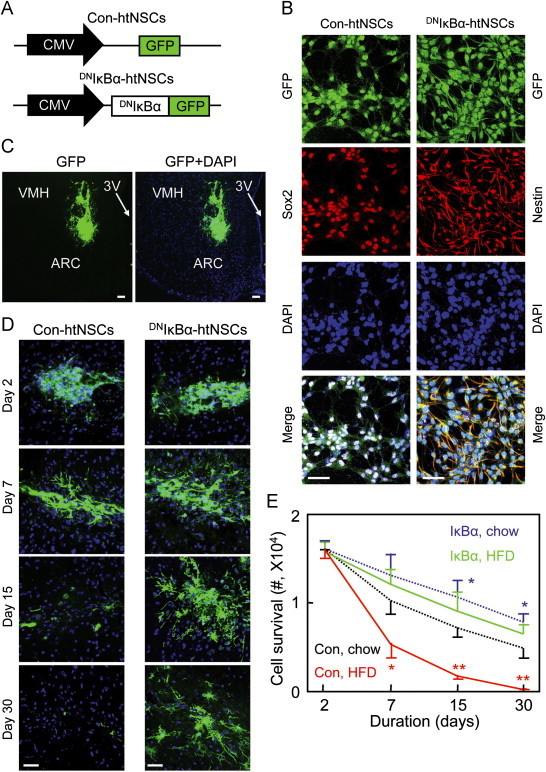
Survival of hypothalamic implanted htNSCs engineered with NF-κB inhibition. Cell lines of Con-htNSCs (Con) and DNIκBα-htNSCs (IκBα) were generated through lentiviral induction (A), examined for stemness and morphology via GFP and immunostaining of a NSC marker (B), and injected bilaterally in the MBH of HFD-fed vs. chow-fed male C57BL/6 mice (C–E). Images show technical success of delivering cells in the MBH at Day 3 post-injection (C), and longitudinal follow-up of indicated htNSCs in the MBH of HFD-fed mice (D). DAPI staining reveals nuclei of all cells in the sections. Scale bar=50 µm. Curves show longitudinal survival of htNSCs in HFD- vs. chow-fed mice (E). *P<0.05, **P<0.01 (compared to matched time points in the group of Con, chow), n=4–6 mice per group. Error bars reflect means±s.e.m.
2.3. NF-κB inhibition promotes survival and neurogenesis of implanted htNSCs
According to the literature, chronic HFD feeding induces hypothalamic inflammation [26,27], and the proinflammatory pathway consisting of nuclear factor-κB (NF-κB) and its upstream IκB kinase-β (IKKβ) is a key mediator that links hypothalamic inflammation to disease outcomes [28–32]. Because obesity-associated hypothalamic IKKβ/NF-κB activation was known to disrupt htNSCs [11], we asked if NF-κB inhibition could help in vivo survival of implanted htNSCs. To test this idea, we used a line of htNSCs that was engineered with NF-κB inhibition, generated via lentiviral induction of dominant-negative IκBα (DNIκBα-htNSCs) (Figure 1A and B), and htNSCs containing lentiviral GFP (Con-htNSCs) were used as matched controls. We noted that compared to control cells, DNIκBα-htNSCs showed more neural branches, while their stemness remained as verified with immunostaining (Figure 1B). We implanted these cells into the MBH of mice under HFD feeding condition. Strikingly, survival rate of implanted DNIκBα-htNSCs increased dramatically, as we observed that ~40% of these cells survived at Day 30, at which we did not detect Con-htNSCs. In addition to survival success, implanted DNIκBα-htNSCs migrated and generated projections, implicating that these cells underwent neurogenesis and interactions with host hypothalamic cells. Then, we directly examined if NF-κB inhibition could promote neural differentiation of implanted htNSCs. Data revealed that while some implanted cells maintained stem cell identity (Figure 2A), 16% of these cells became to express NeuN (Figure 2B) at ~1 month post-implantation. Immunostaining data further revealed that a number of DNIκBα-htNSCs differentiated into cells which expressed POMC in chow-fed mice, and this effect was preserved under HFD feeding (Figure 2C and D). Thus, while chronic HFD feeding led to a loss in hypothalamic POMC cells, just as consistently shown in recent literature [10–12], this loss was amended by DNIκBα-htNSCs-induced neurogenesis (Figure 2D). In addition, we found that leptin signaling was induced in some of DNIκBα-htNSCs-derived neural cells (Figure S3A), resulting in a restoration of leptin-induced feeding control in HFD-fed mice (Figure S3B). Altogether, NF-κB inhibition represents a solution for htNSCs to survive from HFD feeding-altered hypothalamic microenvironment.
Figure 2.

Neurogenesis and metabolic effects of implanting NF-κB-inhibited htNSCs. Dissociated DNIκBα-htNSCs (IκBα) vs. Con-htNSCs (Con) were injected in the MBH of chow- vs. HFD-fed C57BL/6 mice, examined for stemness and neurogenesis of htNSCs at ~1 month post-injection via immunostaining of Sox2 (A–C), NeuN (B) and α-MSH (C), and effects of injected htNSCs on metabolic physiology (E–G). Histological data in A–D show MBH sections from HFD-fed mice injected with DNIκBα-htNSCs (A–C), and counting of endogenous (white bars) vs. exogenous htNSCs-derived (black, blue, red and green bars) POMC cells (D). Bar=50 µm. Physiological data show body weight (E), food intake (F) and GTT (G) obtained at indicated days (E) or 2–3 months post-implantation (F, G). *P<0.05, **P<0.01, ***P<0.001, compared between indicated exogenous cells (D) or compared to HFD-fed controls at matched time points (E, G), n=4 (D) and 5–6 (E–G) per group. Error bars reflect means±s.e.m.
2.4. Implanting NF-κB-inhibited htNSCs counteracts obesity and glucose disorder
While physiological implications of htNSCs implantation could be multiple, here we studied if it could exert an effect in counteracting HFD-induced metabolic disorders. For comparison, we included normal chow-fed mice as a dietary control in the study, and found that hypothalamic implantation of DNIκBα-htNSCs or Con-htNSCs did not affect the normal metabolic profiles of these mice, including body weight (Figure 2E), food intake (Figure 2F) and blood insulin levels (Figure S6C). Importantly, implantation of DNIκBα-htNSCs exerted significant effects in reducing body weight (Figure 2E), food intake (Figure 2F), glucose intolerance (Figure 2G) and blood insulin levels (Figure S4C) in HFD-fed mice. Body weight reduction in mice by DNIκBα-htNSCs was attributed specifically to fat loss (Figure S4A and B), further supporting the conclusion that the treatment provided an anti-obesity effect. In contrast, these metabolic effects were not produced by implanting Con-htNSCs (Figures 2E–G and S4C), which is consistent with the fact that these cells did not survive in HFD-fed mice. We also included vehicle injection in HFD-fed mice to provide a basal control, and data showed that the metabolic effects of injecting vehicle and Con-htNSCs were both negligible (Figure S5), again showing that NF-κB inhibition is necessary for the observed effects of htNSCs-based therapy. Overall, these effects of DNIκBα-htNSCs aligned with the data showing that implanted DNIκBα-htNSCs amended the loss of POMC cells in the hypothalamus in HFD-fed mice (Figure 2D). And these effects were also in agreement with the observation demonstrating that some of DNIκBα-htNSCs-derived cells responded to leptin (Figure S3). Based on these data, we concluded that introduction of htNSCs can work as a strategy for controlling obesity and related glucose disorders, and NF-κB inhibition is critical for success of this strategy.
2.5. Development of htNSCs phenotype by using iPS-derived NSCs
One of important goals in this research was to develop a strategy of htNSCs-based cell therapy that can be applicable for human diseases, and since it is unrealistic to collect endogenous human htNSCs, we explored if we could employ inducible pluripotent stem cells (iPS), a model of re-programmed stem cells [33]. Using a protocol established in the literature [34], we successfully induced NSCs from a line of mouse iPS, referred to as NSCsiPS (Figure 3A), as verified by our data showing that NSCsiPS expressed NSC markers Sox2 and nestin (Figure 3B). Also, just like native htNSCs, NSCsiPS differentiated into three neural lineages, including Tuj-1-expressing neurons, GFAP-expressing astrocytes and O4-expressing oligodendrocytes (Figure 3C). Interestingly, we found that implanted NSCsiPS in the MBH not only underwent neurogenesis (Figure S6A) but led to generation of POMC cells (Figure S6B). We speculated that hypothalamic microenvironment might provide necessary molecular stimuli to program NSCsiPS into htNSCs. To test this idea, we studied if treatment with hypothalamic neuropeptides could induce the differentiation of NSCsiPS into htNSCs. As established in the literature [35], ventral hypothalamic neurons are derived from embryonic NSCs, a process that is directed by hypothalamic programming involving gene upregulation of Nkx2.1, Six3 and Vax1, and downregulation of Pax6 in NSCs. Indeed, we observed that native htNSCs were characterized by increased levels in Nkx2.1, Six3 and Vax1, but decreased levels in Pax6 and Rax (Figure 3D). Intriguingly, this pattern of gene expression in htNSCs was reproduced in NSCsiPS through treatment with neuropeptide Y (NPY), but not other hypothalamic neuropeptides like α-MSH or cocaine/amphetamine-regulated transcript (CART). We also examined brain-derived neurotrphic factor (BDNF), a neuropeptide that can promote brain neurogenesis [36], and found that conversion of NSCsiPS into htNSCs was further promoted by NPY and BDNF co-treatment (Figure 3D). Using immunostaining, we confirmed that while NPY treatment increased differentiation of NSCsiPS into Six3-expressing and Vax1-expressing cells, these effects increased when BDNF was added to the treatment (Figure 3E). Finally, since a fundamental role of hypothalamic neurogenesis is to generate cells that produce hypothalamic neuropeptides, we examined if this potential could be induced in the model NPY/BDNF-treated NSCsiPS. Data showed that, compared to native htNSCs, which can indeed be differentiated to produce Agrp, Pomc and Cart mRNAs, these changes were similarly produced in NPY/BDNF-treated NSCsiPS (Figure 3F). Altogether, these data suggest that hypothalamic neuropeptides have important actions in programming naïve NSCs towards htNSCs.
Figure 3.

NSCs derived from iPS and conversion into htNSCs phenotype. Model of iPS-derived NSCs (NSCsiPS) was generated from iPS-derived embryoid bodies (A), verified via neurosphere staining of Sox2 and nestin (B), and assessed for differentiation via Tuj-1, GFAP and O4 immunostaining (C) and programming under neuropeptide treatment (D–F). D–F: NSCsiPS treated with neuropeptides including α-MSH, CART, NPY, or NPY and BDNF (NPY+B) were examined for induction of htNSCs-specific markers via quantitative PCR (D) and immunostaining (E), or subjected to differentiation followed by measurement of hypothalamic neuropeptide gene expression (F). Vehicle treatment was used to provide baseline levels profiles of various parameters. Native htNSCs under basal condition were included to provide positive controls. E: Bar=50 µm, and Six3- and Vax1-positive cells were counted in lower panels. *P<0.05, **P<0.01, ***P<10−3, n=4 per group. Error bars reflect means±s.e.m.
2.6. Obesity potentiates MBH penetration of carotid artery-injected NSCs
Following the in vitro studies in Figure 3, we tested if iPS-derived NSCs could be used to induce hypothalamic neurogenesis; however, just like htNSCs, NSCsiPS also showed poor survival when injected in the MBH of mice with HFD-induced obesity. We then examined if this defect could be prevented when NF-κB inhibition was induced in these cells, and to do so, we generated a line of NSCsiPS with lentiviral induction of DNIκBα (GFP-conjugated), referred to as DNIκBα-NSCsiPS, and the control cell line was generated through lentiviral induction of GFP, termed Con-NSCsiPS. Both of these cell lines were confirmed for stemness through immunostaining (Figure 4A), and we also noted that DNIκBα-NSCsiPS displayed more neural branches than control cells. Using this model, we studied if DNIκBα-NSCsiPS could be delivered to treat obesity in HFD-fed mice, and to increase the application value, we studied if carotid artery injection could be used instead, since this method has been adopted in research to deliver NSCs into the brain of rodents when the vascular system of the brain was altered [37,38]. It is worth mentioning that the blood–brain barrier (BBB) in the MBH of mice is incomplete in normal condition, and becomes more leaky under conditions of obesity or diabetes [12,39], making it possible for the artery-injected cells to penetrate into the MBH. To take this feature into account, we first induced obesity in mice via ~4-month HFD feeding, and then injected DNIκBα-NSCsiPS into these mice via carotid artery. Excitingly, GFP-labeled cells were significantly found in the MBH of these HFD-fed mice (Figure 4B). In this context, we further confirmed that DNIκBα-NSCsiPS underwent neurogenesis (Figure 4C) and led to new generation of POMC cells in the MBH (Figure 4D and E). In contrast, when carotid artery injection was performed in chow-fed mice, penetration of GFP-labeled cells into the MBH was not obvious, further implicating that the pre-existing condition of obesity can potentiate the mediobasal hypothalamic entry of carotid artery-injected NSCs.
Figure 4.

Artery delivery of NF-κB-inhibited NSCiPS and in vivo neurogenesis. Lines of DNIκBα-NSCiPS and Con-NSCiPS were verified via GFP and immunostaining of a NSC marker (A), and delivered into HFD-fed mice via carotid artery injection (B–E). Brain sections were prepared to analyze the hypothalamic transfer of artery-injected DNIκBα-NSCiPS at ~1 month post-injection (B) and hypothalamic neurogenesis at ~3 months post-injection via immunostaining of NeuN (C) and POMC (D, E). Bar graphs show counting of endogenous cells (white bars) vs. exogenous NSCiPS-derived cells (black, blue, red and green bars) that expressed POMC (E). *P<0.05, **P<0.01, ***P<0.001, (comparisons between the columns of exogenous cells), n=4 mice per group. Error bars reflect means±s.e.m. Scale bar=50 µm (A, B) and 25 µm (C).
2.7. Therapeutic effects of carotid artery-delivered NSCs
As described in Figure 4, HFD-fed mice were injected with DNIκBα-NSCsiPS via carotid artery, and for the control, we injected Con-NSCsiPS in matched HFD-fed mice. In the control group, Con-NSCsiPS failed to survive, and thus as expectedly, we did not see an anti-obesity effect. However, delivery of DNIκBα-NSCsiPS led to ~30% decrease in HFD intake (Figure 5A), and this feeding control was associated with a moderate effect in weight reduction which lasted although only a short period (Figure 5B). When mice continued to be fed on the HFD for long period, the anti-obesity therapeutic effect of DNIκBα-NSCsiPS declined and diminished; however, the extent of weight gain in these mice under HFD feeding was much less substantial, compared to the levels of weight gain in control mice (Figure 5B). We also examined if hypothalamic leptin resistance, a key event in HFD-induced obesity and T2D, could be reduced by this cell therapy in these mice. Data showed that while control mice showed impaired action of leptin in suppressing food intake over 24-h experimental period, the effect of leptin in controlling feeding was significantly restored in mice treated with DNIκBα-NSCsiPS (Figure 5C). Furthermore, consistent with the effects on feeding and weight control, treatment with DNIκBα-NSCsiPS led to improved glucose tolerance in HFD-fed mice (Figure 5D). In summary, NSCsiPS can be developed as a practical source of cells for controlling obesity and glucose disorders, and artery injection as a delivery approach.
Figure 5.
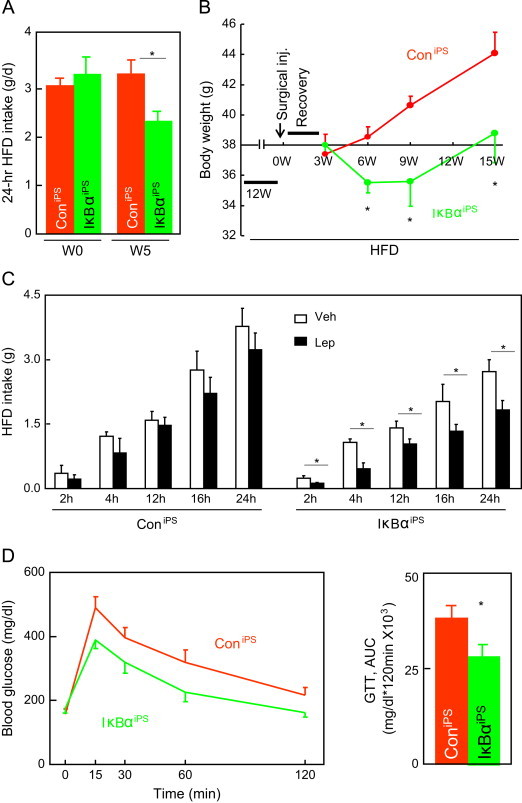
Metabolic effects from artery injection of NF-κB-inhibited NSCiPS. Male C57BL/6 mice were induced to develop dietary obesity through 3-month HFD feeding, and then received intra-carotid artery injection of 2×105 DNIκBα-NSCiPS (IκBαiPS, IκBα) or Con-NSCiPS (ConiPS, Con). Mice continued to be maintained on the HFD for the entire experiment. Data show food intake (A), body weight (B), and leptin-induced feeding inhibition in response to an i.c.v. injection of 5 µg leptin (Lep, L) vs. vehicle (Veh, V) following 6-h fasting (C), and blood glucose levels during GTT (D). Data were obtained at the indicated weeks (W) (A, B) or 15 weeks (C) post-surgical recovery. *P<0.05, **P<0.01, n=5 mice per group (A, B and D), and n=4 mice per group (C). Error bars reflect means±s.e.m.
2.8. Metabolic effects of NSCs mechanistically via neurogenesis
Supported by the potential application value of NSCsiPS, we further studied how these cells could work to generate metabolic effects in mice. In addition to the actions of these cells in inducing neuropeptides (Figure 3F), we investigated if neurotransmitter pathway(s) could be introduced by neurogenesis of these cells. Thus, for the experiment described in Figure 3F, we further analyzed molecules in GABAergic and glutamatergic pathways, including glutamic acid decarboxylase 67 (GAD67) and GAD65 (encoded by Gad1 and Gad2, respectively), vesicular GABA transporter (vGAT, encoded by Slc32a1), vesicular glutamate transporters 2 (vGLUT2, encoded by Slc17a6), and glial glutamate transporter 1 (GLT-1, encoded by Slc1a2). First, using differentiated htNSCs as positive control, we found a pattern of changes in GABAergic pathway, including increases in Gad1 and Slc32a1 mRNA levels but a decrease in Gad2 mRNA levels (Figure 6A). Molecules in glutamatergic pathway also changed, including Slc1a2 and Scl17a6 mRNA levels, but to a lesser degree compared to changes in Slc32a1 mRNA levels (Figure 6A). Interestingly, when treated with NPY and BDNF, differentiated NSCsiPS showed similar molecular changes in GABAergic and glutamatergic pathways (Figure 6A). In this background, we then asked if the metabolic effects of DNIκBα-NSCsiPS were attributed to a neuropeptide or a neurotransmitter or both. To address this question, we designed a “rescue” experiment that is illustrated in Figure 6B; in this study, we used lentiviral RNAi to knockdown various neuropeptides or neurotransmitter-regulatory molecules in DNIκBα-NSCsiPS, and then implanted these cells into the MBH of HFD-fed mice. Data revealed that the metabolic effects of DNIκBα-NSCsiPS in feeding, body weight and glucose tolerance were abrogated by ablation of either Pomc or Gad67 (Figure 6C–E). Therefore, NSCs-derived neural cells can lead to at least two different types of differentiated neural cells which express Pomc and Gad67, respectively, and these cell types can work together to co-mediate the therapeutic effects of NSCs (Figure 6F), a model which is in agreement with the understanding [40] that GABAergic neurons and POMC neurons may have independent actions in regulating body weight.
Figure 6.

Neurogenesis in mediating the metabolic effects of NF-κB-inhibited NSCiPS. (A) NSCiPS were treated with indicated neuropeptides, and subjected to differentiation followed by mRNA measurement of GABAergic and glutamatergic biomarkers. Native htNSCs under the basal condition were included to provide positive controls. (B–E) DNIκBα-NSCiPS (IκBαiPS) and Con-NSCiPS (ConiPS, C) received lentiviral induction of POMC shRNA (POMC sh), GAD67 shRNA (GAD sh) or control scramble shRNA (Con sh), injected in the MBH of HFD-fed mice as illustrated (B), and followed up for food intake (C), body weight (D), and GTT (E) at the indicated weeks (C, D) or 3 months (E) post-injection. *P<0.05, **P<0.01, n=4 (A) and 5–6 (B–E) per group. Error bars reflect means±s.e.m. (F) Proposed model for using NSCs therapy to control obesity and related diseases such as T2D.
3. Discussion
Adult NSCs are a small population of cells with slow dividing rate in the brain [41], and because of this feature, it remains largely unclear regarding the potential physiological role of these cells. More recent in vitro and in vivo studies suggested that the hypothalamus is a brain region that contains adult NSCs [10,11,17,18,42]. In the current work, using site-specific ablation of htNSCs in the mediobasal hypothalamus of adult mice, we found that these animals developed overeating, weight gain and glucose intolerance, a collection of disorders which can be similarly induced by chronic HFD feeding. As this defect of htNSCs was recently revealed as a neurodegenerative basis for obesity and pre-diabetes [10–12], htNSCs-based cell therapy may have a potential as a long-term solution for these metabolic diseases, which can fix the involved central dysregulation in a comprehensive manner which may not be offered by a drug chemical which often targets a specific signaling cascade. Indeed, recent research in neuroscience has begun to suggest that NSCs have therapeutic potential for treating neurological diseases [43–47]. In this background, here we studied if htNSCs-based therapy could be developed to control obesity and metabolic diseases. However, we found that hypothalamic microenvironment of obesity condition is hostile for implanted htNSCs, and this condition leads to poor survival and neurogenesis of these cells. To solve this problem, we discovered that engineering of NSCs with NF-κB inhibition renders implanted htNSCs to survive and undergo neurogenesis under obesity condition. This success is related to the fact that obesity is closely associated with hypothalamic inflammation [26,27], and NF-κB inhibition is strongly counter-inflammatory and thereby protects implanted htNSCs from being disrupted by hypothalamic inflammation [11]. According to our recent understandings, we also reasoned that NF-κB in neurogenesis engages a programmatic switch from neuronal genesis to gliogenesis, a process which can be similarly induced by Notch pathway [48], and agreeably, our recent work has demonstrated that Notch pathway is responsible for the role of NF-κB in NSCs-induced neurogenesis [11]. Interestingly, obesity-related hypothalamic inflammation was recently shown to be associated with gliosis [12], and thus, if NF-κB-inhibited htNSCs can be provided to mediate programmatic switch, this treatment may lead to suppression in gliosis and therefore improvement of hypothalamic functions from another biological perspective.
In this research, we also considered that it is unrealistic to obtain human htNSCs for an application. To address this limitation, we explored if iPS could be used as an alternative source for generating NSCs. Indeed, we successfully derived NSCs from iPS, and further found that conversion of iPS-derived NSCs into htNSCs was induced by treatment with NPY and BDNF. Moreover, survival and neurogenesis of iPS-derived NSCs were improved by NF-κB inhibition to induce protective effects against obesity and glucose intolerance. Hence, iPS-derived NSCs have a potential for being developed as a source of htNSCs to treat or control metabolic diseases. In addition to solving cell source issue, we further considered that the method of hypothalamic implantation is not practical for human application, and knowing that the BBB in the MBH becomes penetrable under obesity/diabetic conditions [12,39], we performed an initial study to test if NSCs could be delivered via carotid artery to exert an therapeutic effect, and the overall findings were supportive. Nevertheless, we recognized that the anti-obesity therapeutic effect which we obtained was rather short-term, indicating that it is necessary to optimize the protocol, perhaps even in combination with other approaches, in order to increase the therapeutic efficacy. In light of the potential mechanism for the metabolic effects of NSCs therapy, we provided data showing that neurogenesis of these cells towards POMCergic and GABAergic lineages was at least accountable. On the other hand, we also want to point out that many of these NSCs-derived cells may represent neuronal precursors rather than mature neurons, and it is possible that the observed metabolic effects of NSCs therapy were mostly attributed to the secretory factors rather than synaptic actions of their derived neural cells. In conclusion, despite many limitations which remain to be addressed, data which we presented can lend a rationale and biotechnological basis for future interest in developing NSCs to control obesity and related diseases.
4. Methods
4.1. Lentiviruses and htNSCs
The cDNAs for DNIκBα and control GFP have been described previously [11,26,30,49]. Lentiviral vectors containing POMC shRNA, GAD67 shRNA or scramble shRNA were obtained from Sigma. Lentiviruses were produced by co-transfecting viral vectors with the package plasmids into HEK 293 FT cells, as previously described [11,26,49]. Lines of htNSCs were generated according to our protocol described previously [11]. Briefly, the hypothalamus was dissected from mice and cut into small pieces (~1 mm3), and digested with 0.25% papain (Worthington) for 30 min at 30 °C and gently triturated approximately 10 times using fire-polished tips. The desired cell population was suspended in growth medium containing Neurobasal-A (Invitrogen), 2% B27 without vitamin A (Invitrogen), 10 ng/ml EGF (Sigma) and 10 ng/ml bFGF (Invitrogen), seeded in ultralow adhesion 6-well plates (Corning) at a density of 100,000 cells per well and incubated in 5% CO2 at 37 °C. On day 7, neurospheres were collected through low-speed centrifugation, dissociated into single cells by trypsinization using TrypLE express media, and processed for passaging or cryopreservation. Lentivirus-induced lines of htNSCs were prepared by subjecting htNSCs in growth medium with lentiviruses for 3 days, followed by selection process through adding nucleoside antibiotic blasticidin (1 μg/ml) to culture medium. Cells transduced with lentiviruses were maintained in blasticidin-containing selection growth medium over several passages until gene knockdown was stably established.
4.2. Pluripotent stem cells (iPS) and derived NSCs
Stemgent® Mouse Primary iPS Cells-NNeo was purchased from STMGENT Company. iPS were maintained in standard embryonic stem cell culture conditions. Briefly, irradiated mouse embryonic fibroblast were plated at a density of 2.5×104 cells per cm2 as feeder cells in gelatin-coated 6-well plates, and iPS were maintained on the feeder cells with standard embryonic stem cells culture medium containing knock-out DMEM, 10% knock-out serum, 2 mM L-Glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol and 10 ng ml−1 LIF (all from Invitrogen). For embryoid body (EB) formation, dissociated iPS cells (via 0.05% trypsin-EDTA solution) were cultured at 5×105 cells per ml in the EB formation medium (standard embryonic stem cells culture medium without LIF). Following 3 days of EB formation, cultured cells were stimulated with 5 µM RA (Sigma) for 7 days with culture medium changed every day. At Day 11, EBs were dissociated into single cells and transfer to NSC culture medium (the growth medium described above). Spheres formed after 2 passages of culture were mainly neurospheres, as confirmed by immunostaining of NSC markers, and were also examined for multi-potent differentiation into 3 neural cell lineages. To generate iPS-derived NSC with stable gene manipulation, dissociated neurospheric cells were infected with various lentiviruses (containing fluorescent marker GFP), and selected over passages using the blasticidin-containing selection medium. Neuropeptide treatment: iPS-derived NSCs were cultured in NSC culture medium in the presence of α-MSH (20 ng/ml), CART (20 ng/ml), NPY (20 ng/ml) or BDNF (20 ng/ml) either individually or in combination for 7 days, with medium replaced every 2 to 3 days. Cells were collected for real-time PCR analysis or immunofluorescence microscopy.
4.3. MBH implantation
Bilateral injections of mediobasal hypothalamus were performed in adult C56BL/6 mice as described previously [26,30,49]. Briefly, under an ultra-precise stereotax, cells at the number of 8000 suspended in 0.5 µl PBS was injected into each side of the mediobasal hypothalamus via guide cannula directed to the coordinates of 0.17 cm posterior to the bregma, 0.03 cm lateral to the middle line, and 0.50 cm below the skull surface of mice.
4.4. Carotid artery injection
C57BL/6 mice were anesthetized and a longitudinal incision is made on the right site of the neck. Under a dissecting microscope, the common, internal, and external carotid arteries are dissected, and the external carotid artery and the proximal part of the common carotid artery were clamped by a microclamp. Cell injections were performed by a 33-gauge microneedle punctured into common carotid artery. For each injection, 2×105 cells suspended in 100-µl PBS are injected with a preserved flow. After cell injection, needle was withdrawn, and a pressure in the injection site was maintained until bleeding stopped. After surgical procedure, mice were kept in a warm environment until recovery.
4.5. Metabolic phenotyping
C57BL/6 mice were obtained from Jackson Laboratory, and were housed in standard conditions. High-fat diet was obtained from Research Diets, Inc. Body weight of individually housed mice was measured twice per week and food intake was recorded on daily basis. MRI measurement of lean vs. fat mass was performed at the core facility at Albert Einstein College of Medicine. For GTT, overnight fasted mice were injected with glucose (2 g kg−1 body weight) intraperitoneally, and blood glucose levels at various time points were measured using a Glucometer (Bayer). Fasting blood insulin levels were measured using serum or plasma samples obtained from mice following 14 to 20 h of fasting. All procedures were approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine.
4.6. Immunostaining
For tissue sections, mice under anesthesia were trans-heart perfused with 4% PFA, and the brains were removed, post-fixed in 4% PFA for 4 h, and infiltrated in 20–30% sucrose. Brain sections (20 μm) were made using a cryostat at −20 °C. Cultured cells on coverslips were fixed with 4% PFA for 10 min at room temperature. For immunostaining of culture cells, cells were prepared onto cover slides and fixed with 4% PFA. Fixed tissue sections/cells were blocked with serum of appropriate species, penetrated with 0.2% Triton-X 100, treated with primary antibodies and followed by reaction with Alexa Fluor® 488 or 555 or 633 secondary antibodies (Invitrogen). Naïve IgGs of appropriate species were used as negative controls. Primary antibodies included rabbit anti-POMC (Phoenix Pharmaceuticals), mouse anti-GFAP, anti-O4, anti-NeuN antibodies (Millipore), mouse anti-Sox2 antibody (R&D Systems), sheep anti-α-MSH (Millipore), rabbit anti-POMC (Phoenix), rabbit anti-AGRP (Phoenix), and rabbit anti-pSTAT3 (Cell Signaling). Rabbit anti-Six3 and anti-Vax1 antibodies were kindly provided by Guillermo Oliver and Greg Lemke, respectively. DAPI (Vector) staining revealed the nuclei of all cells in the slides. Images of immunostaining were captured under a con-focal microscope. Cell counting of immunostaining: serial hypothalamus sections across the MBH were made at the thickness of single cell (10 µm), and every 5 sections were represented by one section with staining and cell counting. The numbers in representative sections were multiplied by 5 to indicate the total numbers. Cell counting in cultured cell slides was performed by including at least 4 to 6 random fields per slide under microscopy.
4.7. Quantitative RT-PCR
Total RNA was extracted using TRIzol (Invitrogen) following the manual. cDNA was synthesized using the M-MLV reverse transcriptase (Promega). Real-time PCR was performed using the StepOnePlus™ Real-Time PCR System and SYBR Green PCR Master Mix (Applied Biosystems). Relative gene expression levels were normalized against the mRNA level of the house-keeping gene β-actin.
4.8. Statistical analyses
Kolmogorov–Smirnov test was used to determine parametric distribution of data sets. ANOVA and appropriate post-hoc analyses were used for comparisons involving more than two groups, and two-tailed Student׳s t tests were used for comparisons involving only two groups. Data are presented as mean±s.e.m. P<0.05 was considered significant.
Conflict of interest
The authors state that they have no competing financial interests.
Acknowledgments
The authors thank G. Oliver and G. Lemke for providing antibodies, and thank Cai׳s laboratory members for technical assistance and discussion. This study was kindly supported by Albert Einstein College of Medicine internal funds, and NIH R01 DK078750, R01 AG031774 and R01 HL113180 (all to D. Cai).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at 10.1016/j.molmet.2014.01.012.
Appendix A. Supplementary materials
Supplementary data.
References
- 1.Niswender K.D., Baskin D.G., Schwartz M.W. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends in Endocrinology and Metabolism. 2004;15(8):362–369. doi: 10.1016/j.tem.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Munzberg H., Myers M.G., Jr. Molecular and anatomical determinants of central leptin resistance. Nature Neuroscience. 2005;8(5):566–570. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- 3.Flier J.S. Neuroscience. Regulating energy balance: the substrate strikes back. Science. 2006;312(5775):861–864. doi: 10.1126/science.1127971. [DOI] [PubMed] [Google Scholar]
- 4.Coll A.P., Farooqi I.S., O׳Rahilly S. The hormonal control of food intake. Cell. 2007;129(2):251–262. doi: 10.1016/j.cell.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dietrich M.O., Horvath T.L. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends in Neurosciences. 2013;36(2):65–73. doi: 10.1016/j.tins.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Dietrich M.O., Horvath T.L. Synaptic plasticity of feeding circuits: hormones and hysteresis. Cell. 2011;146(6):863–865. doi: 10.1016/j.cell.2011.08.031. [DOI] [PubMed] [Google Scholar]
- 7.Olofsson L.E., Unger E.K., Cheung C.C., Xu A.W. Modulation of AgRP-neuronal function by SOCS3 as an initiating event in diet-induced hypothalamic leptin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(8):E697–E706. doi: 10.1073/pnas.1218284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath T.L., Sarman B., Garcia-Caceres C., Enriori P.J., Sotonyi P., Shanabrough M. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(33):14875–14880. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi C.X., Habegger K.M., Chowen J.A., Stern J., Tschop M.H. A role for astrocytes in the central control of metabolism. Neuroendocrinology. 2011;93(3):143–149. doi: 10.1159/000324888. [DOI] [PubMed] [Google Scholar]
- 10.McNay D.E., Briancon N., Kokoeva M.V., Maratos-Flier E., Flier J.S. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. Journal of Clinical Investigation. 2012;122(1):142–152. doi: 10.1172/JCI43134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Tang Y., Cai D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nature Cell Biology. 2012;14(10):999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thaler J.P., Yi C.X., Schur E.A., Guyenet S.J., Hwang B.H., Dietrich M.O. Obesity is associated with hypothalamic injury in rodents and humans. Journal of Clinical Investigation. 2012;122(1):153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Souza F.S., Santangelo A.M., Bumaschny V., Avale M.E., Smart J.L., Low M.J. Identification of neuronal enhancers of the proopiomelanocortin gene by transgenic mouse analysis and phylogenetic footprinting. Molecular and Cellular Biology. 2005;25(8):3076–3086. doi: 10.1128/MCB.25.8.3076-3086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bumaschny V.F., de Souza F.S., Lopez Leal R.A., Santangelo A.M., Baetscher M., Levi D.H. Transcriptional regulation of pituitary POMC is conserved at the vertebrate extremes despite great promoter sequence divergence. Molecular Endocrinology. 2007;21(11):2738–2749. doi: 10.1210/me.2006-0557. [DOI] [PubMed] [Google Scholar]
- 15.Padilla S.L., Carmody J.S., Zeltser L.M. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nature Medicine. 2010;16(4):403–405. doi: 10.1038/nm.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kokoeva M.V., Yin H., Flier J.S. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 2005;310(5748):679–683. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]
- 17.Kokoeva M.V., Yin H., Flier J.S. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. Journal of Comparative Neurology. 2007;505(2):209–220. doi: 10.1002/cne.21492. [DOI] [PubMed] [Google Scholar]
- 18.Pierce A.A., Xu A.W. De novo neurogenesis in adult hypothalamus as a compensatory mechanism to regulate energy balance. Journal of Neuroscience. 2010;30(2):723–730. doi: 10.1523/JNEUROSCI.2479-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pencea V., Bingaman K.D., Wiegand S.J., Luskin M.B. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. Journal of Neuroscience. 2001;21(17):6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee D.A., Bedont J.L., Pak T., Wang H., Song J., Miranda-Angulo A. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nature Neuroscience. 2012;15(5):700–702. doi: 10.1038/nn.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dietrich M.O., Horvath T.L. Fat incites tanycytes to neurogenesis. Nature Neuroscience. 2012;15(5):651–653. doi: 10.1038/nn.3091. [DOI] [PubMed] [Google Scholar]
- 22.Garcia A.D., Doan N.B., Imura T., Bush T.G., Sofroniew M.V. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nature Neuroscience. 2004;7(11):1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- 23.Tiberghien P. Suicide gene for the control of graft-versus-host disease. Current Opinion in Hematology. 1998;5(6):478–482. doi: 10.1097/00062752-199811000-00021. [DOI] [PubMed] [Google Scholar]
- 24.Caruso M., Panis Y., Gagandeep S., Houssin D., Salzmann J.L., Klatzmann D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(15):7024–7028. doi: 10.1073/pnas.90.15.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Culver K.W., Ram Z., Wallbridge S., Ishii H., Oldfield E.H., Blaese R.M. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256(5063):1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X., Zhang G., Zhang H., Karin M., Bai H., Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Souza C.T., Araujo E.P., Bordin S., Ashimine R., Zollner R.L., Boschero A.C. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146(10):4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 28.Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases 2250. Trends in Endocrinology and Metabolism. 2013;24(1):40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng Q., Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. Journal of Biological Chemistry. 2011;286(37):32324–32332. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purkayastha S., Zhang H., Zhang G., Ahmed Z., Wang Y., Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(7):2939–2944. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purkayastha S., Zhang G., Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nature Medicine. 2011;17(7):883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. 2013;497(7448):211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 34.Okada Y., Shimazaki T., Sobue G., Okano H. Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Developmental Biology. 2004;275(1):124–142. doi: 10.1016/j.ydbio.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 35.Wataya T., Ando S., Muguruma K., Ikeda H., Watanabe K., Eiraku M. Minimization of exogenous signals in ES cell culture induces rostral hypothalamic differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(33):11796–11801. doi: 10.1073/pnas.0803078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterhouse E.G., An J.J., Orefice L.L., Baydyuk M., Liao G.Y., Zheng K. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. Journal of Neuroscience. 2012;32(41):14318–14330. doi: 10.1523/JNEUROSCI.0709-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chua J.Y., Pendharkar A.V., Wang N., Choi R., Andres R.H., Gaeta X. Intra-arterial injection of neural stem cells using a microneedle technique does not cause microembolic strokes. Journal of Cerebral Blood Flow and Metabolism. 2011;31(5):1263–1271. doi: 10.1038/jcbfm.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guzman R., De Los A.A., Cheshier S., Choi R., Hoang S., Liauw J. Intracarotid injection of fluorescence activated cell-sorted CD49d-positive neural stem cells improves targeted cell delivery and behavior after stroke in a mouse stroke model. Stroke. 2008;39(4):1300–1306. doi: 10.1161/STROKEAHA.107.500470. [DOI] [PubMed] [Google Scholar]
- 39.Yi C.X., Tschop M.H., Woods S.C., Hofmann S.M. High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Disease Models and Mechanisms. 2012;5:686–690. doi: 10.1242/dmm.009464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong D., Tong Q., Ye C., Koda S., Fuller P.M., Krashes M.J. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell. 2012;151(3):645–657. doi: 10.1016/j.cell.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morshead C.M., Reynolds B.A., Craig C.G., McBurney M.W., Staines W.A., Morassutti D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13(5):1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 42.Markakis E.A., Palmer T.D., Randolph-Moore L., Rakic P., Gage F.H. Novel neuronal phenotypes from neural progenitor cells. Journal of Neuroscience. 2004;24(12):2886–2897. doi: 10.1523/JNEUROSCI.4161-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pluchino S., Zanotti L., Rossi B., Brambilla E., Ottoboni L., Salani G. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436(7048):266–271. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- 44.Martino G., Pluchino S. The therapeutic potential of neural stem cells. Nature Reviews Neuroscience. 2006;7(5):395–406. doi: 10.1038/nrn1908. [DOI] [PubMed] [Google Scholar]
- 45.Wernig M., Brustle O. Fifty ways to make a neuron: shifts in stem cell hierarchy and their implications for neuropathology and CNS repair. Journal of Neuropathology and Experimantal Neurology. 2002;61(2):101–110. doi: 10.1093/jnen/61.2.101. [DOI] [PubMed] [Google Scholar]
- 46.Koch P., Kokaia Z., Lindvall O., Brustle O. Emerging concepts in neural stem cell research: autologous repair and cell-based disease modelling. Lancet Neurology. 2009;8(9):819–829. doi: 10.1016/S1474-4422(09)70202-9. [DOI] [PubMed] [Google Scholar]
- 47.Lindvall O., Kokaia Z. Stem cells in human neurodegenerative disorders--time for clinical translation? Journal of Clinical Investigation. 2010;120(1):29–40. doi: 10.1172/JCI40543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrison S.J., Perez S.E., Qiao Z., Verdi J.M., Hicks C., Weinmaster G. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 2000;101(5):499–510. doi: 10.1016/s0092-8674(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 49.Zhang G., Bai H., Zhang H., Dean C., Wu Q., Li J. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron. 2011;69(3):523–535. doi: 10.1016/j.neuron.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data.