

ABSTRACT
The findings presented here demonstrate the role of α-catenin in cadherin-based adhesion and mechanotransduction in different mechanical contexts. Bead-twisting measurements in conjunction with imaging, and the use of different cell lines and α-catenin mutants reveal that the acute local mechanical manipulation of cadherin bonds triggers vinculin and actin recruitment to cadherin adhesions in an actin- and α-catenin-dependent manner. The modest effect of α-catenin on the two-dimensional binding affinities of cell surface cadherins further suggests that force-activated adhesion strengthening is due to enhanced cadherin–cytoskeletal interactions rather than to α-catenin-dependent affinity modulation. Complementary investigations of cadherin-based rigidity sensing also suggest that, although α-catenin alters traction force generation, it is not the sole regulator of cell contractility on compliant cadherin-coated substrata.
KEY WORDS: Adhesion, α-Catenin, Cadherin, Mechanotransduction
INTRODUCTION
Cadherins are essential adhesion proteins that regulate intercellular cohesion in soft tissues (Gumbiner, 2005; Takeichi, 1991). Cadherins are also signaling proteins, but one of their principal functions is to maintain cell–cell cohesion. The magnitudes and distributions of intercellular stresses influence many tissue properties and processes, including cell shape, morphogenetic movements, neural tube closure, wound healing, cell segregation and the regulation of tissue barriers (Cavey et al., 2008; Diz-Muñoz et al., 2010; Kasza and Zallen, 2011; Krieg et al., 2008; Lecuit and Lenne, 2007; Lecuit et al., 2011; Paluch and Heisenberg, 2009; Papusheva and Heisenberg, 2010; Rauzi et al., 2008). Passive intercellular linkages would, in principle, be sufficient to support many of these functions, but adhesion alone would not account for the instructive cues inherent in the tissue mechanics and the transduction of those cues into biochemical signals that regulate cell functions. Findings increasingly demonstrate the importance of forces in development, tissue homeostasis and disease, and mechanotransduction is the vehicle by which cells sense and respond to their mechanical environment.
In contrast to the mechano-sensitivity of focal adhesions, which are prototypical force-sensitive adhesion complexes (Geiger et al., 2009), earlier reports did not classify cadherin complexes as force sensors (Vogel and Sheetz, 2006). Integrin-based mechanosensing was initially identified on the basis of two measurements. First, direct external mechanical manipulation of ligand-coated beads (e.g. fibronectin or RGD peptides) bound to cell surface integrins were shown to trigger integrin bond strengthening and cytoskeletal remodeling (Choquet et al., 1997; Wang et al., 1993). Second, integrin-based traction forces and cell migration on matrix proteins increases with the substratum rigidity (Beningo and Wang, 2002; Wang et al., 2001). In cells on compliant fibronectin-coated substrata, focal adhesion size, stress fiber formation and integrin-mediated traction forces increase with substratum rigidity. Similar mechanisms were thought to regulate both acute integrin-based force sensation and substratum rigidity sensing (Beningo et al., 2002; Discher et al., 2005; Guo et al., 2006; Pelham and Wang, 1997; Vogel and Sheetz, 2006).
Recent measurements that are analogous to those obtained in mechanical studies of focal adhesions have demonstrated that cadherin complexes are also mechanosensitive. Specifically, cadherin-based traction forces increase with substratum rigidity (Ladoux et al., 2010; Tabdili et al., 2012), and bead twisting on cell surface cadherins triggers actin and E-cadherin-dependent cell stiffening (le Duc et al., 2010; Tabdili et al., 2012). Intercellular stress is altered indirectly by myosin II (MyII, also known as MYH2) activation (le Duc et al., 2010; Liu et al., 2010; Miyake et al., 2006; Twiss et al., 2012; Yonemura et al., 2010), which increases tension between cells (Maruthamuthu et al., 2011), and directly by pulling on cell doublets with dual pipettes (Thomas et al., 2013). MyII activation stimulates both vinculin recruitment to cell–cell junctions and the thickening of adhesion zones between endothelial cells (le Duc et al., 2010; Liu et al., 2010; Miyake et al., 2006; Twiss et al., 2012; Yonemura et al., 2010). Direct tugging on cell pairs also triggers intercellular adhesion strengthening and vinculin recruitment to cell contacts (Thomas et al., 2013).
The previous studies demonstrated that force triggers intercellular junction remodeling, but whether the different results reflect identical or different mechanisms is an open question. In contrast to specifically tugging on cadherin bonds, force transmitted generally throughout intercellular contacts could involve proteins other than cadherins. For example, the main fluid shear sensor at endothelial junctions is platelet endothelial cell adhesion molecule 1 (PECAM-1), which forms a functional complex with VE-cadherin and VEGF receptor 2 (Schwartz and DeSimone, 2008; Tzima et al., 2005). Pharmacological activation of MyII could also stimulate kinases and GTPases that can regulate junctional proteins (Braga et al., 1999; Brunton et al., 2004; Dudek and Garcia, 2001; McLachlan and Yap, 2007; Petrova et al., 2012; Waschke et al., 2004).
Remaining key questions concern the mechanism(s) underlying different manifestations of cadherin-based mechanotransduction, the identity of the mechanosensor(s), and the force-dependent protein cascades underlying the experimental outcomes. Biochemical and biophysical evidence strongly suggest that α-catenin is a force-activated protein in cadherin complexes. α-Catenin is an actin-binding protein, which also binds β-catenin associated with the cadherin cytoplasmic domain, and it is required for mechanical coupling between cadherin and actomyosin (Cavey et al., 2008; Desai et al., 2013; Gumbiner and McCrea, 1993; Imamura et al., 1999). Yonemura et al. (Yonemura et al., 2010) first demonstrated that MyII activation stimulated both vinculin recruitment to cadherin adhesions and increased α-catenin reactivity towards an epitope specific antibody. Based on those observations and mapping studies of α-catenin domains, Yonemura et al. (Yonemura et al., 2010) postulated that force triggers the exposure of a cryptic vinculin-binding-site in α-catenin that in turn recruits vinculin and actin to cadherin junctions. This model is supported by cadherin-based adhesion strengthening and by vinculin recruitment to stressed intercellular junctions, which both require α-catenin and its vinculin-binding-site (Thomas et al., 2013; Twiss et al., 2012). Studies of α-catenin mutants in Drosophila are also consistent with the proposed mechanism (Desai et al., 2013). However, whether the same mechanism(s) accounts for all putative cadherin-based mechanotransduction behavior, such as adhesion strengthening, junctional remodeling, and cell stiffening, has yet to be established.
This study investigated the role of α-catenin in cadherin-based adhesion and mechanotransduction in different mechanical contexts. Bead-twisting measurements in conjunction with imaging, and the use of different cell lines and α-catenin mutants directly tested whether the mechanical manipulation of cadherin bonds triggers vinculin and actin recruitment in an actin- and α-catenin-dependent manner. Traction force measurements further identified differences between acute mechanotransduction and rigidity sensing. Finally, cadherin affinity measurements tested whether α-catenin modulates cadherin affinity (adhesion) through inside-out signaling. These findings demonstrate the role of α-catenin in cadherin-specific mechanotransduction, verify features of the proposed force-transduction mechanism, and reveal aspects of cadherin-based mechanosensing that differ from expected behavior.
RESULTS
α-Catenin is required for acute cadherin-mediated mechanotransduction
To test the impact of α-catenin on cadherin mechanotransduction, we performed experiments with stable cell lines that either express or lack expression of α-catenin. Specifically, we used MDCK cells, which expressed endogenous α-E-catenin (MDCK WT), MDCK cells in which α-catenin was stably knocked down (MDCK KD, from James Nelson, Stanford University, Stanford, CA), and MDCK KD cells with restored α-catenin expression (MDCK Rescued) (Fig. 1, left). Experiments were also performed with DLD-1 cells, with the α-catenin-null subclone of the DLD-1 cell line (R2/7) and with R2/7 cells rescued with GFP–α-catenin (R2/7 Rescued) (Watabe-Uchida et al., 1998; Yonemura et al., 2010). α-Catenin expression levels are shown in Fig. 1 (right). quantitative flow cytometry confirmed that the DLD-1 and R2/7 cell lines expressed membrane-bound E-cadherin at similar levels (data not shown).
Fig. 1.

Western blots of α-catenin expression in MDCK and DLD-1 cell lines. Whole-cell lysates from MDCK WT (parental), MDCK KD (clone number 1) and MDCK Rescued (clone number 10) cells (left) and DLD-1 (parental), R2/7 and R2/7 Rescued cells (right) were separated by SDS-PAGE and blotted for α-catenin, GAPDH and tubulin.
Magnetic twisting cytometry (MTC) measurements (Fig. 2A) of cell surface cadherin complexes probed with ferromagnetic beads modified with Fc-tagged extracellular domains of canine E-cadherin (E-cad-Fc) demonstrated that α-catenin was obligatory for acute cadherin-dependent mechanotransduction. MTC measurements apply shear directly to cadherin bonds at the cell surface, and thus differ from indirect methods that alter tension on intercellular junctions. With MTC, force-activated remodeling alters the junction and possibly the overall cell stiffness, as reflected by altered bead displacement amplitudes.
Fig. 2.
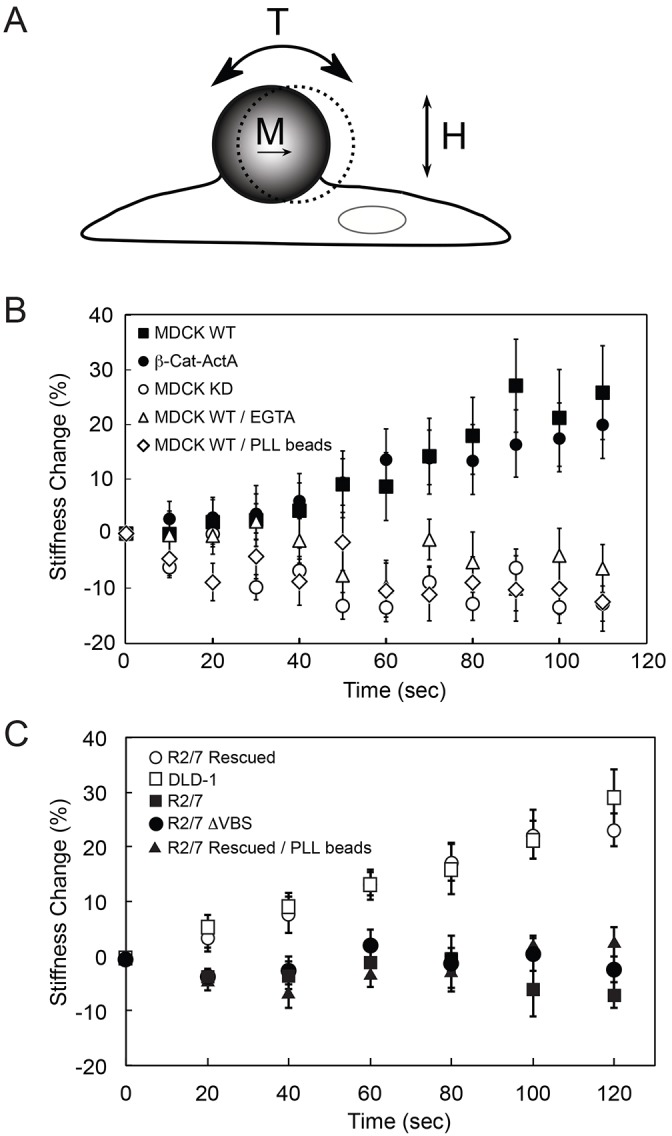
α-Catenin is required for acute cadherin-dependent mechanotransduction. (A) Schematic of the magnetic twisting cytometry experiment. Ligand-coated ferromagnetic beads are magnetized with a magnetic moment (M) parallel to the substrate and subjected to an oscillating field (H). The orthogonal applied field generates a torque (T) on the bead, causing a bead displacement. (B) MTC measurements of force-induced cell stiffening were performed using canine E-cad-Fc-coated beads to probe MDCK WT cells in the absence (black squares) and presence of 4 mM EGTA (white triangles). In controls, MDCK WT cells were probed with PLL-coated beads (white diamonds). E-cad-Fc-coated beads also probed MDCK WT cells in which α-catenin has been stably knocked down (MDCK KD, white circles), and MDCK KD cells expressing β-cat-ActA (black circles). (C) MTC measurements using E-cad-Fc-coated beads to probe DLD-1 cells (white squares), α-catenin-null cells (R2/7, black squares), R2/7 cells rescued with mouse GFP–α-catenin (R2/7 Rescued, white circles) and R2/7 cells rescued with mouse GFP–α-catenin lacking the vinculin-binding site (R2/7 ΔVBS, black circles). In controls, R2/7 Rescued cells were probed with PLL-coated beads (black triangles). Each time point represents the mean±s.d. of >200 beads (one bead per cell).
In MTC measurements with MDCK WT cells (Fig. 2B), bond shear increased the cadherin junction stiffness ∼28%, relative to unperturbed cells. Conversely, MDCK KD cells probed with identical E-cad-Fc-coated beads failed to induce any stiffening response, and the junction modulus decreased slightly (Fig. 2B), possibly due to some bead detachment. Because the E-cadherin expression levels were the same on both cell types, the difference in the mechanoresponse of the MDCK KD and MDCK WT cells can be attributed to α-catenin loss. In control measurements with either poly(L-lysine) (PLL)-coated beads or E-cad-Fc-coated beads in the presence of EGTA, which removes Ca2+ ions required for cadherin activity, junction stiffness was unaltered or decreased in response to bond shear (Fig. 2B).
Cytosolic α-catenin dimers affect actin dynamics at the leading edge (Benjamin et al., 2010) and could indirectly affect cadherin-based mechanotransduction, by altering global cell contractility or actin organization at cadherin adhesions. Measurements with β-cat-ActA MDCK cells (Benjamin et al., 2010) tested this. In these cells (from James Nelson), the mitochondrial-targeting protein ActA is fused to the α-catenin-binding region of β-catenin. This sequesters a proportion of cytosolic α-catenin to mitochondria, while cadherin-bound α-catenin is retained at the membrane (Benjamin et al., 2010). Immunofluorescence measurements confirmed that the β-cat-ActA construct does not affect α-catenin levels at cell–cell contacts (supplementary material Fig. S1A,B). Consistent with the hypothesis that α-catenin affects mechanotransduction locally at cadherin adhesions, the mechanoresponse of β-cat-ActA MDCK cells was statistically identical to MDCK WT cells (Fig. 2B).
Mechanical perturbation of DLD-1 and R2/7 Rescued cells with E-cad-Fc beads triggered a ∼30% increase in junction stiffness (Fig. 2C), but α-catenin-deficient R2/7 cells did not show this response (Fig. 2C). The responses of the R2/7 Rescued cells and DLD-1 cells were statistically similar. Additionally, R2/7 cells rescued with a GFP–α-catenin construct lacking the vinculin-binding-site (GFP–α-catenin-ΔVBS) (Twiss et al., 2012) also failed to trigger cell stiffening, in agreement with the proposed role of the vinculin-binding-site in mechanotransduction. Controls with PLL-coated beads failed to induce junction remodeling in R2/7 Rescued cells (Fig. 2C).
α-Catenin depletion modestly alters cadherin affinity
The influence of α-catenin on cadherin-mediated mechanotransduction could arise from inside-out modulation of cadherin binding-affinity and altered bead-cell adhesion. Alternatively, α-catenin depletion could disrupt mechanical linkages required to transmit force. Micropipette measurements quantified the effect of α-catenin on the intrinsic two-dimensional E-cadherin affinity, in the native context of the cell membrane, by comparing the affinities of E-cadherin expressed on MDCK KD and MDCK Rescued cells. Importantly, bead-twisting measurements used to quantify mechanotransduction are on the same time scale as these kinetic measurements (<2 min).
In these measurements, an MDCK cell expressing full-length E-cadherin was repetitively brought into contact with a red blood cell (RBC) that was modified with oriented E-cadherin-Fc (Fig. 3A). Fig. 3B shows the binding probability P [the number of cell-cell binding events (nb) divided by the total number of cell–cell touches (NT)] as a function of contact time between the modified E-cad-Fc RBCs and either MDCK KD or MDCK Rescued cells. The two-stage kinetic profile observed with both cell types is similar to previously reported cadherin-binding kinetics (Chien et al., 2008; Langer et al., 2012). A fast rise to an initial plateau at P1 ∼0.51 (MDCK KD) is followed by a 2–4-s lag and a slower rise to a steady-state probability at P2 ∼0.8 after ∼20 s. The density of E-cad-Fc on the RBCs was 29 cadherins/µm2, and the E-cadherin densities on the MDCK Rescued and MDCK KD cells were 40 and 44 cadherins/µm2, respectively, as determined by flow cytometry (Chien et al., 2008).
Fig. 3.
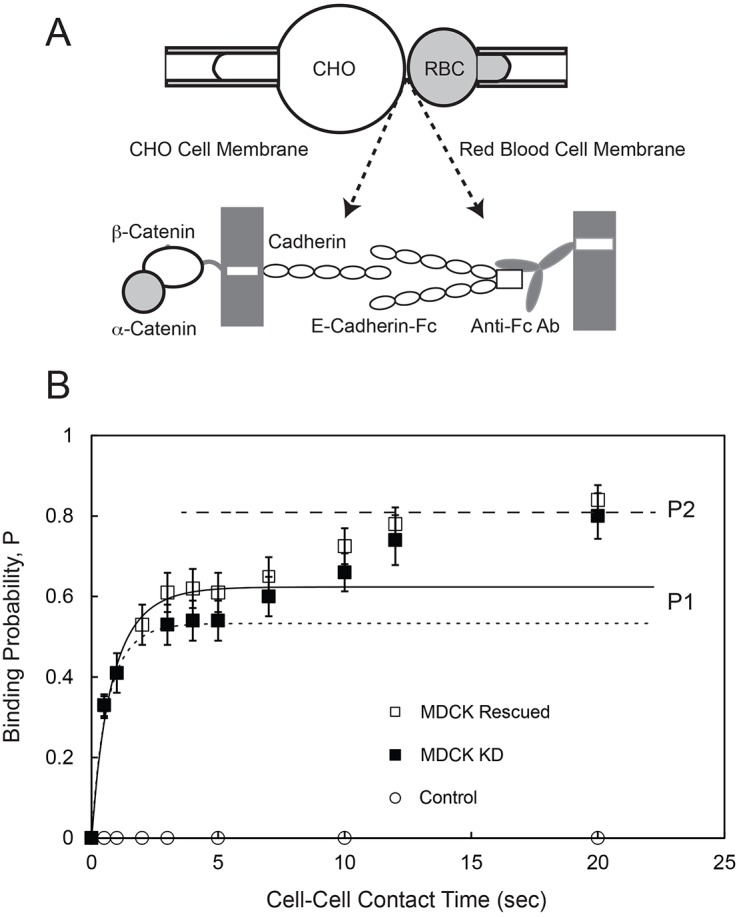
Kinetics of E-cadherin-mediated binding between an MDCK cell and an RBC modified with E-cad-Fc. (A) Schematic of the micropipette aspiration experiment. A cell expressing full-length cadherin is aspirated into a pipette (left) and repetitively brought into contact with an RBC modified with Fc-tagged canine E-cadherin extracellular domains (E-cad-Fc), which are captured and oriented by anti-Fc antibody covalently bound to the RBC (right). (B) The time-dependent binding probability (P) versus cell–cell contact time measured between RBCs modified with E-cad-Fc and MDCK KD cells (black squares) or MDCK Rescued cells (white squares). The solid line is the nonlinear least squares fit of Eqn 1 to data for the first binding step obtained with MDCK Rescued cells, with best-fit parameters given in the text. The dotted line is the fit to data obtained with MDCK KD cells, with best-fit parameters given in the text. The dashed line indicates the limiting binding probability P2 determined with both MDCK Rescued and MDCK KD cells. Control data (white circles) were measured between MDCK Rescued cells and RBCs modified with anti-human IgG (Fc) antibody without bound E-cad-Fc.
The two-dimensional binding affinities and dissociation rates for the EC1-dependent trans dimerization (first binding step) were obtained from data fits to a kinetic model for trans dimerization (Equation 1), using non-linear regression. The best-fit, two-dimensional affinity and dissociation rate for E-cadherin on MDCK Rescued cells were (1.82±0.23)×10−4 µm2 and 0.86±0.15 s−1 (means±s.e.m.), respectively. With α-catenin KD cells, the best-fit affinity and dissociation rate were (1.29±0.17)×10−4 µm2 and 1.2±0.18 s−1. The affinities are slightly different at the 95% confidence level (P = 0.07), and this suggests that α-catenin has a modest effect on the cadherin affinity.
Force-dependent actin and vinculin recruitment requires α-catenin
Immunofluorescence imaging following bead twisting demonstrated that vinculin and actin recruitment to cadherin junctions coincides spatially and temporally with local mechanical stimulation of cadherin bonds and junction reinforcement. Laser scanning confocal microscopy was used to quantify the levels of actin, α-catenin, vinculin and E-cadherin at beads before and after bond shear under identical conditions (culture conditions, twisting time, and shear stress) to those used to obtain data in Fig. 2B,C. In unperturbed cell monolayers, α-catenin and actin localized at the basolateral membrane (Fig. 4A). R2/7 cells do not express α-catenin, but R2/7 Rescued and R2/7 ΔVBS cells overexpress GFP–α-catenin and GFP–α-catenin-ΔVBS, respectively.
Fig. 4.

Force-dependent distributions of α-catenin, F-actin and vinculin imaged at cell–cell and bead–cell junctions. (A) Immunostained R2/7 Rescued, R/27 and R2/7 ΔVBS cells before applying shear stress through E-cad-Fc-coated beads. R2/7 Rescued and R2/7 ΔVBS cells overexpress GFP–α-catenin and GFP–α-catenin-ΔVBS, respectively. R2/7 cells are deficient in α-catenin. Representative images reveal α-catenin (green) and F-actin (orange) at cell–cell junctions at the basal plane. Scale bars: 10 µm. (B) α-Catenin and F-actin distributions at PLL-coated beads bound to the apical surface of R2/7 Rescued cells, before or after bond shear. Images of individual beads were cropped and enlarged from the boxed regions of original images (shown in supplementary material Fig. S2A). The first DIC image (top left) indicates the region of interest (yellow circles) used to quantify changes in protein distributions at bead–cell junctions. Images are representative of >35 different bead–cell pairs. (C) R2/7 Rescued cells stained for F-actin, before or after cadherin bond shear. (D) R2/7 Rescued cells stained for vinculin. (E,F) R/27 cells stained for F-actin or vinculin, respectively. (G,H) R2/7 ΔVBS cells stained for F-actin or vinculin, respectively. Original images for C–H are shown in supplementary material Fig. S2B. Images are representative of >35 different bead–cell pairs. Scale bars: 5 µm.
Fig. 4B (top left) illustrates the ring analysis used to quantify changes in protein levels at PLL-coated beads bound to the apical surface of cells (yellow circles), before and after shearing cadherin bonds (original images are shown in supplementary material Fig. S2A). Images before and after bond shear reflect different bead–cell pairs because bead movements during twisting are incompatible with quantitative live-cell imaging of local fluorescence changes. This necessitated comparisons of averages (n>35) of immunofluorescence images of fixed cells with and without shear, from replicate experiments obtained under identical culture conditions. Standard errors in reported fluorescence intensity differences were determined by propagation of errors and use of pooled standard deviations (Calcutt, 1983).
In R2/7 Rescued cells, α-catenin localized around beads, and levels did not change after twisting (Fig. 4C,D). E-cadherin did not increase significantly at beads (Fig. 5A, top, 5B, left). By contrast, F-actin formed visible rings around beads bound to unperturbed R2/7 Rescued cells, and the intensity and thickness of the actin rings increased after 2 min of bond shear (Fig. 4C; supplementary material Fig. S2B). Bead twisting also triggered vinculin accumulation at the beads (Fig. 4D; supplementary material Fig. S2B). Fig. 6A summarizes changes in protein levels relative to unstressed conditions. Quantification of protein localization at beads, before and after shear, revealed significant increases in F-actin (P = 2×10−8, n≥60) and vinculin (P = 2×10−5, n≥85) (Fig. 6B). Controls with PLL-coated beads exhibited neither a stiffening response (Fig. 2C) nor force-dependent accumulation of F-actin, α-catenin or vinculin at the beads (Fig. 4B; Fig. 6A,D; supplementary material Fig. S2A).
Fig. 5.

E-cadherin distributions at cell–cell and bead–cell junctions in R2/7 cells expressing GFP–α-catenin or GFP–α-catenin-ΔVBS. (A) R2/7 Rescued and R2/7 ΔVBS cells immunostained for E-cadherin, before or after applying cadherin bond shear. Cell–cell junctions were imaged at the basal plane of cell monolayers for E-cadherin. Representative DIC images show beads attached to cells, and the corresponding fluorescence images show α-catenin (green) and E-cadherin (orange). Scale bars: 10 µm. (B) Mean fluorescence intensity of α-catenin and E-cadherin at E-cadherin bead–cell junctions in R2/7 Rescued (left; α-catenin, n≥208; E-cadherin, n≥55) and R2/7 ΔVBS cells (right; α-catenin, n≥44; E-cadherin, n≥44).
Fig. 6.

Shear-induced changes in α-catenin, vinculin and F-actin at E-cadherin bead–cell junctions. (A) Change in mean fluorescence intensity, relative to non-loading conditions, of proteins within rings extending 1.0–1.5 µm from the bead edge. A mask designating a filled outline of the bead was defined, based on DIC images. Data represent R2/7 Rescued (α-catenin, n≥208; vinculin, n≥85; F-actin, n≥60), R/27 (vinculin, n≥55; F-actin, n≥35) and R2/7 ΔVBS (α-catenin, n≥86; vinculin, n≥42; F-actin, n≥44) cells bound to E-cad-Fc beads. The control was obtained with PLL beads bound to R2/7 Rescued cells (α-catenin, n≥119; vinculin, n≥58; F-actin, n≥61). (B) Fluorescence intensities of proteins before and after applied load in R2/7 Rescued cells bound to E-cad-Fc beads. Left: F-actin, ***P = 2×10−8, n≥60; α-catenin, P = 0.64, n≥208. Right: vinculin, ***P = 2×10−5, n≥85; α-catenin, P = 0.64, n≥208. (C) Intensity levels at E-cad-Fc beads on R2/7 ΔVBS cells, with and without load. Left: F-actin, n≥44; α-catenin, n≥44. Right: vinculin, n≥42; α-catenin, n≥42. (D) Intensity levels at PLL beads on R2/7 Rescued cells. Left: F-actin, n≥61; α-catenin, n≥61. Right: vinculin, n≥58; α-catenin, n≥58. All error bars represent s.e.m.
α-Catenin–GFP overexpression in R2/7 Rescued cells increased the error in shear-dependent α-catenin accumulation because the high cytosolic fluorescence often eclipsed α-catenin localization at the beads. Increasing the number of measurements (n∼75) increased the signal-to-noise ratio. This was not a factor in the analyses of immunostained vinculin, E-cadherin, or actin, for which the background fluorescence and standard errors were low. The background in DLD-1 cells immunostained for α-catenin was similarly low.
With α-catenin-deficient R2/7 cells, there was negligible actin near the beads relative to background, and bond shear did not induce measurable junction reinforcement (Fig. 2C) or either F-actin or vinculin accumulation (Fig. 4E,F). The changes in protein levels after shear are summarized in Fig. 6A. For display purposes, the change in α-catenin in R2/7 cells was not included because the intensities were negligible (Fig. 6A).
Line-scan analyses of spatial variations in α-catenin, actin and vinculin intensities relative to the bead center, before and after bond shear (supplementary material Fig. S3A–C), also illustrate force-dependent remodeling at bead–cell junctions. The line scans represent regions of greatest change in the mean fluorescence intensity surrounding the beads, and show the non-uniform protein distributions around beads. Because of the high cytosolic background from overexpressed GFP–α-catenin relative to the dark bead center, the relative intensity at the bead center is negative in some cases.
The vinculin-binding site of α-catenin is required for force-dependent actin recruitment
Fig. 4G,H shows immunofluorescence images of GFP–α-catenin-ΔVBS at beads before and after loading. GFP–α-catenin-ΔVBS localized to cell–cell and bead–cell junctions (Fig. 5A, bottom), as expected, because this construct harbors the domain that binds cadherin-associated β-catenin at adhesions. However, bond shear had no effect on E-cadherin, F-actin, GFP–α-catenin ΔVBS, or vinculin levels at the beads (Fig. 5B, right; Fig. 6A,C).
α-Catenin and its vinculin-binding site modulate cadherin-mediated traction forces
Traction force measurements were used to assess the capacity of cells to sense matrix rigidity through cadherin adhesions. α-Catenin loss significantly decreased the root mean square (RMS) traction forces (Pa) exerted by MDCK Rescued cells on polyacrylamide gels with moduli of 1 kPa and 34 kPa (Fig. 7A,B). The 1-kPa stiffness is comparable to lung tissue, and the 34-kPa stiffness is similar to bone (Engler et al., 2007). As observed with N-cadherin (Ladoux et al., 2010; Tabdili et al., 2012), the traction forces of MDCK Rescued cells on E-cad-Fc substrata decreased from 162±11 Pa on 34 kPa gels to 43±9 Pa on 1 kPa gels (means±s.e.m.; P<0.001) (Fig. 7A). The E-cad-Fc density is independent of the gel modulus (Tabdili et al., 2012). Loss of α-catenin reduced traction forces on 34 kPa gels from 162±11 Pa for MDCK Rescued cells to 117±8 Pa for MDCK KD cells (P = 0.002) (Fig. 7A, Table 1). On 1-kPa gels, MDCK Rescued and MDCK KD cells exerted traction forces of 43±9 Pa and 13±4 Pa, respectively (Fig. 7A). Interestingly, α-catenin loss reduced but did not eliminate the rigidity dependence of the traction forces.
Fig. 7.

α-Catenin modulates cadherin-mediated traction forces. (A) Root mean square (RMS) traction forces (Pa) exerted by MDCK KD and MDCK Rescued cells on soft (1 kPa) and rigid (34 kPa) hydrogels with covalently immobilized and oriented E-cad-Fc. For each condition, n≥4; *P<0.05, **P<0.01, ***P<0.001. Error bars represent s.e.m. (B) Representative traction force maps of MDCK KD and MDCK Rescued cells on soft and rigid hydrogels. (C) RMS traction forces exerted by R2/7, R2/7 ΔVBS, R2/7 Rescued and DLD-1 cells on soft (1 kPa), semi-rigid (9 kPa) and rigid (34 kPa) hydrogels with covalently bound E-cad-Fc. All measurements were done in the presence of integrin-blocking antibodies. For each condition, n≥10. Representative traction force maps are in supplementary material Fig. S4. (D) RMS traction forces exerted by MDCK Rescued and MDCK KD cells on 34-kPa gels coated with E-cad-Fc, after treatment with blebbistatin or cytochalasin D (Cyto D). For each condition, n≥5; *P<0.05, **P<0.01, ***P<0.001. All error bars represent s.e.m. (E) Immunofluorescence images of paxillin and F-actin at the basal plane of R2/7 and R2/7 Rescued cells on 34 kPa gels modified with E-cad-Fc. Scale bars: 10 µm.
Table 1. Summary of root mean square traction forces exerted by cells on E-cad-Fc-modified polyacrylamide gels.

Results are the root mean square traction force in Pa and numbers in parenthesis are the s.e.m.; n.d., not determined; Cyto D, cytochalasin D.
Traction forces exerted by DLD-1, R2/7, R2/7 Rescued and R2/7 ΔVBS cells on E-cad-Fc-coated polyacrylamide gels were compared using gels with moduli of 1, 9 and 34 kPa (Fig. 7C; supplementary material Fig. S4). The DLD-1 and R2/7 Rescued cells exerted greater traction forces on rigid gels than on softer gels. In addition, loss of α-catenin reduced the traction force generated by R2/7 cells, relative to DLD-1 and R2/7 Rescued cells on 9 kPa and 34 kPa gels. The rigidity sensing of R2/7 ΔVBS and R2/7 cells were similar. The lack of α-catenin in R2/7 cells also did not eliminate the dependence of traction force on substrate rigidity.
There are differences between the dependence of traction forces and bead twisting results on F-actin and MyII. In acute MTC measurements, cytochalasin D, which disrupts actin microfilaments, abolished the stiffening response. Blebbistatin, which inhibits MyII, reduced stiffening by ∼50% at E-cadherin adhesions on F9 cells (le Duc et al., 2010). With MDCK KD and MDCK Rescued cells on E-cad-Fc-coated 34 kPa gels, cytochalasin D reduced the traction forces to 94±11 Pa and 95±8 Pa, respectively (Fig. 7D; Table 1), and blebbistatin reduced the traction forces to 90±11 Pa and 108±6 Pa, respectively (Fig. 7D; Table 1).
Defined focal adhesions containing paxillin were absent in both the α-catenin-deficient and α-catenin-expressing cells. Traction measurements performed in the presence of integrin-blocking antibodies revealed no evidence of paxillin at the basal plane of R2/7 and R2/7 Rescued cells adhering to 34 kPa gels modified with E-cad-Fc, as determined by immunofluorescence imaging (Fig. 7E). This confirmed that focal adhesions did not contribute to the traction forces.
DISCUSSION
Different experimental approaches used to investigate cadherin-based mechanotransduction previously confirmed that mechanical perturbations trigger biochemical changes at intercellular junctions, but there was no general view of how α-catenin regulates cadherin-mediated adhesions in the different mechanical contexts investigated. The results presented here unify several findings and identify mechanistic similarities and differences between observed behaviors.
Immunofluorescence measurements before and after mechanical stimulation were used to visualize the molecular cascades triggered by localized force at cadherin-specific bonds, and demonstrated that directly pulling on cadherin bonds triggers both vinculin and actin recruitment to stressed cadherin complexes by an α-catenin- and actin-dependent mechanism. This result directly confirmed that the cadherin-specific remodeling events involve molecular changes analogous to those at cell–cell junctions following MyII activation or tugging on cell doublets (Thomas et al., 2013; Yonemura et al., 2010).
Spatial and temporal correlation between applied force, cytoskeletal remodeling and measured cell stiffening also demonstrated that mechanical changes triggered by cadherin bond shear (le Duc et al., 2010; Tabdili et al., 2012) are due to force-activated actin and vinculin accumulation, rather than to nonspecific effects of bead twisting. This is similar to integrin-dependent adhesion stiffening mediated by actin recruitment (Icard-Arcizet et al., 2008). In this case, the absence of junctional stiffening and vinculin or actin accumulation at E-cad-Fc beads in cells lacking α-catenin confirmed that α-catenin is essential for acute cadherin-based mechanotransduction. These data support the hypothesis that α-catenin is the obligate force sensor in this complex, because the loss of its vinculin-binding site abolished tension-dependent responses, while otherwise preserving the mechanical connection between cadherin and actin (Twiss et al., 2012).
The complete abrogation of acute force sensing by the α-catenin ΔVBS mutant differs from the 40–50% decrease in the stiffening response of vinculin-deficient F9 cells (le Duc et al., 2010). Vinculin-knockout F9 cells might have expressed low levels of endogenous vinculin, although none was detected. Alternatively, the vinculin-binding domain also harbors sites for the actin-binding proteins afadin, zonula occludens-1 (ZO-1), formin and α-actinin that could also contribute to actin recruitment (Knudsen et al., 1995; Kobielak et al., 2004; Pokutta et al., 2002; Provost and Rimm, 1999). So far, however, there is no evidence that the latter proteins accumulate at stressed junctions (Twiss et al., 2012).
The increased width and intensity of the actin zone around the beads is qualitatively similar to the thickening of endothelial cell junctions subject to endogenous tugging forces (Liu et al., 2010), and the co-accumulation of vinculin and radial actin fibers at stressed intercellular junctions (Huveneers et al., 2012; le Duc et al., 2010; Twiss et al., 2012). In those studies, the mechanism underlying the zonal thickening was not addressed (Liu et al., 2010), but similar cytoskeletal remodeling following bead twisting, MyII stimulation and cell tugging (Twiss et al., 2012) suggests that vinculin and actin recruitment in these different contexts involves the same α-catenin-dependent pathway.
The increased force to detach cell doublets after initial tugging, with concurrent junctional accumulation of vinculin, also suggests that there is cadherin-based mechanotransduction (Thomas et al., 2013). Our results addressed whether α-catenin might contribute to force-activated adhesion strengthening by enhancing cadherin anchorage to the cytoskeleton (Maître and Heisenberg, 2011), or by altering the cadherin affinity (adhesion) by inside-out signaling (Bajpai et al., 2008), analogous to integrins (Geiger et al., 2009). The kinetics results (Fig. 3B) demonstrated that α-catenin loss modestly reduces the intrinsic two-dimensional cadherin affinity by ∼30%, similar to the 30% reduction in the apparent strength of cadherin tethers between cells expressing an α-catenin mutant with impaired α-catenin binding (Bajpai et al., 2008). There are differences between force-independent two-dimensional affinity measurements (Fig. 3B) and bond rupture measurements, which could reflect the force to break homophilic cadherin bonds, cadherin–cytoskeletal bonds or the force to form membrane tethers (Evans and Calderwood, 2007; Maître and Heisenberg, 2011). Nevertheless, the limited effect of α-catenin on homophilic cadherin bonds would not account for the complete abrogation of acute mechanotransduction in α-catenin-deficient cells. Signaling cascades might amplify small binding differences, but the latter processes could be slower than the rapid stiffening response observed. The affinity and mechanotransduction measurements were on the same short timescale (<2 min). Additionally, inhibitors of Rac and Src, which are cytoskeletal regulatory proteins at E-cadherin junctions, have similarly modest effects on acute E-cadherin mechanotransduction (Shi, 2009). These findings suggest that α-catenin plays a primarily mechanical role in adhesion strengthening by increasing the extent of cytoskeletal connections and by mechanically anchoring cadherin to the cytoskeleton, as observed in zebrafish progenitor cells (Maître et al., 2012) and in Drosophila (Desai et al., 2013).
Because α-catenin is crucial for acute mechanotransduction, one might also expect it to control sensing of substrate rigidity at cadherin adhesions. It was therefore somewhat surprising that α-catenin loss reduced but did not ablate the dependence of cadherin-based traction forces on substratum stiffness. The absence of focal adhesions suggests that other mechanisms cooperate with adhesion-based force transducers to regulate contractility in different mechanical environments and is consistent with a report that fibroblast traction forces appeared to be modulated by an integrin-independent mechanism (Trichet et al., 2012). Here, α-catenin regulates the tension sustained by cadherin adhesions, but our findings suggest that α-catenin does not solely regulate cell tractions.
Rigidity sensing would require mechanical connectivity between the substratum and cytoskeleton. Besides α-catenin, possible links between cadherins and the cytoskeleton include the microtubule–Nezha–PLEKHA7 complex (Meng et al., 2008) and the vinculin–β-catenin complex (Peng et al., 2011). Intermediate filaments interact with C-cadherin in Xenopus mesendoderm cells (Weber et al., 2012). Unraveling the mechanisms regulating cell pre-stress is beyond the scope of this study, but α-catenin clearly cooperates with such mechanisms, to regulate cell contractility in different mechanical environments.
These findings directly demonstrate the obligatory role of α-catenin and its vinculin-binding site in acute force transduction through cadherin adhesions, consequent cytoskeletal remodeling and force-dependent junction reinforcement. The observed molecular cascades that were triggered by cadherin-specific bead twisting further linked the observed force-dependent changes at intercellular junctions to a common α-catenin-dependent mechanism. The modest effect of α-catenin on cadherin affinity also suggests that force-activated adhesion strengthening is due to enhanced cadherin–cytoskeletal interactions rather than to cadherin affinity modulation. Somewhat unexpectedly, α-catenin affects cadherin-mediated traction forces, but it does not solely determine cell contractility on compliant cadherin-coated substrata.
MATERIALS AND METHODS
Cell lines and protein production
Madin-Darby Canine Kidney (MDCK) II and DLD-1 human colon carcinoma cells from ATCC were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% v/v fetal bovine serum (FBS) and 1% v/v penicillin/streptomycin. The stable knockdown of α-E-catenin was achieved using a hybrid vector provided by Adam Kwiatkowski and James Nelson (Stanford University, Palo Alto, CA) as described previously (Benjamin et al., 2010). The vector contains a short hairpin RNA (shRNA) that specifically targets canine α-E-catenin (Capaldo and Macara, 2007), pEGFP-C1, and it contains the neomycin resistance gene for selection in G418 (400 µg/ml) (Sigma-Aldrich, St. Louis, MO). The α-E-catenin knockdown line (MDCK KD, clone #1) was generated by transfecting MDCK cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Subsequent G418 selection of a single MDCK clone showed >90% reduction in α-catenin by immunoblot analysis (Fig. 1, left). As the EGFP expression in this vector can become ‘uncoupled’ from α-catenin knockdown, this line was periodically re-selected by the serial limiting dilution method. The α-E-catenin-restored MDCK cells (MDCK Rescued) were generated using a vector (from Adam Kwiatkowski and James Nelson) containing both the canine-specific α-E-catenin shRNA and a GFP-tagged murine-αE-catenin cDNA that is refractory to the shRNA. The latter also contains a neomycin selectable marker. Two stable integrants were selected after transfection with Lipofectamine (clones #10 and #15) and selection in G418. A transfection efficiency of 100% for cells expressing GFP-tagged murine α-catenin was periodically maintained by FACS. The R2/7 line is a non-cell–cell adhesive α-catenin-null variant of the DLD-1 parental clone (Watabe-Uchida et al., 1998; Yonemura et al., 2010). MDCK β-cat-ActA cells were a gift from James Nelson (Stanford University, Palo Alto, CA).
Both the DLD-1 subclone R2/7 and the α-catenin-knockdown MDCK cells were transfected with lentiviral EGFP–α-catenin and EGFP–α-catenin-ΔVBS constructs described elsewhere (Huveneers et al., 2012), and selected by puromycin. All R2/7 Rescued cells expressed GFP–α-catenin or GFP–α-catenin-ΔVBS, as was confirmed by GFP fluorescence. To characterize their capacity for forming cell–cell junctions, cells were fixed and stained with antibodies against E-cadherin (clone 36, BD Biosciences), α-catenin (rabbit polyclonal, Sigma) and vinculin (hVin-1, Sigma). Cell–cell junctions were restored in these cells by either EGFP–α-catenin or EGFP–α-catenin-ΔVBS expression, although junction maturation seemed somewhat delayed in EGFP–α-catenin-ΔVBS cells. This was not analyzed further. Vinculin was absent from junctions in EGFP–α-catenin-ΔVBS cells, whereas it was present in a subset of junctions in EGFP–α-catenin cells.
The recombinant canine E-cadherin ectodomain with a C-terminal Fc-tag (E-cad-Fc) was stably expressed in human embryonic kidney cells (HEK293T) as described previously (Prakasam et al., 2006). Cells were routinely maintained in DMEM containing 10% v/v FBS. Protein A Affi-Gel (Bio-Rad, Hercules, CA) was used to affinity-purify the soluble Fc-tagged E-cadherin from the conditioned medium. This was followed by gel-filtration chromatography. SDS-PAGE was used to assess the protein purity.
Hybridoma cells producing the β1-integrin-blocking antibody, AIIB2, or the integrin α6 antibody (GOH3) were from Santa Cruz Biotechnology (Santa Cruz, CA).
Magnetic twisting cytometry
Bead-twisting measurements were conducted with a home-built magnetic twisting cytometer (Wang et al., 1993). The measurements used 4.0–4.9 µm carboxyl ferromagnetic beads (Spherotech, Lake Forest, IL) that were covalently modified with E-cad-Fc or PLL (Sigma) as described previously (le Duc et al., 2010). The protein-coated beads were then allowed to settle on confluent cell monolayers at ∼one bead/cell for 20 min at 37°C and 5% CO2, before applying torque. Cells were grown to confluence (2 days) on 35-mm glass-bottomed dishes coated with 20 µg/ml type I collagen (Sigma).
Cells were maintained on a heated microscope stage (37°C). A 1 Tesla pulse magnetized the beads with a magnetic moment parallel to the cell substrate, and a 0.3 Hz oscillating magnetic field (60 Gauss) applied perpendicular to the substrate for 2 min generated a torque on the beads. The resulting bead displacements were quantified with an inverted microscope (Leica) equipped with a 20× 0.6 NA objective lens and a charge-coupled device camera (Orca2, Hamamatsu Photonics). The complex modulus of the bead–cell junction was calculated from the bead displacements (Wang et al., 1993). The data follow a log normal distribution, and plots display the mean and standard deviation. Each experimental condition represents n>300 cells (∼one bead/cell). P-values were calculated from two-tailed Student's t-tests, with P<0.05 considered to be statistically significant. Only beads at the center of the apical surface, not at cell–cell junctions, were included in the analyses in order to avoid interference from changes in cell–cell junction remodeling.
Combined MTC and immunofluorescence imaging
α-Catenin, vinculin, cadherin and filamentous actin (F-actin) densities at the beads were compared before and after bond shear by confocal laser scanning microscopy. In order to control for differences across cells, only beads bound to a central region on the apical surface – not at cell–cell junctions – were included in the analyses. Immediately after bead twisting, cells were fixed with 4% w/v paraformaldehyde (PFA) for 15 min at room temperature, then permeabilized with 0.1% Triton X-100 for 5 min, blocked in 1% w/v BSA for 20 min, and stained with phalloidin, primary antibodies and secondary antibodies in 1% w/v BSA for 1 h. Primary antibodies included rabbit monoclonal anti-α-catenin antibody (Sigma), mouse monoclonal anti-vinculin antibody (Sigma), and mouse monoclonal anti-E-cadherin antibody (clone 36, BD Transduction Laboratories). Secondary antibodies were coupled to FITC (Sigma) or Alexa Fluor 594 (Invitrogen), and Rhodamine-phalloidin was from Invitrogen. Coverslips were mounted with ProLong Gold (Invitrogen). Images were acquired with ZEN 2008 software (Zeiss) and a laser scanning confocal microscope (LSM 700, Zeiss) equipped with a 40× 1.3 NA oil immersion DIC EC Plan-Neofluar objective lens (Zeiss) and 488 nm and 555 nm lasers.
Confocal image analyses
The mean fluorescence intensities (MFI) of α-catenin, vinculin, E-cadherin and F-actin at bead-cell junctions were quantified with ImageJ (National Institutes of Health) from images acquired at a focal plane at which the beads were in focus. Fluorescence intensities surrounding each bead were quantified by drawing a ring extending 1.0–1.5 µm from the bead edge, and a mask outlining the bead was defined, based on DIC images. The mean fluorescence intensity of α-catenin, vinculin, F-actin and E-cadherin surrounding n≥35 beads (one bead/cell) was determined by subtracting the background fluorescence, which was defined by the mean intensity in a user-defined area near each bead but outside the defined ring. Averages of the MFI surrounding each bead–cell pair, pooled standard deviations and standard errors of the mean were calculated in Microsoft Excel. Although some beads did not appear to engage cell surface cadherins, based on the absence of α-catenin–GFP rings, beads with and without α-catenin rings were included indiscriminately in the analyses.
Protein accumulation was quantified by comparing the averaged MFIs surrounding ∼35 beads per experiment, before and after bond shear. It is not feasible to directly quantify fluorescence changes at the same bead before and after shear, because the torque rotates the bead out of the focal plane. Immunofluorescence was quantified from images of fixed cells. The standard errors in the averaged immunofluorescence intensities for each condition were determined from the pooled standard error of replicate experiments performed under identical conditions, in which a minimum of 35 beads were analyzed per experiment. Thus, the determined errors reflect the measurement variance. The reported error in the change in MFI values, relative to unstressed conditions (Fig. 6A) was determined by propagation of errors (Calcutt, 1983).
The ring analyses do not take into account the non-uniformity of the circumferential protein distributions around the beads; thus, line-scan intensity profiles also depicted spatial variations in fluorescence intensities across bead–cell junctions. Line scans were generated from original images using ImageJ. Lines that were 10 µm in length (extending ∼3 µm from either side of the bead edges) and centered on the bead were drawn in order to quantify regions of greatest change in MFI. The background fluorescence was defined as the mean intensity of a user-defined cytoplasmic region near each bead. The background-subtracted intensity profiles across beads (one bead/condition) were plotted in Microsoft Excel. Because of the non-uniform circumferential protein distributions, quantitative comparisons were based on ring analyses, which is less subjective than line scans.
Gel preparation and traction force microscopy
Polyacrylamide hydrogels were prepared and functionalized as described previously (Beningo et al., 2002; Tse and Engler, 2010). Gels with Young's moduli of 1 kPa, 9 kPa and 34 kPa embedded with fluorescent microspheres (0.2 µm) (Molecular Probes, Eugene, OR) were activated with Sulfo-SANPAH (0.5 mg/ml, 100 mM HEPES, pH 7.5) (Pierce, Rockford, IL). Gels were irradiated twice at 320 nm for 8 min and washed with 100 mM HEPES (pH 7.5). Rabbit anti-human IgG (Fc) antibody (0.2 mg/ml) (Jackson ImmunoResearch, West Grove, PA) was incubated with the activated gels overnight at 4°C. After rinsing gels with PBS to remove unbound antibody, they were incubated for at least 3 h with 0.2 mg/ml E-cad-Fc at 4°C.
Prior to traction measurements, cells were detached from tissue culture flasks using a 1% w/w BSA in PBS containing 3.5 mM EDTA, in order to preserve cell surface cadherins (Takeichi and Nakagawa, 2001). Cells were pelleted by centrifugation and rinsed with buffer lacking EDTA. Then 2000–4000 cells/cm2 were seeded onto the cadherin-coated hydrogels embedded with fluorescent beads and allowed to adhere for 6 h at 37°C in DMEM supplemented with 0.5% v/v FBS. To block focal adhesions, the medium contained both anti-α6 antibody GOH3 (20 µg/ml) and anti-β1 antibody AIIB2 (1∶25 dilution of hybridoma culture medium). Fluorescence images of the beads were acquired before and after cell removal with a solution of 1% w/v SDS, 3.5 mM EDTA and 1% BSA in PBS. The bead positions were imaged with an inverted microscope (Leica) equipped with a 40× 0.6 NA objective and a charge-coupled device camera (Orca2, Hamamatsu Photonics). The constrained traction maps were calculated from the bead displacement field (Butler et al., 2002). The net contractile moment was determined for ∼15 cells, and the reported values are the mean±s.e.m.
Traction forces were analyzed for MDCK KD, MDCK Rescued, DLD-1, R2/7, R2/7 Rescued, and R2/7 cells restored with GFP–α-catenin-ΔVBS (R2/7 ΔVBS). Transfected cells were identified by GFP fluorescence. Where relevant, only data from transfected cells were analyzed.
Confocal immunofluorescence imaging of paxillin at the basal plane was performed after a 6-h incubation of R2/7 and R2/7 Rescued cells with the E-cadherin modified gels. Cells were fixed with 4% w/v PFA for 15 min at room temperature, permeabilized with 0.1% Triton X-100 for 5 min, blocked for 20 min in 1% w/v BSA, and then stained with mouse anti-paxillin antibody (BD Transduction Laboratories). The secondary antibody was Alexa Fluor 647 goat anti-mouse (Invitrogen). F-actin was stained with Rhodamine–phalloidin (Invitrogen), in 1% w/v BSA for 1 h. For display purposes, brightness and contrast of all images were enhanced equally using ImageJ.
Cadherin immobilization on RBCs for kinetic analyses
RBCs were isolated from human whole blood collected from healthy consenting donors using Histopaque 1119 (Sigma) following manufacturer's instructions. The isolated RBCs were stored in EAS45 solution (2 mM adenine, 110 mM dextrose, 55 mM mannitol, 50 mM NaCl, 10 mM glutamine and 20 mM Na2HPO4, pH 8.0) (Dumaswala et al., 1996) at 4°C for up to 3 weeks.
Polyclonal goat anti-human IgG Fc-specific antibody (Sigma) was covalently coupled to RBCs via CrCl3 activation (Gold and Fudenberg, 1967; Kofler and Wick, 1977). Around 2×106 RBCs were washed with 0.85% w/v NaCl. Then, 250 µl of ∼0.01% w/v CrCl3 was used per reaction and mixed with an equal volume of RBC-antibody mixture. After 5 min, the reaction was stopped with 500 µl of ‘stop solution’ (5 mM EDTA in PBS with 1% w/v BSA). Approximately 100,000 RBCs were labeled by incubation with 4 µg E-cad-Fc in 100 µl of PBS buffer (5 mM EDTA, 1% w/v BSA), on an orbital shaker at 4°C. Binding probabilities were then determined between endogenous canine E-cadherin expressed on MDCK cells (MDCK Rescued or MDCK KD) and adjacent RBCs modified with immobilized, oriented canine E-cad-Fc.
Flow cytometry quantified the cadherin densities on the CHO and RBC surfaces (Chesla et al., 1998; Chien et al., 2008). Canine E-cadherin on MDCK cells was labeled with rat anti-E-cadherin (DECMA-1, Sigma), and then with goat anti-rat IgG-FITC. Approximately 100,000 cells were used for each sample, and 3 µg/ml antibody was used for each labeling step. The fluorescence intensities of labeled cells and bead standards (Bangs Laboratories, Fishers, IN) were quantified with an LSR II flow cytometer (BD Biosciences, San Jose, CA). Calibration curves relating the fluorescence intensity to total surface bound fluorophores were generated with fluorescent bead standards (Chien et al., 2008; Zhang et al., 2005).
Micropipette measurements of cell-binding kinetics
For the micropipette measurements (Chien et al., 2008; Langer et al., 2012), MDCK cells were brought repetitively into contact with an E-cad-Fc-coated RBC for specified times. Cells were maintained in L-15 medium with 1% w/v BSA. Cell–cell contact was observed with a Zeiss Axiovert 200M microscope equipped with a 100× oil objective lens (Zeiss). Adhesion events were identified from RBC deformations during cell separation and the recoil at bond rupture. The contact area was controlled at 4.6 µm2 (∼2.4 µm diameter). The binding probability P is the ratio of binding events (nb) to the total number of cell–cell touches (NT) (Chesla et al., 1998). Binding probabilities at each contact time represent ∼50 cell–cell touches measured with at least two cell pairs (n>100). The reported probabilities are the mean and standard deviation of the mean. The binding probability was plotted versus cell–cell contact times for up to 20 s, beyond which the probability does not change for at least 60 s (Chien et al., 2008).
Analysis of kinetic data
The kinetics of type I classical cadherin extracellular domains display a fast initial rise in the binding probability to P1, followed by a short lag, and a second rise to a limiting plateau at P2 (see Fig. 3B) (Chien et al., 2008). The fast initial step is due to binding between the N-terminal domains, but the lag and second rise involves the full-length extracellular domain (Chien et al., 2008). The first step is described by a trans-dimerization reaction.
Eqn 1 is the analytical expression for the time-dependent binding probability P(t) for the above reaction 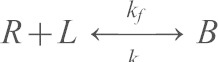 (Chien et al., 2008):
(Chien et al., 2008):
| (1) |
Here, mL and mR are the receptor and ligand surface densities (#/µm2) on the two cells, Ac is the contact area (µm2), Ka is the two-dimensional binding affinity (µm2), and kr is the dissociation rate (s−1). The ligand densities and contact areas (number per µm2) are known, and Ka and kr are determined from nonlinear least-squares fits of the first EC1-dependent binding step to Eqn 1.
A nonlinear lack-of-fit test parsed the data into the two distinct kinetic stages leading to plateaus P1 and P2 (Langer et al., 2012). This test compares the least squares residuals of the model (Eqn 1) to the intrinsic variability of the data. If the test statistic exceeds the critical value for a given time point, then the model does not describe the data in question. Nonlinear least squares fits (OriginLab, Northampton, MA) of the maximum number of time points in each data set that did not fail the lack-of-fit test to Eqn 1 determined the dissociation rate and two-dimensional affinity for the first EC1-dependent step.
Supplementary Material
Acknowledgments
We thank Saiko Rosenberger for technical assistance.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
A.K.B., N.S., I.M., and H.T. designed, conducted, and analyzed experiments. A.K.B. and H.T. wrote the paper; G.G. and A.Y. assisted with imaging studies; C.J.G. and J.d.R. generated stable cell lines; N.W. and D.E.L. designed experiments, analyzed data and wrote the paper.
Funding
This work was supported by the National Institutes of Health [grant numbers 5RO1 GM0974 to D.E.L. and N.W., 1RO1 GM076561 to C.J.G.]; a National Science Foundation [grant numbers CMMI 10-29871 to A.K.B. and I.M.]; a Beckman Institute Graduate Fellowship (to A.K.B.); an American Heart Association Predoctoral Fellowship [grant number 10PRE3840004 to A.K.B.]; and an NSF Integrative Graduate Education and Research Traineeship [grant number IGERT 0965918 to H.T.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.139014/-/DC1
References
- Bajpai S., Correia J., Feng Y., Figueiredo J., Sun S. X., Longmore G. D., Suriano G., Wirtz D. (2008). alpha-Catenin mediates initial E-cadherin-dependent cell-cell recognition and subsequent bond strengthening. Proc. Natl. Acad. Sci. USA 105, 18331–18336 10.1073/pnas.0806783105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo K. A., Wang Y. L. (2002). Flexible substrata for the detection of cellular traction forces. Trends Cell Biol. 12, 79–84 10.1016/S0962-8924(01)02205-X [DOI] [PubMed] [Google Scholar]
- Beningo K. A., Lo C. M., Wang Y. L. (2002). Flexible polyacrylamide substrata for the analysis of mechanical interactions at cell-substratum adhesions. Methods Cell Biol. 69, 325–339 10.1016/S0091-679X(02)69021-1 [DOI] [PubMed] [Google Scholar]
- Benjamin J. M., Kwiatkowski A. V., Yang C., Korobova F., Pokutta S., Svitkina T., Weis W. I., Nelson W. J. (2010). AlphaE-catenin regulates actin dynamics independently of cadherin-mediated cell-cell adhesion. J. Cell Biol. 189, 339–352 10.1083/jcb.200910041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga V. M., Del Maschio A., Machesky L., Dejana E. (1999). Regulation of cadherin function by Rho and Rac: modulation by junction maturation and cellular context. Mol. Biol. Cell 10, 9–22 10.1091/mbc.10.1.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunton V. G., MacPherson I. R., Frame M. C. (2004). Cell adhesion receptors, tyrosine kinases and actin modulators: a complex three-way circuitry. Biochim. Biophys. Acta 1692, 121–144 10.1016/j.bbamcr.2004.04.010 [DOI] [PubMed] [Google Scholar]
- Butler J. P., Tolić-Nørrelykke I. M., Fabry B., Fredberg J. J. (2002). Traction fields, moments, and strain energy that cells exert on their surroundings. Am. J. Physiol. 282, C595–C605 10.1152/ajpcell.00270.2001 [DOI] [PubMed] [Google Scholar]
- Calcutt R. B. (1983). Statistics for Analytical Chemistry New York, NY: Chapman and Hall [Google Scholar]
- Capaldo C. T., Macara I. G. (2007). Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol. Biol. Cell 18, 189–200 10.1091/mbc.E06-05-0471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavey M., Rauzi M., Lenne P. F., Lecuit T. (2008). A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature 453, 751–756 10.1038/nature06953 [DOI] [PubMed] [Google Scholar]
- Chesla S. E., Selvaraj P., Zhu C. (1998). Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 75, 1553–1572 10.1016/S0006-3495(98)74074-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y. H., Jiang N., Li F., Zhang F., Zhu C., Leckband D. (2008). Two stage cadherin kinetics require multiple extracellular domains but not the cytoplasmic region. J. Biol. Chem. 283, 1848–1856 10.1074/jbc.M708044200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D., Felsenfeld D. P., Sheetz M. P. (1997). Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell 88, 39–48 10.1016/S0092-8674(00)81856-5 [DOI] [PubMed] [Google Scholar]
- Desai R., Sarpal R., Ishiyama N., Pellikka M., Ikura M., Tepass U. (2013). Monomeric α-catenin links cadherin to the actin cytoskeleton. Nat. Cell Biol. 15, 261–273 10.1038/ncb2685 [DOI] [PubMed] [Google Scholar]
- Discher D. E., Janmey P., Wang Y. L. (2005). Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143 10.1126/science.1116995 [DOI] [PubMed] [Google Scholar]
- Diz-Muñoz A., Krieg M., Bergert M., Ibarlucea-Benitez I., Muller D. J., Paluch E., Heisenberg C. P. (2010). Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 8, e1000544 10.1371/journal.pbio.1000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek S. M., Garcia J. G. (2001). Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91, 1487–1500 [DOI] [PubMed] [Google Scholar]
- Dumaswala U. J., Wilson M. J., José T., Daleke D. L. (1996). Glutamine- and phosphate-containing hypotonic storage media better maintain erythrocyte membrane physical properties. Blood 88, 697–704 [PubMed] [Google Scholar]
- Engler A. J., Rehfeldt F., Sen S., Discher D. E. (2007). Microtissue elasticity: measurements by atomic force microscopy and its influence on cell differentiation. Methods Cell Biol. 83, 521–545 10.1016/S0091-679X(07)83022-6 [DOI] [PubMed] [Google Scholar]
- Evans E. A., Calderwood D. A. (2007). Forces and bond dynamics in cell adhesion. Science 316, 1148–1153 10.1126/science.1137592 [DOI] [PubMed] [Google Scholar]
- Geiger B., Spatz J. P., Bershadsky A. D. (2009). Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 10, 21–33 10.1038/nrm2593 [DOI] [PubMed] [Google Scholar]
- Gold E. R., Fudenberg H. H. (1967). Chromic chloride: a coupling reagent for passive hemagglutination reactions. J. Immunol. 99, 859–866 [PubMed] [Google Scholar]
- Gumbiner B. M. (2005). Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6, 622–634 10.1038/nrm1699 [DOI] [PubMed] [Google Scholar]
- Gumbiner B. M., McCrea P. D. (1993). Catenins as mediators of the cytoplasmic functions of cadherins. J. Cell Sci. 1993 Suppl. 17, 155–158 10.1242/jcs.1993.Supplement_17.22 [DOI] [PubMed] [Google Scholar]
- Guo W. H., Frey M. T., Burnham N. A., Wang Y. L. (2006). Substrate rigidity regulates the formation and maintenance of tissues. Biophys. J. 90, 2213–2220 10.1529/biophysj.105.070144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S., Oldenburg J., Spanjaard E., van der Krogt G., Grigoriev I., Akhmanova A., Rehmann H., de Rooij J. (2012). Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 196, 641–652 10.1083/jcb.201108120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icard-Arcizet D., Cardoso O., Richert A., Hénon S. (2008). Cell stiffening in response to external stress is correlated to actin recruitment. Biophys. J. 94, 2906–2913 10.1529/biophysj.107.118265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura Y., Itoh M., Maeno Y., Tsukita S., Nagafuchi A. (1999). Functional domains of alpha-catenin required for the strong state of cadherin-based cell adhesion. J. Cell Biol. 144, 1311–1322 10.1083/jcb.144.6.1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasza K. E., Zallen J. A. (2011). Dynamics and regulation of contractile actin-myosin networks in morphogenesis. Curr. Opin. Cell Biol. 23, 30–38 10.1016/j.ceb.2010.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen K. A., Soler A. P., Johnson K. R., Wheelock M. J. (1995). Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J. Cell Biol. 130, 67–77 10.1083/jcb.130.1.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak A., Pasolli H. A., Fuchs E. (2004). Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat. Cell Biol. 6, 21–30 10.1038/ncb1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R., Wick G. (1977). Some methodologic aspects of the chromium chloride method for coupling antigen to erythrocytes. J. Immunol. Methods 16, 201–209 10.1016/0022-1759(77)90198-3 [DOI] [PubMed] [Google Scholar]
- Krieg M., Arboleda-Estudillo Y., Puech P. H., Käfer J., Graner F., Müller D. J., Heisenberg C. P. (2008). Tensile forces govern germ-layer organization in zebrafish. Nat. Cell Biol. 10, 429–436 10.1038/ncb1705 [DOI] [PubMed] [Google Scholar]
- Ladoux B., Anon E., Lambert M., Rabodzey A., Hersen P., Buguin A., Silberzan P., Mège R. M. (2010). Strength dependence of cadherin-mediated adhesions. Biophys. J. 98, 534–542 10.1016/j.bpj.2009.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer M. D., Guo H. B., Shashikanth N., Pierce M., Leckband D. (2012). N-Glycosylation alters the dynamics of N-cadherin junction assembly. J. Cell Sci. 125, 2478–2485 [DOI] [PubMed] [Google Scholar]
- le Duc Q., Shi Q., Blonk I., Sonnenberg A., Wang N., Leckband D., de Rooij J. (2010). Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 189, 1107–1115 10.1083/jcb.201001149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit T., Le Goff L. (2007). Orchestrating size and shape during morphogenesis. Nature 450, 189–192 10.1038/nature06304 [DOI] [PubMed] [Google Scholar]
- Lecuit T., Lenne P. F. (2007). Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Natl. Rev. Mol. Cell Biol. 8, 633–644 10.1038/nrm2222 [DOI] [PubMed] [Google Scholar]
- Lecuit T., Lenne P. F., Munro E. (2011). Force generation, transmission, and integration during cell and tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 27, 157–184 10.1146/annurev-cellbio-100109-104027 [DOI] [PubMed] [Google Scholar]
- Liu Z., Tan J. L., Cohen D. M., Yang M. T., Sniadecki N. J., Ruiz S. A., Nelson C. M., Chen C. S. (2010). Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 107, 9944–9949 10.1073/pnas.0914547107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maître J. L., Heisenberg C. P. (2011). The role of adhesion energy in controlling cell-cell contacts. Curr. Opin. Cell Biol. 23, 508–514 10.1016/j.ceb.2011.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maître J. L., Berthoumieux H., Krens S. F., Salbreux G., Jülicher F., Paluch E., Heisenberg C. P. (2012). Adhesion functions in cell sorting by mechanically coupling the cortices of adhering cells. Science 338, 253–256 10.1126/science.1225399 [DOI] [PubMed] [Google Scholar]
- Maruthamuthu V., Sabass B., Schwarz U. S., Gardel M. L. (2011). Cell-ECM traction force modulates endogenous tension at cell-cell contacts. Proc. Natl. Acad. Sci. USA 108, 4708–4713 10.1073/pnas.1011123108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan R. W., Yap A. S. (2007). Not so simple: the complexity of phosphotyrosine signaling at cadherin adhesive contacts. J. Mol. Med. 85, 545–554 10.1007/s00109-007-0198-x [DOI] [PubMed] [Google Scholar]
- Meng W., Mushika Y., Ichii T., Takeichi M. (2008). Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell 135, 948–959 10.1016/j.cell.2008.09.040 [DOI] [PubMed] [Google Scholar]
- Miyake Y., Inoue N., Nishimura K., Kinoshita N., Hosoya H., Yonemura S. (2006). Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp. Cell Res. 312, 1637–1650 10.1016/j.yexcr.2006.01.031 [DOI] [PubMed] [Google Scholar]
- Paluch E., Heisenberg C. P. (2009). Chaos begets order: asynchronous cell contractions drive epithelial morphogenesis. Dev. Cell 16, 4–6 10.1016/j.devcel.2008.12.011 [DOI] [PubMed] [Google Scholar]
- Papusheva E., Heisenberg C. P. (2010). Spatial organization of adhesion: force-dependent regulation and function in tissue morphogenesis. EMBO J. 29, 2753–2768 10.1038/emboj.2010.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham R. J., Jr, Wang Y. (1997). Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 94, 13661–13665 10.1073/pnas.94.25.13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X., Cuff L. E., Lawton C. D., DeMali K. A. (2010). Vinculin regulates cell-surface E-cadherin expression by binding to beta-catenin. J. Cell Sci. 123, 567–577 10.1242/jcs.056432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova Y. I., Spano M. M., Gumbiner B. M. (2012). Conformational epitopes at cadherin calcium-binding sites and p120-catenin phosphorylation regulate cell adhesion. Mol. Biol. Cell 23, 2092–2108 10.1091/mbc.E11-12-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S., Drees F., Takai Y., Nelson W. J., Weis W. I. (2002). Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J. Biol. Chem. 277, 18868–18874 10.1074/jbc.M201463200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakasam A. K., Maruthamuthu V., Leckband D. E. (2006). Similarities between heterophilic and homophilic cadherin adhesion. Proc. Natl. Acad. Sci. USA 103, 15434–15439 10.1073/pnas.0606701103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost E., Rimm D. L. (1999). Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr. Opin. Cell Biol. 11, 567–572 10.1016/S0955-0674(99)00015-0 [DOI] [PubMed] [Google Scholar]
- Rauzi M., Verant P., Lecuit T., Lenne P. F. (2008). Nature and anisotropy of cortical forces orienting Drosophila tissue morphogenesis. Nat. Cell Biol. 10, 1401–1410 10.1038/ncb1798 [DOI] [PubMed] [Google Scholar]
- Schwartz M. A., DeSimone D. W. (2008). Cell adhesion receptors in mechanotransduction. Curr. Opin. Cell Biol. 20, 551–556 10.1016/j.ceb.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q. (2009). Cadherin specificity in adhesion and mechanotransduction. Chemical Engineering. PhD thesis University of Illinois: Urbana, IL, USA [Google Scholar]
- Tabdili H., Langer M., Shi Q., Poh Y-C., Wang N., Leckband D. (2012). Cadherin-dependent mechanotransduction depends on ligand identity but not affinity. J. Cell Sci. 125, 4362–4371 10.1242/jcs.105775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. (1991). Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455 10.1126/science.2006419 [DOI] [PubMed] [Google Scholar]
- Takeichi M., Nakagawa S. (2001). Cadherin-dependent cell-cell adhesion. Curr. Protoc. Cell Biol. 9, Unit 9.3 [DOI] [PubMed] [Google Scholar]
- Thomas W. A., Boscher C., Chu Y. S., Cuvelier D., Martinez-Rico C., Seddiki R., Heysch J., Ladoux B., Thiery J. P., Mege R. M. et al. (2013). α-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 288, 4957–4969 10.1074/jbc.M112.403774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trichet L., Le Digabel J., Hawkins R. J., Vedula S. R., Gupta M., Ribrault C., Hersen P., Voituriez R., Ladoux B. (2012). Evidence of a large-scale mechanosensing mechanism for cellular adaptation to substrate stiffness. Proc. Natl. Acad. Sci. USA 109, 6933–6938 10.1073/pnas.1117810109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse J. R., Engler A. J. (2010). Preparation of hydrogel substrates with tunable mechanical properties. Curr. Protoc. Cell Biol Chapter 10, Unit 10.16 10.1002/0471143030.cb1016s47 [DOI] [PubMed] [Google Scholar]
- Twiss F., le Duc Q., Van Der Horst S., Tabdili H., Van Der Krogt G., Wang N., Rehmann H., Huveneers S., Leckband D., de Rooij J. (2012). Vinculin-dependent cadherin mechanosensing regulates efficient epithelial barrier formation. Biol. Open. 1, 1128–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E., Irani-Tehrani M., Kiosses W. B., Dejana E., Schultz D. A., Engelhardt B., Cao G., DeLisser H., Schwartz M. A. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431 10.1038/nature03952 [DOI] [PubMed] [Google Scholar]
- Vogel V., Sheetz M. (2006). Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 7, 265–275 10.1038/nrm1890 [DOI] [PubMed] [Google Scholar]
- Wang N., Butler J. P., Ingber D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124–1127 10.1126/science.7684161 [DOI] [PubMed] [Google Scholar]
- Wang H. B., Dembo M., Hanks S. K., Wang Y. (2001). Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA 98, 11295–11300 10.1073/pnas.201201198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschke J., Baumgartner W., Adamson R. H., Zeng M., Aktories K., Barth H., Wilde C., Curry F. E., Drenckhahn D. (2004). Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am. J. Physiol. 286, H394–H401 10.1152/ajpheart.00221.2003 [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M., Uchida N., Imamura Y., Nagafuchi A., Fujimoto K., Uemura T., Vermeulen S., van Roy F., Adamson E. D., Takeichi M. (1998). alpha-Catenin-vinculin interaction functions to organize the apical junctional complex in epithelial cells. J. Cell Biol. 142, 847–857 10.1083/jcb.142.3.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. F., Bjerke M. A., DeSimone D. W. (2012). A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev. Cell 22, 104–115 10.1016/j.devcel.2011.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S., Wada Y., Watanabe T., Nagafuchi A., Shibata M. (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12, 533–542 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
- Zhang F., Marcus W. D., Goyal N. H., Selvaraj P., Springer T. A., Zhu C. (2005). Two-dimensional kinetics regulation of alphaLbeta2-ICAM-1 interaction by conformational changes of the alphaL-inserted domain. J. Biol. Chem. 280, 42207–42218 10.1074/jbc.M510407200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.