

ABSTRACT
The permeability transition pore (PT-pore) mediates cell death through the dissipation of the mitochondrial membrane potential (ΔΨm). Because the exact composition of the PT-pore is controversial, it is crucial to investigate the actual molecular constituents and regulators of this complex. We found that mitochondrial creatine kinase-1 (CKMT1) is a universal and functionally necessary gatekeeper of the PT-pore, as its depletion induces mitochondrial depolarization and apoptotic cell death. This can be inhibited efficiently by bongkrekic acid, a compound that is widely used to inhibit the PT-pore. However, when the ‘classical’ PT-pore subunits cyclophilin D and VDAC1 are pharmacologically inhibited or their expression levels reduced, mitochondrial depolarization by CKMT1 depletion remains unaffected. At later stages of drug-induced apoptosis, CKMT1 levels are reduced, suggesting that CKMT1 downregulation acts to reinforce the commitment of cells to apoptosis. A novel high-molecular-mass CKMT1 complex that is distinct from the known CKMT1 octamer disintegrates upon treatment with cytotoxic drugs, concomitant with mitochondrial depolarization. Our study provides evidence that CKMT1 is a key regulator of the PT-pore through a complex that is distinct from the classical PT-pore.
KEY WORDS: Mitochondrial membrane potential, Creatine kinase, Apoptosis, Permeability transition pore
INTRODUCTION
The dissipation of the mitochondrial membrane potential (ΔΨm) is a crucial event in the intrinsic apoptosis pathway, and it is caused by a sudden increase in the inner mitochondrial membrane (IMM) permeability, known as the mitochondrial permeability transition (MPT) (Zamzami et al., 2005; Zamzami et al., 1995). This is thought to be mediated by a protein complex known as the permeability transition pore (PT-pore), which spans the outer mitochondrial membrane (OMM) and the IMM at contact sites (Brdiczka, 1991; Crompton, 2000), and which is converted into an unspecific channel to allow IMM permeabilization. These changes result in the loss of ΔΨm, a distorted balance of osmotic pressure and the swelling of the mitochondrial matrix (Kroemer and Reed, 2000; Susin et al., 1998). The concomitant unfolding of the IMM induces the rupture of the OMM, causing the release of intermembrane space (IMS) proteins such as cytochrome c and AIF, finally leading to the induction of apoptosis (Zamzami and Kroemer, 2001).
The PT-pore is involved in numerous scenarios of cell death, in particular in pathological conditions, such as ischemia-reperfusion injury (Crompton, 1999). By contrast, its inhibition seems to contribute to tumorigenesis (Brenner and Grimm, 2006). It is believed that the core PT-pore complex consists of voltage-dependent anion channel (VDAC, an OMM protein), adenine nucleotide translocator (ANT, which spans the IMM), and cyclophilin D (CypD, also known as PPIF, which binds to ANT in the matrix) (Crompton et al., 1998). In addition, hexokinase-II (HKII, also known as HK2) and peripheral benzodiazepine receptor (PBR, also known as TSPO) associate with the OMM, and mitochondrial creatine kinase1 (CKMT1) is localized in the IMS. HKII, PBR and CKMT1 have all been reported to interact with the PT-pore complex (Verrier et al., 2004).
Importantly, the exact composition of the PT-pore is still a subject of scientific debate. Knockout studies have demonstrated that cells that are deficient in ANT1 (also known as SLC25A4) and ANT2 (also known as SLC25A5) are still able to induce MPT in response to increased concentrations of Ca2+, and these cells show only minor alterations in the induction of cell death (Kokoszka et al., 2004). Also, it has been claimed that VDACs are dispensable for mitochondrial-dependent cell death: mitochondria from mice that are null for VDAC1 or VDAC3 and from cells in which VDAC2 has been knocked down exhibit a Ca2+- and oxidative-stress-induced MPT that is indistinguishable from that of wild-type mitochondria (Baines et al., 2007). The involvement of CypD in PT-pore regulation has been studied in mitochondria from CypD knockout mice, which were found to be resistant to mitochondrial swelling caused by MPT, even though PT-pore opening could still be observed when a stronger stimulus, such as higher Ca2+ concentrations, was applied (Baines et al., 2005; Basso et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005). These studies corroborated the crucial role of CypD in MPT, but failed to prove its unequivocal involvement in apoptosis regulation: Nakagawa et al. claim that CypD is only involved in necrotic cell death, whereas Baines et al. show that CypD-deficient cells also show an altered apoptotic response (Baines et al., 2005; Nakagawa et al., 2005).
CKMT1 is a protein that has consistently been found as a PT-pore component. In vitro reconstituted complexes containing CKMT1, ANT and VDAC have been shown to display many features of the PT-pore, such as Ca2+-dependent pore opening and release of intravesicular contents (Beutner et al., 1998; Beutner et al., 1996). CKMT1 is believed to induce the formation of contact sites between the OMM and IMM by binding to both membranes, as demonstrated by resistance against detergent-induced lysis (Speer et al., 2005).
Considering the controversial data on the validation of the PT-pore subunits, it is crucial to investigate the actual molecular constituents and the regulators of the PT-pore. Because numerous previous studies suggest that CKMT1 is involved in the regulation of mitochondrial apoptosis through PT-pore regulation, we addressed the role of CKMT1 by downregulating the protein. This resulted in MPT and commitment to apoptosis, which we found is mediated by a complex that is different from the classical PT-pore.
RESULTS
Depletion of CKMT1 results in MPT
In order to address the function of CKMT1, we first made use of ASB9 (ankyrin repeat and suppressor of cytokine signaling box protein 9), which has recently been shown to interact with and induce the ubiquitylation of CKMT1 (Kwon et al., 2010). We hypothesized that ASB9 overexpression would mediate ubiquitylation and proteasomal degradation of CKMT1. Indeed, ASB9 transfection resulted in an upshift of CKMT1 complexes in a blue native gel at 24 h post transfection, indicative of CKMT1 polyubiquitylation (Fig. 1A). ASB9 overexpression caused the downregulation of CKMT1 protein levels after 48 h and 72 h (Fig. 1B). This was concomitant with the dissipation of ΔΨm and the induction of apoptotic cell death (Fig. 1C,D). ASB9 was able to cause caspase 3 and Bax activation as well as annexin-V-positive staining in transfected cells (Fig. 1E,F,G). The co-transfection of wild-type (WT) CKMT1 failed to reduce cell death, probably because the WT CKMT1 was still efficiently ubiquitylated (supplementary material Fig. S1A,B), and transfection of the ASB9-interaction-deficient mutant CKMT1ΔBS (Kwon et al., 2010) induced apoptosis (supplementary material Fig. S1C,D). As an additional and more specific tool to target CKMT1, we employed siRNA-mediated knockdown. The transfection efficiency, as assayed by measuring the uptake of Alexa-Fluor-647-labeled siRNA, proved to be comparable in the siCK1- and control-transfected Hela cells, reaching ∼85% (data not shown). We validated the depletion of endogenous CKMT1 on the mRNA level by using semi-quantitative reverse transcription (RT)-PCR for up to 72 h post transfection (Fig. 2A). CKMT1 protein expression started to be reduced by 48 h post transfection, and it further decreased after 72 h and 96 h (Fig. 2B). From 48 h post transfection onwards, we also detected cleavage of PARP and activation of caspase 3, two general signs of apoptosis (Fig. 2B). Because we initially assumed that this effect is mediated by the PT-pore, a complex that has often been implicated in necrosis (Crompton, 1999), we investigated additional features of apoptosis. Indications of this type of cell death could be observed upon CKMT1 knockdown from 48 h post siRNA transfection, by using subG1-G0 analysis and annexin-V and propidium-iodide (PI) staining. At 96 h after siRNA transfection, ∼60–70% of the cells showed DNA fragmentation or externalization of phosphatidylserine, compared with ∼10% in the control population (Fig. 2C,D). Necrosis, as indicated by cells that were positive for PI only, was absent. Cells with apoptotic morphology (reduced volume and membrane blebbing) were observed from 48 h post transfection onwards (data not shown). Confocal immunofluorescence microscopy revealed the presence of cleaved caspase 3 and activated Bax in cells harboring CKMT1 siRNA (data not shown). Immunofluorescence staining of activated caspase 3 and Bax were quantified using FACS analysis, which confirmed significant activation of both proteins upon knockdown of CKMT1 (Fig. 2E). The apoptotic cell death upon CKMT1 downregulation was dependent on caspase activity, as demonstrated by the inhibition of cell death in the presence of the pan-caspase inhibitor Z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone, referred to here as zVAD) (Fig. 2F).
Fig. 1.
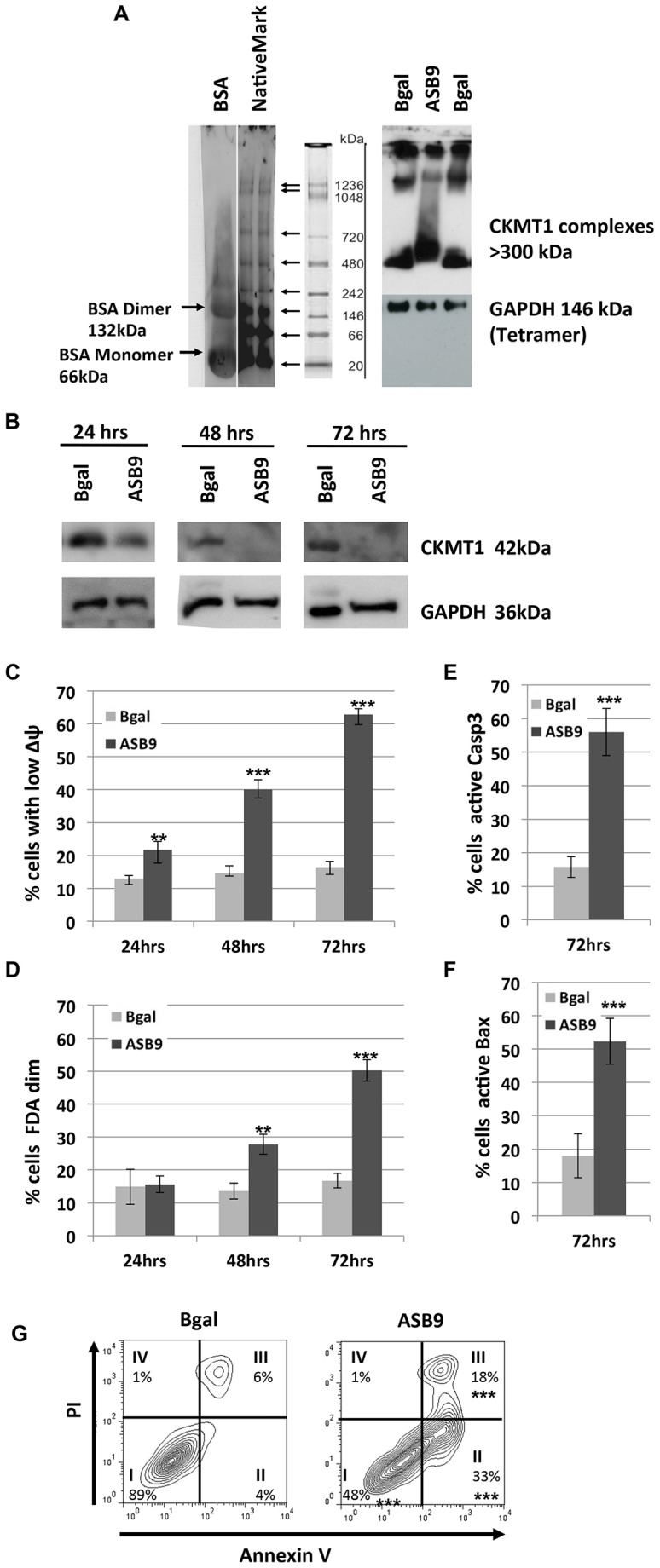
ASB9-mediated ubiquitylation and degradation of CKMT1 coincides with loss of ΔΨm and induction of apoptosis. (A) Analysis of endogenous CKMT1 complexes using native blue gel electrophoresis and immunoblotting. BSA and NativeMark (Invitrogen) markers were visualized by using Coomassie staining and were used as molecular-mass markers ranging from 20 kDa to 1236 kDa. High-molecular-mass CKMT1 complexes were detected by using an anti-CKMT1 antibody for immunoblotting. At 24 h post transfection, ASB9 transfection results in an upshift in the smear of CKMT1 complexes, suggesting polyubiquitylation. (B) ASB9 overexpression results in CKMT1 protein degradation, as shown by western blot analysis for CKMT1 expression at 24 h, 48 h and 72 h post transfection. (C) ASB9 overexpression results in the collapse of the ΔΨm, as analyzed by DiOC6 staining and subsequent FACS. (D) ASB9 overexpression results in cell death, as shown by the fluorescein diacetate (FDA) assay for enzymatic hydrolysis of FDA. (E,F) Activation of caspase 3 and Bax was assayed by immunostaining for cleaved caspase 3 (E) and activated Bax (F) and subsequent FACS analysis using Alexa-Fluor-488- and Alexa-Fluor-633-conjugated secondary antibodies, respectively. Data show the mean±s.d. **P<0.01; ***P<0.001 (Student's t-test). (G) PI and annexin V co-staining and subsequent FACS analysis at 72 h post transfection with ASB9. The four quadrants represent (I) healthy viable cells, (II) early apoptotic cells, (III) late apoptotic cells and (IV) necrotic cells. Representative FACS density blots are shown, with the percentages representing the means of three independent experiments. ***P<0.001 (Student's t-test).
Fig. 2.
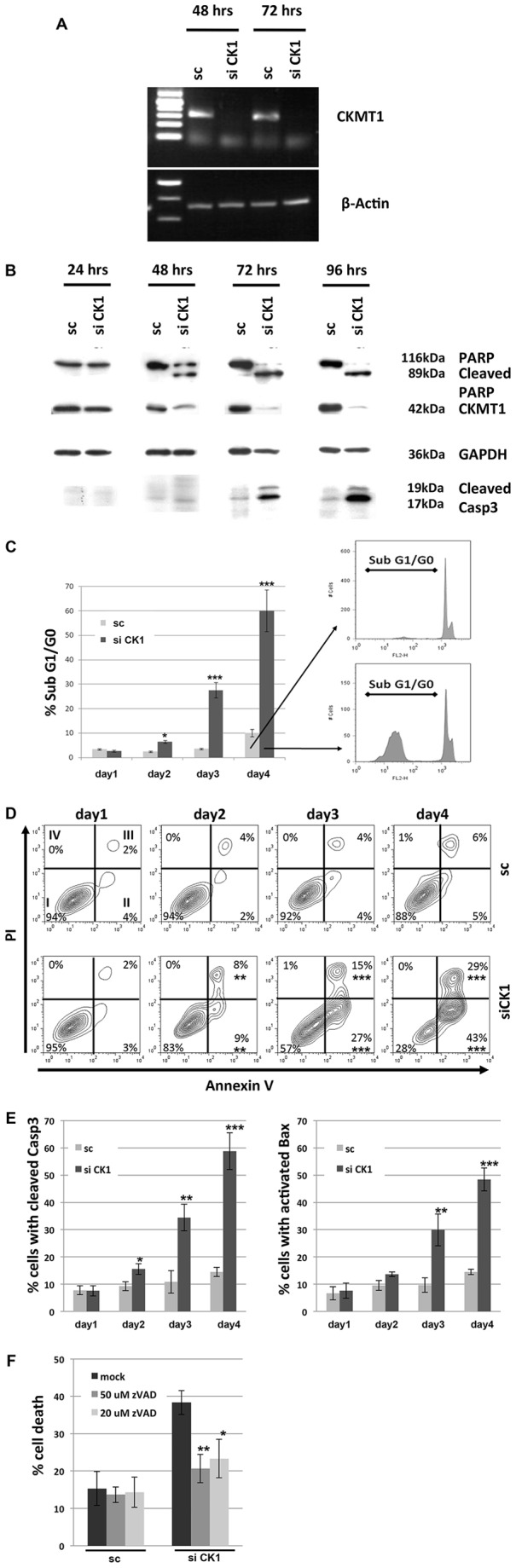
Knockdown of endogenous CKMT1 results in apoptosis. (A) Semi-quantitative RT-PCR at 48 h and 72 h post siRNA transfection using primers specific for human CKMT1 and β-actin in HeLa cells. (B) Western blot analysis for CKMT1 expression. HeLa cells were transfected with siRNA against CKMT1 (siCK1) or with a scrambled control (sc) sequence. PARP cleavage and activation of caspase 3 were observed after siRNA transfection in HeLa cells. The antibody against caspase 3 recognises only the cleaved product. (C) PI staining and subsequent subG1-G0 analysis by FACS. Representative FACS profiles and subG1-G0 gating are shown on the right. Data show the mean±s.d. of three independent experiments. (D) PI and annexin V co-staining and subsequent FACS analysis post transfection with siCK1. Representative FACS density blots are shown, with the percentages representing the means of three independent experiments. (E) Quantification of caspase 3 and Bax activation by immunostaining and subsequent FACS analysis using Alexa-Fluor-488- and Alexa-Fluor-633-conjugated secondary antibodies monitored over 4 days post transfection. (F) Inhibition of the induction of cell death that is mediated by siCK1 by treatment of cells with the pan-caspase inhibitor. HeLa cells were transfected with siCK1 or sc, and treated with the indicated concentration of zVAD at 24 h and 48 h post transfection. Cell death was assessed by PI exclusion staining and subsequent FACS analysis. Data show the mean±s.d. of three independent experiments. *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
In order to assess whether CKMT1 knockdown resulted in the dissipation of ΔΨm, live-cell confocal microscopy and FACS analysis using JC1 dye as an indicator of ΔΨm were performed. At 96 h post transfection, knockdown of CKMT1 led to mitochondrial depolarization, whereas control transfected cells maintained their ΔΨm (Fig. 3A,B). To ascertain that the dissipation of ΔΨm was not a secondary event after induction of apoptosis through a mitochondria-independent pathway, we monitored ΔΨm by using DiOC6 (3,3′-dihexyloxacarbocyanine iodide) and FACS analysis at various time points (24–96 h post transfection, Fig. 3C). Knockdown of endogenous CKMT1 resulted in the depolarization of mitochondria, the onset of which coincided with the disappearance of the CKMT1 protein (compare with Fig. 2B). We also found caspase 9 activation when CKMT1 was targeted (Fig. 3D). Inhibiting caspases with zVAD had no significant effect on the collapse of ΔΨm, suggesting that the initial events in cell death are mitochondrial changes (Fig. 3E). Dissipation of ΔΨm and the induction of apoptosis upon depletion of endogenous CKMT1 was found in numerous cell lines, including HeLa, 293T, MCF7, HCT116, MDA231 and LNCaP cells, and was also observed in primary human HFF1 cells, as well as in mouse N2A and MLE12 cells, as listed in supplementary material Fig. S2A–E. This indicates that the induction of cell death upon knockdown of CKMT1 is a general effect.
Fig. 3.

Downregulation of CKMT1 results in ΔΨm dissipation and caspase 9 activation. (A) Confocal live-cell imaging using JC1 as an indicator of mitochondrial membrane polarization at 96 h post transfection with siRNA against CKMT1 (siCK1) in HeLa cells. Note that 4′,6-diamidino-2-phenylindole (DAPI) is not cell permeable in viable cells, whereas it enters apoptotic cells. Mon, monomer; agg, aggregate. (B) FACS analysis of mitochondrial membrane polarization using JC1 at 96 h post transfection with siCK1 or a scrambled control sequence (sc). Representative ratiometric contour plots are shown. (C) Kinetics of mitochondrial depolarization after the downregulation of CK1 by siRNA, as monitored by DiOC6 and PI co-staining and subsequent FACS analysis. Representative multiparametric contour plots are shown on the right. (D) Caspase 9 activation was assessed by live-cell staining with CaspaTag9 and subsequent FACS analysis. (E) HeLa cells were transfected with siCK1 or sc and treated with the indicated concentrations of zVAD at 24 h and 48 h post transfection. The ΔΨm was assessed by using DiOC6 staining and subsequent FACS analysis. Data show the mean±s.d. of three independent experiments. *P<0.05; **P<0.01; ***P<0.001; ns, not significant (Student's t-test).
MPT that is induced by knockdown of CKMT1 can be inhibited by bongkrekic acid but is independent of cyclophilin D and VDAC1
We hypothesized that the dissipation of ΔΨm that was observable after siRNA-mediated knockdown of CKMT1 was due to the activation of the PT-pore complex and that, consequently, pharmacological inhibition of the PT-pore would counteract mitochondrial depolarization and the induction of apoptosis. In order to address this question, we used bongkrekic acid, a compound that has been widely used to inhibit the PT-pore (Klingenberg et al., 1970; Marchetti et al., 1996). Both the MPT and the apoptotic cell death observed after knockdown of CKMT1 could be completely abrogated by the treatment of cells with bongkrekic acid (Fig. 4A,B). It has been postulated that CypD is a key regulator or component of the PT-pore. Both overexpression and downregulation of CypD can repress mitochondrial apoptosis (Kato et al., 2009; Li et al., 2004; Lin and Lechleiter, 2002; Schubert and Grimm, 2004; Zhang and Armstrong, 2007). Thus, we investigated whether apoptosis that was induced by depletion of endogenous CKMT1 could be modulated by siRNA-mediated downregulation of CypD. Reduction of CypD expression, in either a transient or a stable manner, did not impact on siCK1-induced ΔΨm dissipation (Fig. 5A). Likewise, MPT by depletion of endogenous CKMT1 could not be inhibited by overexpression of CypD, whereas ANT1-induced ΔΨm dissipation was significantly reduced (Fig. 5A). We also investigated ΔΨm dissipation by siCK1 under pharmacological inhibition of CypD by treatment with cyclosporin A, sanglifehrin A, and compound BC544 (Moss, 2011). However, none of these CypD inhibitors had any effect on siCK1-induced mitochondrial membrane depolarization (Fig. 5B). Because these inhibitors might not be efficient or specific enough, and because CypD RNAi downregulation was not complete (Fig. 5A), we hypothesized that the remaining CypD could have obscured any changes in MPT. Consequently, we examined CypD knockout cells (SV40-immortalized murine adult fibroblasts) for its involvement in siCK1-mediated MPT. Various short hairpin (sh)RNA constructs targeting the murine CKMT1 (mCKMT1) coding sequence were tested for their ability to downregulate mCKMT1. Six different constructs, shHUSH14, shHUSH15, shmCK1, TRC25, TRC27 and TRC703 showed downregulation of mCKMT1 and ΔΨm collapse in mouse N2A cells (data not shown). The RNAi consortium (TRC) viral constructs were used to infect murine MLE12 cells and likewise elicited cell death (data not shown). WT and CypD−/− cells were investigated for their response to the knockdown of CKMT1 (Fig. 5C,D). The CypD expression status did not have any effect on the loss of the ΔΨm or on the induction of apoptosis after downregulation of mCKMT1 (Fig. 5E,F; data not shown). By contrast, CypD−/− cells were more resistant against other apoptotic stimuli that induce MPT through a mechanism involving the PT-pore, including ANT1 overexpression, ionomycin, H2O2, As2O3 and menadione (Bauer et al., 1999; Takeyama et al., 2002; Tazawa et al., 2009) (Fig. 5E,F; data not shown). It has been suggested that CKMT1 and VDAC1 are constituents of the PT-pore (Beutner et al., 1998), and they have been shown to directly interact (Schlattner et al., 2001). Therefore, we speculated that this interaction regulates apoptosis. By using co-immunoprecipitation experiments, we found that endogenous CKMT1 and VDAC1 interacted in untreated cells, and that this interaction remained stable upon induction of apoptosis by various concentrations of doxorubicin and As2O3 (Fig. 6A; data not shown). To test whether VDAC1 functioned in CKMT1-downregulation-mediated cell death, we knocked down endogenous VDAC1 using RNAi with two constructs, and assessed mitochondrial depolarization and apoptosis upon depletion of endogenous CKMT1. A reduction in the level of VDAC1 did not significantly impact on the collapse of the ΔΨm or the number of annexin-V-positive cells upon CKMT1 depletion (Fig. 6B–C). Additionally, pharmacological inhibition of VDACs by 4,4′-diisothiocyanatostilbene-2,2′-disulfonate (DIDS) also did not inhibit siCK1-induced ΔΨm dissipation and apoptosis, although it could significantly reduce the ΔΨm loss and apoptosis induced by VDAC1 overexpression (Fig. 6E,F). This indicated that VDAC1 function is dispensable for the induction of apoptosis by siCK1, and that the interaction of VDAC1 and CKMT1 is not relevant for the regulation of apoptosis under the treatment conditions tested.
Fig. 4.
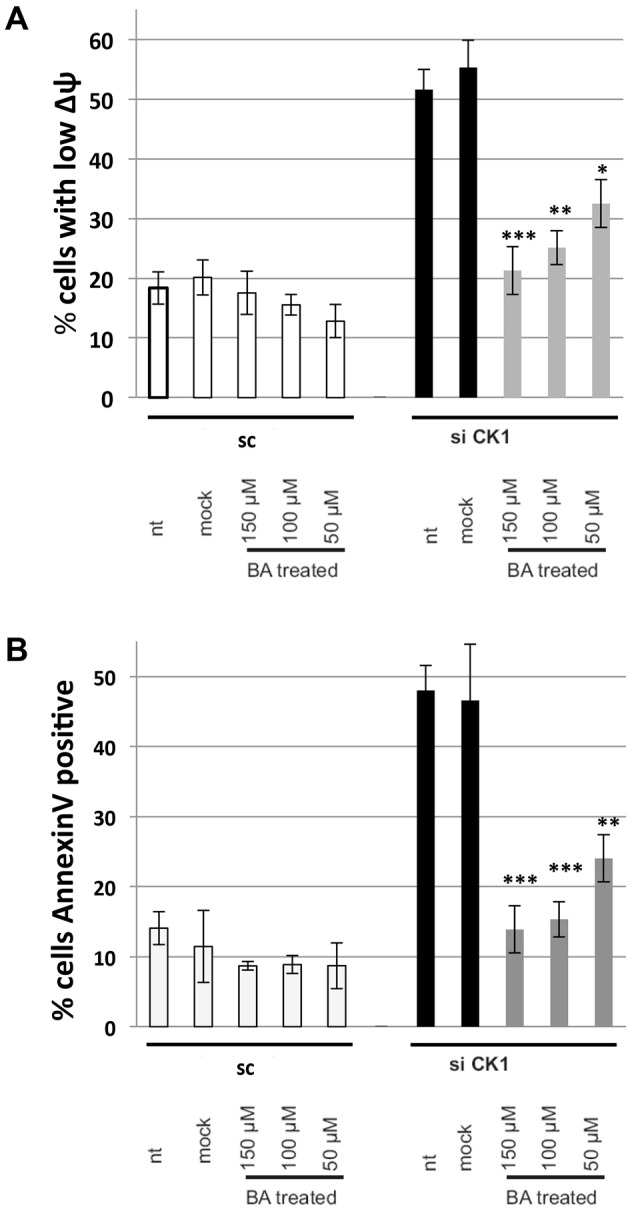
Bongkrekic acid inhibits MPT induced by downregulation of endogenous CKMT1. HeLa cells were transfected with siRNA against CKMT1 (siCK1) or a scrambled sequence (sc), and were treated with the indicated amount of bongkrekic acid (BA) after 24 h. (A) MPT was assessed by using DiOC6 and PI co-staining, and FACS analysis at 72 h post transfection. (B) Apoptosis was assessed by annexin-V staining and subsequent FACS analysis. nt, non-treated. The data show the mean±s.d. *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
Fig. 5.
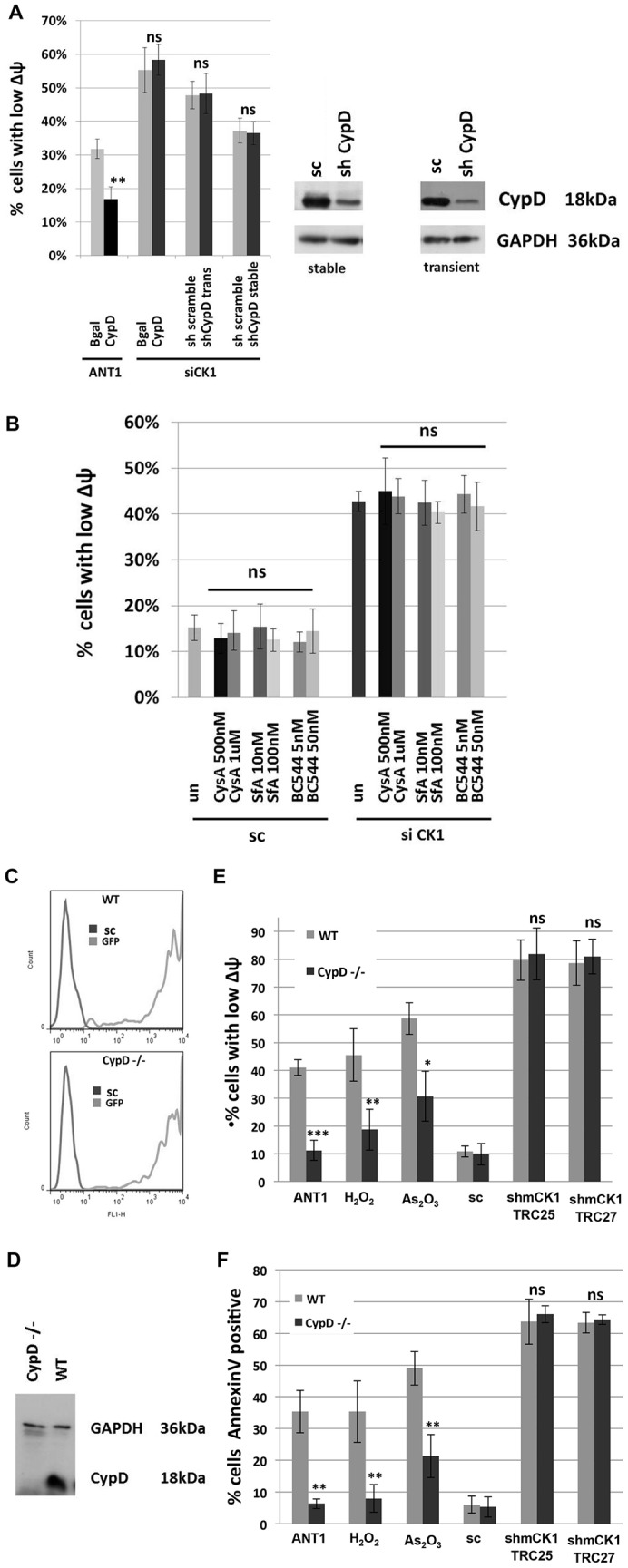
Induction of apoptosis caused by downregulation of CKMT1 is independent of CypD. (A) The overexpression of CypD significantly inhibits ANT1-induced apoptosis, whereas apoptosis induced by siRNA against CKMT1 (siCK1) remains unchanged. Equally, neither transient nor stable downregulation of CypD has any effect on apoptosis induced by the depletion of endogenous CKMT1. The histograms on the left show the assessment of the ΔΨm after transfection with siCK1 combined with overexpression or downregulation of CypD. The endogenous expression level of CypD was assessed by western blotting (right panel). sc, scrambled control sequence. (B) Pharmacological inhibition of CypD activity by cyclosporin A (CysA), sanglifehrin A (SfA) and the compound BC544 do not affect siCK1-induced apoptosis. Cells were transfected with the indicated siRNA constructs and treated with inhibitors at 20 h post transfection. The ΔΨm was assessed by DiOC6 staining and subsequent FACS analysis at 72 h post transfection. un, untreated. (C) GFP expression as assessed by FACS Fl1 analysis in WT and CypD−/− cells at 48 h after infection with viral particles encoding GFP. (D) The absence of CypD expression in CypD−/− cells was verified by using western blotting. (E,F) Analysis of the ΔΨm by DiOC6 staining and apoptosis by annexin-V staining and subsequent FACS analysis. CypD−/− cells are resistant to ΔΨm dissipation (E) as well as to apoptosis (F) induced by ANT1 overexpression, H2O2 (500 µM for 24 h) and As2O3 (12 µM for 24 h), whereas the CypD expression status does not have any impact on apoptosis induction by viral particles encoding shmCK1 TRC25 and TRC27. The data were acquired at 6 days post infection. The data show the mean±s.d. *P<0.05; **P<0.01; ***P<0.001; ns, non-significant (Student's t-test).
Fig. 6.
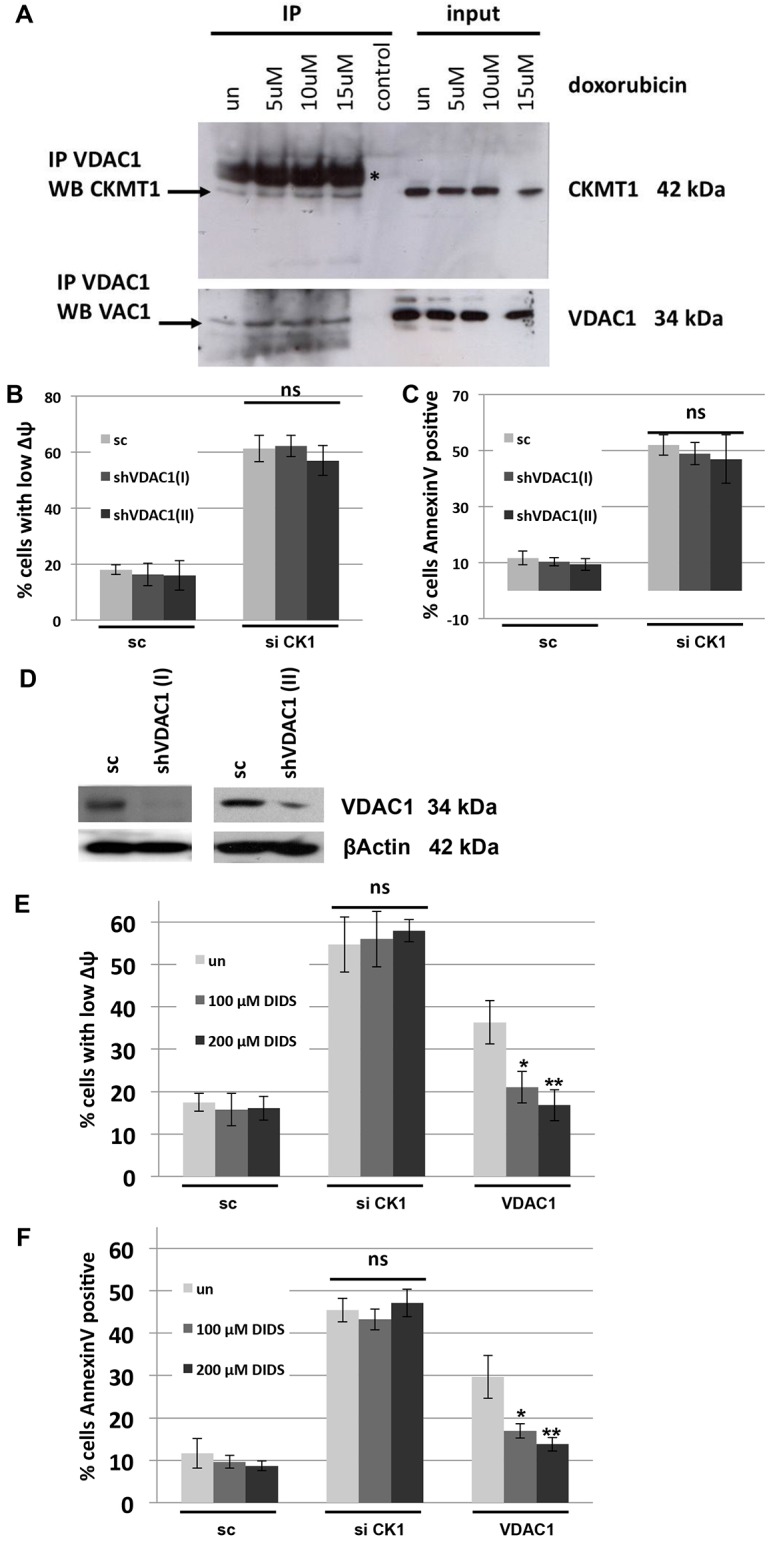
VDAC1 does not mediate the regulation of apoptosis by CKMT1. (A) Co-immunoprecipitation of CKMT1 with VDAC1 in untreated (un) and doxorubicin-treated MCF7 cells (the cells were treated with the indicated doxorubicin concentrations and lysates were obtained after 12 h). The asterisk (*) indicates the 50-kDa heavy chain fragment of the antibody used for the immunoprecipitation (IP). (B,C) Knockdown of VDAC1 by using either of two shRNA constructs specific for VDAC1 does not affect mitochondrial depolarization or the induction of apoptosis that results from the knockdown of endogenous CKMT1 at 72 h and 96 h post transfection. sc, scrambled control sequence; siCK1, siRNA against CKMT1. (D) Validation of VDAC1 knockdown by western blot analysis. (E,F) VDAC inhibition by DIDS does not impact on mitochondrial depolarization (E) or on apoptosis induction (F) caused by knockdown of CKMT1 (72 h post siCK1 transfection), whereas VDAC1-overexpression-induced mitochondrial depolarization (E) and apoptosis (F) can be significantly inhibited by DIDS. The data show the mean±s.d. *P<0.05; **P<0.01; ns, non-significant (Student's t-test).
CKMT1 depletion does not affect the interactions of constituents of the classical PT-pore, and is a cellular response to cytotoxic reagents
We hypothesized that depletion of CKMT1 would change the interactions of the molecular constituents of the PT-pore and thereby cause MPT. Therefore, we immunoprecipitated ANT1 (as a central subunit of the PT-pore) from protein lysates of CKMT1-knockdown and control cells, and assayed for changes in its protein interaction partners. As can be seen in Fig. 7, endogenous ANT1 interacted with endogenous CKMT1, VDAC1, CypD and HKII in control cells. Knockdown of endogenous CKMT1 did not impact on the level of these co-immunoprecipitated PT-pore components, indicating that their interaction with ANT1 remained stable under this condition. We hypothesized that a reduction in the level of endogenous CKMT1 could be part of the physiological apoptotic response, and we investigated whether changes in CKMT1 expression are linked to mitochondrial depolarization and subsequent induction of apoptosis by the cytotoxic agent As2O3, which has been demonstrated to mediate mitochondrial depolarization through MPT (Cai et al., 2000). A titration of As2O3 revealed that, in Hela cells, the onset of CKMT1 degradation could be observed after mitochondria started to become depolarized and annexin-V-positive, and the caspase substrate PARP was cleaved. This was a specific effect, because GAPDH levels remained unaffected at these time points (Fig. 8A,B). Assessing mitochondrial depolarization, annexin-V staining and the presence of the respective proteins over time supported these findings (Fig. 8C,D). By contrast, transient expression (by using plasmid transfection) of CKMT1 and the enzymatic activity mutant CKMT1mut, although not having an effect on the cell cycle and proliferation (data not shown), was able to counteract cell death and MPT when the cells were challenged with the MPT inducers H2O2 and As2O3 (supplementary material Fig. S2F). The two CKMT1 forms were found to be equally expressed and exclusively localized to mitochondria when overexpressed (data not shown). The independence of the apoptosis inhibition effect of CKMT1 from its enzymatic activity is in line with our finding that ΔΨm dissipation upon CKMT1 depletion could not be rescued by exogenous phosphocreatine addition (supplementary material Fig. S3). With CKMT1 not being released from mitochondria during apoptosis (data not shown), we hypothesized that CKMT1 controls MPT through changes in its oligomeric configuration and/or its association with other mitochondrial proteins. Native blue gel electrophoresis coupled with immunoblotting revealed that, in 293T cells, endogenous CKMT1 formed high-molecular-mass complexes (Fig. 8E). Comparison with purified CKMT1, which readily forms octamers in solution (Schlegel et al., 1988), indicated that the main native complex corresponds to octameric CKMT1 (336 kDa). No dimeric CKMT1 was found in native lysates from untreated 293T cells, even after prolonged exposure (not shown). However, an additional band could be detected in native cell lysates (CKMT1* complex). We examined whether the CKMT1 complexes changed upon the induction of apoptosis and could functionally be connected to apoptosis induction. Native cell lysates from As2O3- and H2O2-treated 293T cells were assayed for changes in CKMT1 quaternary organization. As can be observed in Fig. 8F,G the octamer of CKMT1 remained unaffected by As2O3 and H2O2 treatment, despite the fact that strong effects on the ΔΨm as well as on the induction of cell death were recorded. The higher-molecular-mass complex CKMT1*, by contrast, disappeared in an As2O3- and H2O2-concentration-dependent manner: Fig. 8H shows the correlation between mitochondrial depolarization and the percentage change in the ratio of the CKMT1 complexes (ratio of CKMT1* to the CKMT1 octamer) for As2O3- and H2O2-treated 293T cells, as determined by densitometric quantification of immunoblots.
Fig. 7.

CKMT1 depletion does not change the interaction of major constituents of the classical PT-pore model. 293T cells were infected with viral particles encoding shRNA against CKMT1 (shCK1) or a scrambled sequence (sc), and protein lysates were obtained at 6 days post infection. Immunoprecipitation (IP) of ANT1 resulted in co-immunoprecipitation of hexokinase II, CKMT1, VDAC1 and cyclophilin D. These interactions remained stable in the shRNA-induced absence of CKMT1.
Fig. 8.

The CKMT1 complex disappears during apoptosis. (A) HeLa cells were treated with different concentrations of As2O3 for 18 h. The ΔΨm and induction of apoptosis were assessed by DiOC6 staining or annexin-V staining, and subsequent FACS analysis. un, untreated. (B) Protein lysates were assayed for CKMT1 expression and PARP cleavage, both of which were quantified by using densitometric analysis of immunoblots and were plotted against the loss of mitochondrial polarization shown in A. (C,D) HeLa cells were treated with 16 µM As2O3 and assessed for mitochondrial depolarization and induction of apoptosis (C). Protein lysates were assessed for PARP cleavage and CKMT1 content over 24 h at 6 h intervals (D). (E) Blue native gel electrophoresis was used to analyze the migration behavior (size and quaternary structure) of purified CKMT1 and CKMT1 complexes present in native 293T cell lysates. Purified CKMT1 readily forms dimers and octamers. In native cell lysates, the majority of CKMT1 forms complexes displaying migration behavior identical to that of the CKMT1 octamer. Also, an additional CKMT1 band is visible, which runs at an apparently higher molecular mass (CKMT1* complex). (F) Analysis of CKMT1 complexes upon the induction of apoptosis by using various concentrations of As3O3 (1–20 µM) and H2O2 (200 and 1000 µM) for 16 h. The higher-molecular-mass complex of CKMT1 disappears in a concentration-dependent manner. (G) Blue native gel electrophoresis and immunoblotting of native 293T cell lysates upon induction of apoptosis by treating cells with 20 µM As3O3 for 16 h. The higher-molecular-mass CKMT1 complex disappears in apoptotic cells. Mitochondrial depolarization (shown by DiOC6 staining) and cell death (shown by annexin-V staining) are quantified in the panels on the right. (H) Correlation of mitochondrial depolarization (as assessed by DiOC6 staining and subsequent FACS analysis) with the disappearance of the upper band in the CKMT1 blot from F upon treatment with As3O3 and H2O2. The histogram shows the percentage of cells with depolarized mitochondria and the CKMT1 complex ratio (the ratio of the higher-molecular-mass complex to the octamer complex, as determined by densitometric quantification of immunoblots using ImageJ). The CKMT1 complex ratio that was observed in healthy cells was set to 100. nt, not treated. The data shown the mean±s.d. *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
DISCUSSION
In this study, we present evidence that the mitochondrial creatine kinase CKMT1 plays an important role in regulating apoptosis, by acting as a crucial gatekeeper for MPT. The depletion of CKMT1 induced MPT and subsequent apoptosis in a way that seemed to be independent of the classical PT-pore model. A number of specific reagents and interventions that inhibit this protein complex were unable to prevent the consequences of CKMT1 depletion. Such conditions include CypD knockdown and knockout, CypD overexpression, VDAC1 knockdown, and the compounds cyclosporin A, BC544, sanglifehrin A and DIDS; none of these strategies were able to impact on the cellular consequences of CKMT1 depletion. Only bongkrekic acid potently repressed the effects of CKMT1 downregulation on both MPT and the induction of cell death (Fig. 4). Bongkrekic acid is reported to function as an inhibitor of the PT-pore and MPT, by acting as a ligand of ANT, fixing its conformation so that it is open to the mitochondrial matrix (i.e. fixing it in the ‘m-state’) and thus inhibiting PT-pore opening (Bernardi et al., 2006; Halestrap and Brenner, 2003). However, the involvement of ANT in MPT, and, in particular, the requirement of ANT for PT-pore opening are controversial (Halestrap, 2004). Moreover, the specificity of the inhibitory effect of bongkrekic acid on the PT-pore has been re-addressed recently, when it was discovered that this compound also targets unidentified anion or chloride channels present in mitochondrial membranes (Malekova et al., 2007). This is especially relevant because there is increasing evidence that ion channels located in the IMM and OMM are key regulators of cellular signaling for cell death (Ryu et al., 2010).
In corroboration with the view that CKMT1 is regulating cell death through a route other than the classical PT-pore, we observed that its knockdown induced all tested features of apoptosis (Fig. 2C–F), whereas the PT-pore has been suggested to be involved in necrotic cell death only (Nakagawa et al., 2005). The classical PT-pore has been implicated in apoptosis and necrosis (Crompton, 1999). All our experiments indicate that reduction in the level of CKMT1 causes apoptosis. Hence, it is possible that there is a specific form of the PT-pore that is responsible for one, but not the other, form of cell death.
Because the depletion of endogenous CKMT1 through RNAi induces MPT and subsequent apoptosis, the downregulation of CKMT1 could constitute a physiologic mechanism of apoptotic induction. Examining this hypothesis, we observed that CKMT1 was reduced during the later stages of drug-induced apoptosis, subsequent to the onset of PARP cleavage (Fig. 8A,B). This was not a consequence of an unspecific proteolytic cleavage of cellular substrates by lysosomal proteases in late apoptosis, as GAPDH expression was not affected at these time points. Therefore, we propose that CKMT1 downregulation upon the induction of apoptosis by the signals tested, although not the priming event, acts as a positive feedback loop to reinforce the commitment of cells to apoptotic execution. Such changes in the expression level of CKMT1 also seem to be involved in pathological scenarios of apoptotic induction. Transcriptional profiling has detected significant downregulation of CKMT1 mRNA in neurons after spinal cord contusion (Aimone et al., 2004), and in neurodegeneration following spinal cord root avulsion (Swanberg et al., 2006). In line with this, downregulation of CKMT1 expression by RNAi in N2A neuronal cells also resulted in apoptosis (data not shown). By contrast, some studies have suggested that CKMT1 is upregulated in several malignancies with poor prognoses, such as lung cancer, gastric cancer and prostate cancer (DeLuca et al., 1981; Kanemitsu et al., 1984; Pratt et al., 1987; Tsung, 1983). By using microarray data available in the OncomineTM database (Compendia Bioscience, Ann Arbor, MI), CKMT1 can be shown to be overexpressed at the mRNA level in diverse tumors compared with the respective normal tissues, including tumors of the bladder, breast, lung, pancreas, ovaries and prostate (data not shown).
Because several other authors suggested that the dissociation of the CKMT1 octamer could induce MPT and apoptosis (Beutner et al., 1998; Soboll et al., 1999), we assayed the stability of the CKMT1 complexes. Although the octamer of CKMT1 was stable under all conditions, a novel CKMT1* complex, apparently of higher molecular mass than the octamer, was detected in blue native gel electrophoresis. This complex disappeared upon treatment with cytotoxic drugs in a dose-dependent manner, which correlated with mitochondrial depolarization (Fig. 8H). These experiments, together with the data presented on CKMT1 stability (Fig. 8A,B), support the view that the reduction in the amount of CKMT1* complex is a key change that allows MPT and the induction of apoptosis. Hence, the identity and composition of the apoptosis-sensitive CKMT1* complex is of interest. It could correspond to currently unidentified multimeric isoforms of CKMT1, to CKMT1 octamers formed of unprocessed CKMT1 (in which the mitochondrial import sequence has not been cleaved) or to (post-translationally) modified CKMT1 octamers. In particular, the CKMT1* complex could correspond to CKMT1 molecules (including, but not restricted to, octameric CKMT1) complexed with another protein that guards the MPT.
Creatine kinases catalyze the reversible trans-phosphorylation of phosphocreatine and ADP to creatine and ATP. Rapidly diffusible phosphocreatine can function as a temporal and spatial buffer for cellular ATP levels, and it links the site of energy production (mitochondria) to sites of energy consumption (various cellular ATPases) through creatine and phosphocreatine shuttling between cellular subcompartments (Dolder et al., 2003). The effects of CKMT1 depletion (i.e. loss of the ΔΨm and subsequent induction of apoptosis) are likely not due to the lack of mitochondrial creatine kinase activity (i.e. decreased phosphocreatine levels), as supplementation of cells with the enzymatic end product, phosphocreatine, did not rescue the CKMT1 RNAi phenotype (supplementary material Fig. S3). Supporting evidence comes from data relating to creatine deficiency syndromes, which arise from genetic errors in the biosynthesis or transport of creatine [including guanidinoacetate methyltransferase (GMAT) deficiency, l-arginine:glycine amidinotransferase (AGAT or GATM) deficiency and the creatine transporter (SLC6A8) deficiency]. These genetic disorders are not lethal.
The independence of the cellular effects from the enzymatic activity of CKMT1 could also explain why the CKMT1 downregulation is lethal, in contrast to the CKMT1-knockout phenotype, which is viable (Steeghs et al., 1995). Apart from this difference being due to metabolic and morphological compensations (Steeghs et al., 1998) and/or due to the difference in the physiology of the human and the murine system used in the knockout experiments, the knockout of CKMT1 in the original publication (Steeghs et al., 1995) was only verified using zymogram analysis for enzymatic creatine kinase activity, and the knockout was established by substituting exons seven and eight with a selection marker, making it possible that a shortened, but enzymatically inactive, CKMT1 transcript could still be expressed (Steeghs et al., 1995).
Although CKMT1 is a known component of the PT-pore, our study provides evidence for a classical PT-pore-independent role of CKMT1 in MPT and apoptosis. Hence, rather than the concept of one protein complex mediating MPT and apoptosis, a more accurate model might involve a variety of protein complexes being responsible for MPT. These complexes could coexist in cells, and their compositions could change depending on the stimuli for cell death.
MATERIALS AND METHODS
Reagents and antibodies
The commercial expression vectors pcDNA3 (Invitrogen), pEGFP-N1 (Clontech), pIRESpuro2 (Clontech) and pIRES2-EGFP (Clontech) containing the gene of interest were used for the transient and stable expression of genes by plasmid transfection. pLenti7.3 (Invitrogen) was used for the production of lentiviruses and pLKO.1 (Sigma-Aldrich) was used for the production of lentiviruses or for the expression of shRNA constructs. Apoptosis-inducing genes (ANT1 and BAX) were isolated from a genetic screen for apoptosis inducers (Albayrak and Grimm, 2003; Grimm and Kachel, 2002; Grimm et al., 1998) and were subsequently tagged C-terminally with hemagglutinin (HA-tag). A list of all siRNAs used to target human and/or murine CKMT1 can be found in the supplementary data set (supplementary material Table S1). The ASB9 expression plasmid was a kind gift from Hyung-Joo Kwon (Department of Microbiology, College of Medicine, Hallym University, Gangwon-do, Republic of Korea), and contains the full-length human ASB9 coding sequence in the pcDNA3.1/Myc-His (Invitrogen) expression vector. The shCypD pLKO.1-puro and the shVDAC1 pLKO.1-puro plasmid were obtained from Sigma-Aldrich, and correspond to the official identifiers TRCN0000232681, and TRCN0000029126 [shVDAC(I)] and TRCN0000123937 [shVDAC1(II)], respectively. The human CypD pCMV6 was obtained from Origene (UK), and contains the full-length human peptidylprolyl isomerase F coding sequence. CaspaTag Caspase 9 In Situ Assay Kit, Fluorescein was from Millipore.
DiOC6 and PI co-staining
Supernatant and trypsinized cells were transferred to a FACS tube and centrifuged for 5 min at 240 g. The supernatant was discarded, and the cells resuspended in PBS. Mitochondria were loaded with DiOC6 at a final concentration of 50 nM for 30 min in a humidified incubator (5% CO2, 37°C) and for another 30 min at room temperature. PI was used as a cell viability dye at a final concentration of 5 mg/ml. Forward and side scatter were used to characterize the cell population. DiOC6 fluorescence was acquired in Fl1, PI fluorescence in Fl2. FlowJo 7.25 (Stanford University, Stanford, CA) was used for subsequent analysis.
CaspaTag9 staining
Trypsinized cells were incubated with fluorescent-labeled inhibitor of caspases (FLICA, FAM-DEVD-FMK) reagent for 1 h as per the manufacturer's instructions, and were immediately assayed for FL1 fluorescence by FACS analysis.
JC1 staining
Mitochondria of living cells were loaded with JC1 diluted in medium to a final concentration of 10 µg/ml. Fluorescence of JC1 aggregates was acquired in Fl2, emission of monomeric JC1 was recorded in Fl1 and the ratio between Fl1 and Fl2 staining was used as an indicator of ΔΨm. The same staining protocol was used for live-cell confocal imaging.
Plasmid and siRNA transfections
Effectene (Qiagen, UK) was used for transfection of HeLa cells. 293T, N2A and MLE12 cells were transfected using Xfect (Clontech). β-galactosidase or a scrambled shRNA construct were used as control plasmids, and a co-transfected GFP plasmid was used to determine the transfection efficiency. siRNA transfections were performed using Effectene.
Protein extraction and quantification
Cell pellets were resuspended in 100 µl RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0) supplemented with complete protease inhibitor cocktail (Thermo Fisher Scientific) and incubated on ice for 30 min. The lysate was then centrifuged at 19,100 g for 30 min at 4°C. The supernatant containing the protein lysate was collected. Protein concentrations were measured using either the Bradford protein assay reagent (Bio-Rad) or the BCA protein assay kit (Sigma-Aldrich). Protein lysates were diluted in 5× loading buffer (Thermo Fisher Scientific) containing 0.005% bromophenol blue as a loading dye, 5% β-mercaptoethanol (Sigma-Aldrich) as a reducing agent, 10% SDS as a denaturing agent, 45% glycerol for loading and 0.5 M Tris buffer. Samples were then heated to 100°C for 10 min to ensure complete denaturation of the proteins.
Western blotting (SDS-PAGE)
Total cell lysates were separated by SDS-PAGE in TGS running buffer (25 mM Tris-HCl, 192 mM glycine, 0.1% w/v SDS). Proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, UK) by semidry electroblotting (Biorad, UK). Membranes were blocked with 5% bovine serum albumin (BSA) or milk in PBS containing 0.1% Tween (PBS-T), incubated with primary antibody, washed three times with PBS-T, and incubated with horseradish-peroxidase-conjugated secondary antibody. Proteins were visualized by enhanced chemiluminescence with Immobilon ECL reagent (Millipore, UK). Photographic films were from Amersham, UK. For multiple probing, membranes were incubated in Restore Western Blot Stripping Buffer (Pierce, UK) for 15 min at 37°C, and were washed in PBS-T to completely remove primary and secondary antibodies.
Immunostaining for FACS analysis
The supernatant and the adherent cells were harvested, centrifuged (240 g for 5 min) and fixed in 2% paraformaldehyde (Thermo Scientific) in PBS for 20 min at 4°C, then blocked in 10% fetal calf serum (FCS) in PBS for 30 min and permeabilized with 0.02% Triton X-100 for 30 min at 37°C. Cells were then centrifuged (240 g for 5 min), and the primary antibody, diluted in 3% FCS in PBS, was added, and the samples were incubated for 1 h at 37°C. The cells were then washed twice with 2 ml of 3% FCS in PBS. For Alexa-Fluor-labeled primary antibodies, cells were subsequently analyzed in the respective channel for fluorescence intensity (Fl1 for Alexa Fluor 488, Fl4 for Alexa Fluor 643). For staining using unlabeled primary antibodies, cells were centrifuged, and the isotype-matched secondary antibody (conjugated to Alexa Fluor 488 or Alexa Fluor 633), diluted in PBS at 1∶500, was added to the tube and incubated for 1 h at 37°C. Two washing steps with 2 ml of PBS were used to remove unbound secondary antibody. The cells were analysed by FACS using the Fl1 channel or Fl4 channel. FlowJo 7.25 was used for subsequent analysis.
Native blue gel electrophoresis
Native blue gel electrophoresis was performed using the NativePAGETM Novex® Bis-Tris Gel System (Invitrogen), which is based on the Blue Native Polyacrylamide Gel Electrophoresis (BN PAGE) technique for isolating mitochondrial protein complexes (Schägger, 2001). The near-neutral pH 7.5 environment during electrophoresis results in maximum stability of both proteins and gel matrix, providing superior band resolution. Sample preparation and electrophoresis were performed according to the manufacturer's instructions. Briefly, the supernatant and trypsinized cells were harvested by centrifugation. One confluent T175 flask was lysed in 1 ml of NativePAGETM sample buffer supplemented with 2% n-dodecyl β-d-maltoside (Sigma-Aldrich), 0.4% Coomassie G-250 (Invitrogen) and protease inhibitor cocktail (Fermentas). Lysates were incubated for 1 h on ice and then centrifuged at 18,300 g for 1 h to clear cell debris. Precast gels were washed with distilled water and placed into the XCellTM SureLockTM Mini-Cell (Life Technologies). The gel was loaded with 20 µl of native lysates. NativeMarkTM Unstained Protein Standard (Invitrogen) and BSA (Sigma-Aldrich) were used as markers for size estimations. Electrophoresis was performed at a constant voltage of 150 V, corresponding to 12–16 mA at the start of the run. The gel was then removed and soaked in transfer buffer (5 mM Tris, 192 mM glycine and 0.1% w/v SDS). Proteins were transferred onto 0.2-µm pore size PVDF membrane (Millipore, UK) by semidry electroblotting (Biorad, UK). Proteins were subsequently fixed and denatured by shaking in 8% acetic acid solution in PBS for 10 min. Membranes were then processed as described above for staining with primary and secondary antibodies.
Semi-quantitative RT-PCR
RNA was isolated by using 1 ml Trizol (Invitrogen, UK) per well of a six-well plate and resuspended in 20 µl ultrapure H20. The Titan One Tube RT-PCR kit (Roche, UK) was used to carry out RT-PCR with 2 µg total RNA. Specific primers for CKMT1 and β-actin were used for PCR reactions. PCR was performed for 29 cycles (10 s at 94°C, 30 s at 54°C and 50 s at 68°C; followed by 7 min at 68°C). PCR products were separated by gel electrophoresis on a 1% agarose gel.
Co-immunoprecipitation
Cells were washed once with PBS and lysed in 500 µl of RIPA buffer containing protease inhibitors (Fermentas). For immunoprecipitation of endogenous proteins, ∼7 mg of total cell lysate was used per condition. For immunoprecipitation of overexpressed proteins, 500 µg of total cell lysate was used per condition. The cell lysates were placed on ice for 30 min and then centrifuged (18,300 g) for 30 min at 4°C. The supernatant was pre-cleared twice with 50 µl of protein-G–SepharoseTM 4 Fast Flow (GE Healthcare) or with PureProteome magnetic protein G beads (Millipore) for 30 min, each with gentle rotation at 4°C. After pre-clearing, appropriate amounts of antibody (according to the data sheets) were added to the supernatant and were incubated overnight with rotation at 4°C. No antibody was added to the control immunoprecipitations. The antibody–protein complexes were then precipitated with 50 µl of beads and were incubated with rotation for >2 h at 4°C. The beads were washed five times with RIPA lysis buffer and re-suspended in SDS loading dye (Fermentas) supplemented with reducing agent (β-mercaptoethanol, 1%). For co-immunoprecipitation in isolated mitochondria, the mitochondria were lysed in RIPA buffer supplemented with 1% n-dodecyl maltoside.
Statistical analysis
Statistical analysis was performed using unpaired Student's t-tests (one- or two-tailed depending on the hypothesis tested). Samples were regarded to have an equal variance unless the F-test returned P<0.05. Data were regarded as statistically significant if P<0.05 based on the t-test.
Supplementary Material
Acknowledgments
Cyclophilin D knockout cells were kindly provided by Andrea Rasola (University of Padova, Department of Biomedical Sciences, Italy).
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
S.G. designed the study; C.D. performed the experiments that constitute the main body of this work; E.P., A.-L.M.-M. and F.O. designed and performed additional experiments; S.G. took a leadership role in writing the manuscript; and C.D. edited the manuscript.
Funding
C.D. was supported by a fellowship from Breast Cancer Campaign; E.P. was supported by Cancer Research UK; A.-L.M.-M. was supported by Breast Cancer Campaign; F.O. was supported by the Wellcome Trust. W.C. was supported by the Development and Promotion of Science and Technology Talents Project (DPST), Royal Thai Government, Thailand and M-S.H. by a stipend from AstraZeneca Ldt. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.140467/-/DC1
References
- Aimone J. B., Leasure J. L., Perreau V. M., Thallmair M. Christopher Reeve Paralysis Foundation Research Consortium(2004). Spatial and temporal gene expression profiling of the contused rat spinal cord. Exp. Neurol. 189, 204–221 10.1016/j.expneurol.2004.05.042 [DOI] [PubMed] [Google Scholar]
- Albayrak T., Grimm S. (2003). A high-throughput screen for single gene activities: isolation of apoptosis inducers. Biochem. Biophys. Res. Commun. 304, 772–776 10.1016/S0006-291X(03)00653-3 [DOI] [PubMed] [Google Scholar]
- Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen M. R., Gottlieb R. A., Dorn G. W. et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- Baines C. P., Kaiser R. A., Sheiko T., Craigen W. J., Molkentin J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 10.1038/ncb1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. (2005). Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 280, 18558–18561 10.1074/jbc.C500089200 [DOI] [PubMed] [Google Scholar]
- Bauer M. K. A., Schubert A., Rocks O., Grimm S. (1999). Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 147, 1493–1502 10.1083/jcb.147.7.1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P., Krauskopf A., Basso E., Petronilli V., Blachly-Dyson E., Di Lisa F., Forte M. A. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099 10.1111/j.1742-4658.2006.05213.x [DOI] [PubMed] [Google Scholar]
- Beutner G., Ruck A., Riede B., Welte W., Brdiczka D. (1996). Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 396, 189–195 10.1016/0014-5793(96)01092-7 [DOI] [PubMed] [Google Scholar]
- Beutner G., Rück A., Riede B., Brdiczka D. (1998). Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta 1368, 7–18 10.1016/S0005-2736(97)00175-2 [DOI] [PubMed] [Google Scholar]
- Brdiczka D. (1991). Contact sites between mitochondrial envelope membranes. Structure and function in energy- and protein-transfer. Biochim. Biophys. Acta 1071, 291–312 10.1016/0304-4157(91)90018-R [DOI] [PubMed] [Google Scholar]
- Brenner C., Grimm S. (2006). The permeability transition pore complex in cancer cell death. Oncogene 25, 4744–4756 10.1038/sj.onc.1209609 [DOI] [PubMed] [Google Scholar]
- Cai X., Shen Y. L., Zhu Q., Jia P. M., Yu Y., Zhou L., Huang Y., Zhang J. W., Xiong S. M., Chen S. J. et al. (2000). Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemia 14, 262–270 10.1038/sj.leu.2401650 [DOI] [PubMed] [Google Scholar]
- Crompton M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341, 233–249 10.1042/0264-6021:3410233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. (2000). Mitochondrial intermembrane junctional complexes and their role in cell death. J. Physiol. 529, 11–21 10.1111/j.1469-7793.2000.00011.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M., Virji S., Ward J. M. (1998). Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 258, 729–735 10.1046/j.1432-1327.1998.2580729.x [DOI] [PubMed] [Google Scholar]
- DeLuca M., Hall N., Rice R., Kaplan N. O. (1981). Creatine kinase isozymes in human tumors. Biochem. Biophys. Res. Commun. 99, 189–195 10.1016/0006-291X(81)91731-9 [DOI] [PubMed] [Google Scholar]
- Dolder M., Walzel B., Speer O., Schlattner U., Wallimann T. (2003). Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J. Biol. Chem. 278, 17760–17766 10.1074/jbc.M208705200 [DOI] [PubMed] [Google Scholar]
- Grimm S., Kachel V. (2002). Robotic high-throughput assay for isolating apoptosis-inducing genes. Biotechniques 32, 670–672, 674-677 [DOI] [PubMed] [Google Scholar]
- Grimm S., Weinstein E. J., Krane I. M., Leder P. (1998). Neu differentiation factor (NDF), a dominant oncogene, causes apoptosis in vitro and in vivo. J. Exp. Med. 188, 1535–1539 10.1084/jem.188.8.1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A. P. (2004). Mitochondrial permeability: dual role for the ADP/ATP translocator? Nature 430, 1p following 983 [DOI] [PubMed] [Google Scholar]
- Halestrap A. P., Brenner C. (2003). The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 10, 1507–1525 10.2174/0929867033457278 [DOI] [PubMed] [Google Scholar]
- Kanemitsu F., Kawanishi I., Mizushima J., Okigaki T. (1984). Mitochondrial creatine kinase as a tumor-associated marker. Clin. Chim. Acta 138, 175–183 10.1016/0009-8981(84)90232-8 [DOI] [PubMed] [Google Scholar]
- Kato M., Akao M., Matsumoto-Ida M., Makiyama T., Iguchi M., Takeda T., Shimizu S., Kita T. (2009). The targeting of cyclophilin D by RNAi as a novel cardioprotective therapy: evidence from two-photon imaging. Cardiovasc. Res. 83, 335–344 10.1093/cvr/cvp094 [DOI] [PubMed] [Google Scholar]
- Klingenberg M., Grebe K., Heldt H. W. (1970). On the inhibition of the adenine nucleotide translocation by bongkrekic acid. Biochem. Biophys. Res. Commun. 39, 344–351 10.1016/0006-291X(70)90582-6 [DOI] [PubMed] [Google Scholar]
- Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cai J., Jones D. P., MacGregor G. R., Wallace D. C. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 10.1038/nature02229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Reed J. C. (2000). Mitochondrial control of cell death. Nat. Med. 6, 513–519 10.1038/74994 [DOI] [PubMed] [Google Scholar]
- Kwon S., Kim D., Rhee J. W., Park J. A., Kim D. W., Kim D. S., Lee Y., Kwon H. J. (2010). ASB9 interacts with ubiquitous mitochondrial creatine kinase and inhibits mitochondrial function. BMC Biol. 8, 23 10.1186/1741-7007-8-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Johnson N., Capano M., Edwards M., Crompton M. (2004). Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem. J. 383, 101–109 10.1042/BJ20040669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D. T., Lechleiter J. D. (2002). Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J. Biol. Chem. 277, 31134–31141 10.1074/jbc.M112035200 [DOI] [PubMed] [Google Scholar]
- Malekova L., Kominkova V., Ferko M., Stefanik P., Krizanova O., Ziegelhöffer A., Szewczyk A., Ondrias K. (2007). Bongkrekic acid and atractyloside inhibits chloride channels from mitochondrial membranes of rat heart. Biochim. Biophys. Acta 1767, 31–44 10.1016/j.bbabio.2006.10.004 [DOI] [PubMed] [Google Scholar]
- Marchetti P., Castedo M., Susin S. A., Zamzami N., Hirsch T., Macho A., Haeffner A., Hirsch F., Geuskens M., Kroemer G. (1996). Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 184, 1155–1160 10.1084/jem.184.3.1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss S. J. (2011). Sangamides, a new class of cyclophilin-inhibiting host-targeted antivirals for treatment of HCV infection. Med. Chem. Commun. 3, 944–949 10.1039/C1MD00227A [DOI] [Google Scholar]
- Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. (2005). Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 10.1038/nature03317 [DOI] [PubMed] [Google Scholar]
- Pratt R., Vallis L. M., Lim C. W., Chisnall W. N. (1987). Mitochondrial creatine kinase in cancer patients. Pathology 19, 162–165 10.3109/00313028709077128 [DOI] [PubMed] [Google Scholar]
- Ryu S. Y., Peixoto P. M., Teijido O., Dejean L. M., Kinnally K. W. (2010). Role of mitochondrial ion channels in cell death. Biofactors 36, 255–263 10.1002/biof.101 [DOI] [PubMed] [Google Scholar]
- Schägger H. (2001). Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol. 65, 231–244 10.1016/S0091-679X(01)65014-3 [DOI] [PubMed] [Google Scholar]
- Schinzel A. C., Takeuchi O., Huang Z., Fisher J. K., Zhou Z., Rubens J., Hetz C., Danial N. N., Moskowitz M. A., Korsmeyer S. J. (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 102, 12005–12010 10.1073/pnas.0505294102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlattner U., Dolder M., Wallimann T., Tokarska-Schlattner M. (2001). Mitochondrial creatine kinase and mitochondrial outer membrane porin show a direct interaction that is modulated by calcium. J. Biol. Chem. 276, 48027–48030 [DOI] [PubMed] [Google Scholar]
- Schlegel J., Zurbriggen B., Wegmann G., Wyss M., Eppenberger H. M., Wallimann T. (1988). Native mitochondrial creatine kinase forms octameric structures. I. Isolation of two interconvertible mitochondrial creatine kinase forms, dimeric and octameric mitochondrial creatine kinase: characterization, localization, and structure-function relationships. J. Biol. Chem. 263, 16942–16953 [PubMed] [Google Scholar]
- Schubert A., Grimm S. (2004). Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res. 64, 85–93 10.1158/0008-5472.CAN-03-0476 [DOI] [PubMed] [Google Scholar]
- Soboll S., Brdiczka D., Jahnke D., Schmidt A., Schlattner U., Wendt S., Wyss M., Wallimann T. (1999). Octamer-dimer transitions of mitochondrial creatine kinase in heart disease. J. Mol. Cell. Cardiol. 31, 857–866 10.1006/jmcc.1998.0925 [DOI] [PubMed] [Google Scholar]
- Speer O., Bäck N., Buerklen T., Brdiczka D., Koretsky A., Wallimann T., Eriksson O. (2005). Octameric mitochondrial creatine kinase induces and stabilizes contact sites between the inner and outer membrane. Biochem. J. 385, 445–450 10.1042/BJ20040386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeghs K., Oerlemans F., Wieringa B. (1995). Mice deficient in ubiquitous mitochondrial creatine kinase are viable and fertile. Biochim. Biophys. Acta 1230, 130–138 10.1016/0005-2728(95)00044-J [DOI] [PubMed] [Google Scholar]
- Steeghs K., Oerlemans F., de Haan A., Heerschap A., Verdoodt L., de Bie M., Ruitenbeek W., Benders A., Jost C., van Deursen J. et al. (1998). Cytoarchitectural and metabolic adaptations in muscles with mitochondrial and cytosolic creatine kinase deficiencies. Mol. Cell. Biochem. 184, 183–194 10.1023/A:1006811717709 [DOI] [PubMed] [Google Scholar]
- Susin S. A., Zamzami N., Kroemer G. (1998). Mitochondria as regulators of apoptosis: doubt no more. Biochim. Biophys. Acta 1366, 151–165 10.1016/S0005-2728(98)00110-8 [DOI] [PubMed] [Google Scholar]
- Swanberg M., Duvefelt K., Diez M., Hillert J., Olsson T., Piehl F., Lidman O. (2006). Genetically determined susceptibility to neurodegeneration is associated with expression of inflammatory genes. Neurobiol. Dis. 24, 67–88 10.1016/j.nbd.2006.05.016 [DOI] [PubMed] [Google Scholar]
- Takeyama N., Miki S., Hirakawa A., Tanaka T. (2002). Role of the mitochondrial permeability transition and cytochrome C release in hydrogen peroxide-induced apoptosis. Exp. Cell Res. 274, 16–24 10.1006/excr.2001.5447 [DOI] [PubMed] [Google Scholar]
- Tazawa H., Fujita C., Machida K., Osada H., Ohta Y. (2009). Involvement of cyclophilin D in mitochondrial permeability transition induction in intact cells. Arch. Biochem. Biophys. 481, 59–64 10.1016/j.abb.2008.10.033 [DOI] [PubMed] [Google Scholar]
- Tsung S. H. (1983). Creatine kinase activity and isoenzyme pattern in various normal tissues and neoplasms. Clin. Chem. 29, 2040–2043 [PubMed] [Google Scholar]
- Verrier F., Deniaud A., Lebras M., Métivier D., Kroemer G., Mignotte B., Jan G., Brenner C. (2004). Dynamic evolution of the adenine nucleotide translocase interactome during chemotherapy-induced apoptosis. Oncogene 23, 8049–8064 10.1038/sj.onc.1208001 [DOI] [PubMed] [Google Scholar]
- Zamzami N., Kroemer G. (2001). The mitochondrion in apoptosis: how Pandora's box opens. Nat. Rev. Mol. Cell Biol. 2, 67–71 10.1038/35048073 [DOI] [PubMed] [Google Scholar]
- Zamzami N., Marchetti P., Castedo M., Decaudin D., Macho A., Hirsch T., Susin S. A., Petit P. X., Mignotte B., Kroemer G. (1995). Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 182, 367–377 10.1084/jem.182.2.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Larochette N., Kroemer G. (2005). Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Differ. 12, Suppl. 2 1478–1480 10.1038/sj.cdd.4401682 [DOI] [PubMed] [Google Scholar]
- Zhang D., Armstrong J. S. (2007). Bax and the mitochondrial permeability transition cooperate in the release of cytochrome c during endoplasmic reticulum-stress-induced apoptosis. Cell Death Differ. 14, 703–715 10.1038/sj.cdd.4402072 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.