

Abstract
AMP-activated protein kinase (AMPK) is a master regulator of metabolism. While muscle-specific AMPK β1β2 double-knockout (β1β2M-KO) mice display alterations in metabolic and mitochondrial capacity, their severe exercise intolerance suggested a secondary contributor to the observed phenotype. We find that tibialis anterior (TA), but not soleus, muscles of sedentary β1β2M-KO mice display a significant myopathy (decreased myofiber areas, increased split and necrotic myofibers, and increased centrally nucleated myofibers. A mitochondrial- and fiber-type-specific etiology to the myopathy was ruled out. However, β1β2M-KO TA muscles displayed significant (P<0.05) increases in platelet aggregation and apoptosis within myofibers and surrounding interstitium (P<0.05). These changes correlated with a 45% decrease in capillary density (P<0.05). We hypothesized that the β1β2M-KO myopathy in resting muscle resulted from impaired AMPK-nNOSμ signaling, causing increased platelet aggregation, impaired vasodilation, and, ultimately, ischemic injury. Consistent with this hypothesis, AMPK-specific phosphorylation (Ser1446) of nNOSμ was decreased in β1β2M-KO compared to wild-type (WT) mice. The AMPK-nNOSμ relationship was further demonstrated by administration of 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) to β1β2-MKO muscles and C2C12 myotubes. AICAR significantly increased nNOSμ phosphorylation and nitric oxide production (P<0.05) within minutes of administration in WT muscles and C2C12 myotubes but not in β1β2M-KO muscles. These findings highlight the importance of the AMPK-nNOSμ pathway in resting skeletal muscle.—Thomas, M. M., Wang, D. C., D'Souza, D. M., Krause, M. P., Layne, A. S., Criswell, D. S., O'Neill, H. M., Connor, M. K., Anderson, J. E., Kemp, B. E., Steinberg, G. R., and Hawke, T. J. Muscle-specific AMPK β1β2-null mice display a myopathy due to loss of capillary density in nonpostural muscles.
Keywords: necrosis, NO signaling, nitric oxide synthase
AMP-activated protein kinase (AMPK) is a heterotrimeric serine/threonine kinase, consisting of a catalytic α subunit and regulatory β and γ subunits, which each have multiple isoforms (1). AMPK is activated by elevations in cellular ADP and AMP/ATP ratio due to metabolic stress (i.e., hypoxia, glucose deprivation) and acts, in part, to up-regulate catabolic processes and reduce anabolic processes (1). As AMPK is ubiquitously expressed throughout the body, it has been considered to be the “master switch” of cellular and whole-body metabolism (1). Despite the knowledge of its role in whole-body metabolism, other functional roles of AMPK in skeletal muscle health are still being elucidated.
We have generated whole-body AMPK β1- (2) or β2-knockout (KO) (3) mice, as well as skeletal and cardiac muscle-specific AMPK β1β2 double-KO (β1β2M-KO) mice to explore the importance of AMPK in skeletal muscle metabolism and exercise capacity (4). A benefit of this double-KO model is the lack of compensatory up-regulation of remaining isoforms that occurs with targeting single AMPK-subunit isoforms (3, 5).
Previously, Mounier et al. (6) reported that the plantaris muscles of mice lacking the α1 subunit of AMPK displayed significantly accelerated overload-induced muscle hypertrophy compared to controls. This group also reported that skeletal muscle-specific deficient AMPK α1/α2 mouse soleus muscles were higher in mass with myofibers of larger size compared to controls (7). Despite these reports of the importance of AMPK in regulating muscle and myofiber size (6, 7), the soleus and extensor digitorum longus (EDL) muscles of our β1β2M-KO mice displayed no significant differences in mass or in their maximal tetanic force production (4). However, significant reductions in voluntary and forced exercise capacities were observed in these β1β2M-KO mice (4), prompting us to conclude that reductions in skeletal muscle mitochondrial content and contraction-stimulated glucose uptake were primary contributors to the exercise intolerance (4). Nevertheless, the severity of the exercise intolerance made us reconsider whether there might be other contributors to this phenotype. Thus, the purpose of this study was to investigate the effect of an absence of AMPK on overall muscle health in sedentary mice.
The findings of the present study reveal a critical role for AMPK as a key regulator of a signaling pathway linking vascular blood flow to metabolic stress in nonpostural [e.g., tibialis anterior (TA), gastrocnemius] muscles. This model aids in our understanding of the heterogeneous blood perfusion of resting skeletal muscle and proposes that in response to metabolic need, resting myofibers will activate AMPK that then phosphorylates, and activates, the skeletal muscle-specific neuronal nitric oxide synthase (nNOSμ) splice variant, promoting nitric oxide release to the surrounding capillaries, allowing for the delivery of oxygen and nutrients. In the absence of this intricate paracrine cascade, a significant myopathy exists, a condition that likely contributes to the severe exercise restriction noted in these mice (4).
MATERIALS AND METHODS
Animals
Male and female C57BL6 β1β2M-KO mice (14–16 wk) were used for this study. Wild-type (WT) littermates (AMPK β1β2 flox/flox) were used as controls. The generation of the β1β2M-KO model was described previously (4). Mice were housed under controlled environmental conditions (12-h light-dark cycles) and received chow (diet 8664; Harlan Teklad, Madison, WI, USA) and water ad libitum. The McMaster University Animal Research Ethics Board in accordance with the Canadian Council for Animal Care approved all methods and animal numbers used in this study.
Tissue collection
TA, gastrocnemius-plantaris complex, soleus, and diaphragm muscles were harvested from the mice at 14–16 wk of age. The muscles were coated in optimal cutting temperature-embedding compound, frozen in isopentane cooled by liquid nitrogen, and stored at −80°C.
Histochemical and immunofluorescent analysis
Cross sections (8 μm thick) of TA and soleus muscle were cut on a cryostat and mounted on glass slides. Sections underwent the histochemical and immunofluorescent staining procedures described below.
Hematoxylin-and-eosin (H&E)
H&E staining was used for measuring muscle cross-sectional area (TA) and investigating morphological features (TA, soleus) for both AMPK β1β2M-KO and WT mice. More than 100 fibers were analyzed within each muscle section.
Succinate dehydrogenase (SDH) and cytochrome oxidase (COX)
SDH and COX/SDH double staining were used to assess the presence of a mitochondrial myopathy in the TA muscle. The blue precipitate formed from the reduction of tetrazolium salt detected SDH activity, and the brown precipitate formed from the oxidation of diaminobenzidine detected COX activity. The SDH sections were used to assess the presence of fibers with high levels of subsarcolemmal SDH activity, also known as ragged blue fibers. (Ragged blue fibers are analogous to ragged red fibers in a Masson's trichrome stain). The COX/SDH double staining was undertaken to detect fibers that lacked COX activity staining but were positive for SDH staining, a characteristic of mitochondrial myopathies (8). SDH staining density was determined as a marker of mitochondrial enzyme content between β1β2M-KO and WT mice, as described previously (9). Briefly, >100 fibers from each animal were circled using NIS Elements (Nikon, Mississauga, ON, Canada), and the mean density was recorded. Darker-stained fibers imply a greater content of SDH activity and translate to greater mean densities.
Alkaline phosphatase
Alkaline phosphatase was used to determine muscle capillarization between β1β2M-KO and WT using SigmaFAST BCIP/NBT alkaline phosphatase tablets (Sigma-Aldrich, Oakville, ON, Canada). TA and soleus sections were incubated in alkaline phosphatase solution for 15 min at 37°C. Capillary area was determined via thresholding and automated detection from NIS Elements. Capillary density was calculated as a percentage of positively stained areas relative to the area of the entire muscle section. This method of analysis is more conservative than assessing “capillaries per fiber,” as the fiber cross-sectional area was reduced in β1β2M-KO mice (i.e., smaller fibers can erroneously raise capillary density if they are counted as “per fiber”).
Immunofluorescent staining
Sections were prepared for immunofluorescent staining by first allowing cut sections to air dry, fixing with ice-cold 2% paraformaldehyde, and incubating in blocking solution (10% normal goat serum and 1.5% BSA) for 30 min. Laminin (chicken polyclonal laminin antibody, 1:250; ab14055; Abcam, Cambridge, MA, USA) and dystrophin (rabbit polyclonal dystrophin, 1:200; ab15277; Abcam) were costained for a 1-h incubation period. IIB fiber typing was detected by using undiluted mouse IIB myosin heavy-chain antibody (10F5; Developmental Studies Hybridoma Bank, Iowa City, IA, USA), blocked using a mouse-on-mouse (MOM) blocking kit (Vector Laboratories, Burlingame, CA, USA) for 1 h, and incubated overnight. CD41 was used as a measure of platelet aggregation (10) and was detected using a purified rat anti-CD41 antibody (1:100; 553847; BD Pharmingen, San Jose, CA, USA) incubated overnight. Platelet endothial cell adhesion molecule (PECAM; rabbit polyclonal to CD31; ab2834; Abcam) was used at a 1:250 dilution as a measure of skeletal muscle capillaries. The ratio of CD41-positive to PECAM-positive areas was used to ascertain platelet aggregates per capillary number.
Primary antibodies were detected using the appropriate secondary antibodies at a concentration of 1:500 [anti-mouse Alexa Fluor 488, anti-chick Alexa Fluor 488, anti-rabbit Alexa Fluor 488, anti-rat Alexa Fluor 594 (Invitrogen, Carlsbad, CA, USA); anti-chick Texas Red (Abcam)]. Nuclei were visualized using a drop of 1:10,000 4,6-diamidino-2-phenylindole (DAPI) applied for 5 min on each section.
Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL)
Apoptotic nuclei in frozen skeletal muscle cross sections were detected using the Click-iT TUNEL Alexa Fluor 594 kit (Invitrogen) according to the manufacturer's instructions. TUNEL-positive nuclei that costained with DAPI were counted in whole-muscle cross sections. Apoptotic nuclei were quantified as myofiber nuclei if they resided within the muscle fiber (by visual inspection of an overlain phase contrast image) or nonmyofiber nuclei (fibroblastic, endothelial cell nuclei) if they resided outside the myofiber.
Image analysis
Images were taken with a Nikon Eclipse 90i microscope and stitched together using Nikon NIS Elements software. Image analysis included determination of fiber cross-sectional area through manual fiber outlining and determination of platelet-positive and capillary-positive areas through threshold detection method.
Phospho- and total nNOS content
Tissue sample preparation
Nonpostural glycolytic muscles (EDL, plantaris, peroneus, and biceps brachii) from one limb were removed from WT and β1β2-MKO animals and incubated in DMEM at 37°C with 1 mM 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR; Sigma-Aldrich) for 15 min, while the corresponding contralateral limb muscles served as a control and were incubated in DMEM. Muscles from WT and β1β2M-KO animals were ground in liquid nitrogen to a powder. The appropriate amount of TENT++ buffer (0.01 M Tris-HCl, 0.001 M EDTA, 0.1 M NaCl, and protease and phosphatase inhibitors) was added to produce a 5-fold dilution. Samples were sonicated (2×10 s at 10% power), subjected to 3 freeze-thaw cycles, and subsequently spun for 10 min at 14,000 g. Supernatants were transferred to new Eppendorf tubes and stored at −80°C.
Cell culture preparation
C2C12 myoblasts (American Type Culture Collection, Manassas, VA, USA) were seeded in 100-mm culture dishes and maintained at subconfluence in proliferation medium (DMEM, 10% FBS, and 1% penicillin/streptomyocin) at 37°C and 5% CO2. To induce differentiation, proliferation medium was replaced with differentiation medium (DMEM, 2% horse serum, and 1% penicillin/streptomyocin) on confluent myoblasts. Following 7 d of differentiation, AICAR (or an equal volume of PBS in controls) was added to a final concentration of 1 mM for 0 or 15 min. Cells were harvested by scraping in 1 ml of ice-cold 1× PBS. Following centrifugation (10 min at 2300 g), supernatants were discarded, and the pellet was resuspended in TENT++ buffer. Samples were sonicated (5 s at 10% power), and then centrifuged a second time (10 min at 15,700 g). The resulting supernatant was stored at −80°C.
Coimmunoprecipitation (Co-IP)
Co-IP of endogenously expressed total and phosphorylated nNOS was performed using a commercially available kit (Santa Cruz Biotechnology, Dallas, TX, USA). Prepared cell lysates were incubated in a preclearing matrix for 30 min and then spun quickly (30 s at 15,000 g). The resulting supernatant was transferred to a new Eppendorf tube, mixed with a dystrophin antibody (1:20; ab15277; Abcam), and rotated at 4°C for 1 h. Following rotation, the appropriate IP matrix was added to each sample and allowed to rotate at 4°C overnight. Samples were quickly spun the following morning, and the supernatant was removed. The resulting pellet was dissolved in 2× SDS and heated at 95°C for 10 min. Following a quick spin, the remaining supernatants were loaded, resolved by 8% SDS-PAGE gels, and transferred to PVDF membranes. Primary antibodies for phosphorylated nNOS (phospho-nNOS at Ser1446 in mice, corresponding to Ser1417 in humans; 1:250; Abcam) or total nNOS (1:150, Invitrogen), and dystrophin (1:250, Abcam) were used to detect proteins of interest and were conjugated with the appropriate horseradish peroxidase (HRP) secondary antibodies. Signals were visualized using chemiluminescent reagent (Amersham, Piscataway, NJ, USA), and acquired with the CareStream imager and accompanying software (Carestream Health, Rochester, NY, USA).
Measurement of nitric oxide production in C2C12 myotubes
C2C12 myoblasts were seeded in 24-well plates and grown in DMEM supplemented with 10% FBS. When the cells reached ∼60% confluence, they were switched to DMEM supplemented with 2% horse serum to induce differentiation. After 4 d of differentiation, cells were washed once with phenol-red free and serum-free DMEM, and incubated with 10 μM DAF-FM diacetate (Invitrogen) for 30 min at 5% CO2 and 37°C, while protected from light. After loading was completed, cells were rinsed 3 times with phenol red-free, serum-free DMEM and then exposed to medium supplemented with 1 or 3 mM AICAR (Toronto Research Chemicals, Toronto, ON, Canada) or no AICAR (control). Cells were then immediately placed in a SpectraMax M5 multidetection reader (Molecular Devices, Sunnyvale, CA, USA) for background fluorometric analysis using excitation and emission wavelengths of 488 and 520 nm, respectively. At this point, cells were transferred back to the incubator for 15 min before final NO fluorescence was once more assessed. Results represent the difference between the fluorescence seen at 15 min of incubation and the background fluorescence. Separate cultures were treated with AICAR as described above, with the addition of 1 mM L-NG-monomethyl arginine citrate (L-NMMA) beginning 1 h prior to the experiment and continuing throughout the DAF-FM diacetate loading and AICAR incubation periods.
Statistical analysis
All statistical calculations were performed with GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Differences between β1β2M-KO and WT animals were assessed through appropriate Student's t tests. A 2-way ANOVA with a Bonferroni post hoc test was used to detect differences in fiber-specific cross-sectional area bins between β1β2M-KO and WT animals. Values of P < 0.05 were considered significant. Data are presented as means ± sem.
RESULTS
TA muscles of sedentary β1β2M-KO mice display myopathy
β1β2M-KO mice display many morphological characteristics of myopathy. These include a significant increase in the number of muscle fibers containing centrally located nuclei compared to WT mice (β1β2M-KO: 10.6±1.9% of total fibers vs. WT: 0.4±0.2% of total fibers; n=4/group; P<0.01; Fig. 1A, B). Presence of centrally located nuclei in >3% of all muscle fibers is a hallmark indicator of a myopathy (11). In addition, β1β2M-KO mice demonstrated disorganized fiber distribution, excess endomysium, opaque and necrotic fibers, as well as many split fibers (Fig. 1C); all further nonspecific characteristics of skeletal muscle myopathy (12).
Figure 1.

Myopathic features of β1β2M-KO muscles. A) H&E images of WT and β1β2M-KO (M-KO) skeletal muscles. Note the numerous central nucleated (CN) fibers, diverse fiber shapes, and presence of necrotic (N) fibers with excess endomesial tissue. B) When quantified, there was a significant increase in the total number of fibers containing a central nuclei in the β1β2M-KO tibialis anterior muscles. C) Another general characteristic of muscle myopathy is the presence of split fibers. In this H&E image, you can note the presence of a split fiber in the β1β2M-KO muscle, while this feature was absent in WT muscle. This was confirmed with an immunofluorescent stain for dystrophin (green; sarcolemma), laminin (red; basal lamina), and DAPI (blue; nuclei). In a split fiber, the basal lamina will surround the entire (both) fibers, while the dystrophin will surround each fiber separately. Yellow arrow highlights the presence of dystrophin with no overlying basal lamina. Split fibers were consistently observed in the β1β2M-KO muscle cross sections but were not observed in WT muscles. D) Myofiber cross-sectional area of WT and β1β2M-KO TA muscles were measured and binned according to size. Note the significant increase in smaller fibers and significant decrease in larger fibers in β1β2M-KO muscles. On average, the mean cross-sectional area of β1β2M-KO muscle fibers was significantly reduced compared to WT (inset). Data are expressed as means ± se; n = 3 or 4. Scale bars = 50 μm (A); 60 μm (C). *P < 0.05 vs. WT.
A 20% decrease in mean myofiber cross-sectional area was measured in β1β2M-KO TA muscles compared to WT (P<0.01; Fig. 1D, inset), which was largely due to a significantly greater proportion of smaller fibers (800–1199 μm2) and lower proportion of larger fibers (2000–2399 μm2) in the β1β2M-KO compared to WT littermates (Fig. 1D).
β1β2M-KO myopathy is not mitochondrially based or fiber-type specific
Investigations using SDH and COX/SDH double-histochemical staining showed no evidence of mitochondrial abnormalities. On the basis of histochemical analysis, neither the intensity nor the distribution of SDH staining was significantly different between β1β2M-KO and WT mice (β1β2M-KO: 0.30±0.02 AU vs. WT: 0.34±0.03; n=4/group; Fig. 2A), indicating no mitochondrial proliferation or ragged blue fibers (analogous to ragged-red fibers); the COX/SDH double staining did not contain any COX−/SDH+ fibers (not shown). The presence of ragged blue fibers and/or COX−/SDH+ fibers is indicative of mitochondrial dysfunction (8).
Figure 2.
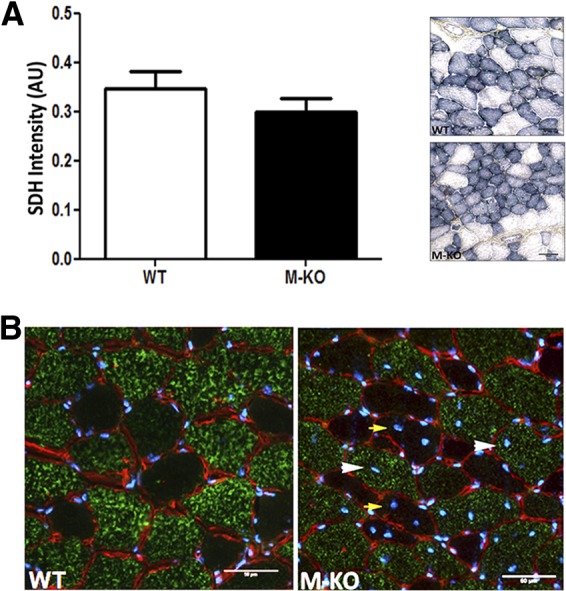
Absence of a mitochondrial basis to the myopathic features observed in β1β2M-KO muscles. A) Lack of significant difference in SDH staining density between WT and β1β2M-KO (M-KO) TA muscles. No distinct patterns of staining are visible, which would indicate a mitochondria-based etiology to the myopathy (e.g., darkly stained fibers characteristic of mitochondrial proliferation, or ragged blue fibers that are analogous to ragged red fibers observed with modified Gomori trichrome stain). B) Myosin IIB immunofluorescent stain (green; laminin, red; DAPI, blue) demonstrating the distribution of regenerating fibers (determined by the presence of centrally located nuclei) was not different between type IIB, glycolytic muscle fibers (white arrowheads; 57%), and the more oxidative IIX/IIA muscle fibers (yellow arrows; 43%). No regenerating fibers observed in WT muscle image. Data are expressed as means ± se; n = 4. Scale bars = 50 μm.
Given previous impairments observed in contraction-stimulated glucose uptake (4), immunofluorescent staining for glycolytic type IIB fibers was used to determine the possibility of a glycolytic (IIB) fiber-type-specific myopathy. The distribution of regenerating fibers in the β1β2M-KO (determined by the presence of centrally located nuclei) was not different between type IIB, glycolytic muscle fibers (57%) and the more oxidative IIX/IIA muscle fibers (43%; Fig. 2B).
β1β2M-KO TA muscles display reduced nNOSμ phosphorylation, increased platelet aggregation, and decreased muscle capillarization
β1β2M-KO TA muscles had normal nNOSμ expression within the dystrophin complex, but phosphorylation at the AMPK site (Ser1446) was significantly reduced in resting muscle (P<0.05; Fig. 3A, B). To verify the relationship between AMPK and nNOSμ, we used C2C12 cells with intact functional AMPK and found that just 15 min of incubation with AICAR (an AMPK activator) could stimulate significant increases in nitric oxide production in C2C12 myotubes (Fig. 3D) that coincided with significant increases in phospho-nNOSμ (but not total nNOSμ; Fig. 3E). Further support for this relationship is observed when AICAR is administered to WT and β1β2M-KO EDL muscles. AICAR-induced increases in phospho-nNOSμ were observed in WT muscles, but AICAR incubation failed to increase phospho-nNOSμ levels in β1β2-MKO mice (Fig. 3C).
Figure 3.

Phosphorylated nNOS is decreased in β1β2M-KO muscles. A, B) Immunoblotting of TA muscles demonstrates no significant difference between groups in total nNOS associated with the dystrophin complex (A), but a significant reduction in AMPK phosphorylation of nNOS at Ser1446 (B) was measured in the β1β2M-KO (M-KO) muscles. Total and phospho-nNOS measures came from the same mice and are expressed relative to dystrophin, which was used as a loading control. *P < 0.05. C) Incubation of muscles in AICAR (an AMPK activator) for 15 min significantly increased dystrophin-associated phospho-nNOS in WT muscles but not in β1β2M-KO muscles. Levels of phospho-nNOS in AICA-treated muscles have been normalized to phospho-nNOS in muscles from the untreated contralateral limb. Dystrophin levels were similar between all groups. *P < 0.05. D) To demonstrate the importance of AMPK phosphorylation in regulating nNOS activity, a functional NO production assay was performed. Within 15 min of incubation, both 1 and 3 mM AICAR significantly increased NO production (DAF fluorescence) in C2C12 myotubes; this response could be inhibited by the NOS inhibitor, L-NMMA. *P < 0.05 vs. control; #P < 0.05. E) Immunoblotting of dystrophin-associated nNOS in C2C12 myotubes with and without 15 min of 1 mM AICAR treatment. As expected, 15 min of AICAR did not affect total nNOS levels, but it did significantly increase the phosphorylation of nNOS at Ser1446. Data are expressed as means ± se; n = 2–7. *P < 0.05.
Given the role of nitric oxide signaling in skeletal muscle capillary flow, we investigated whether impaired nitric oxide signaling would lead to increased thrombus formation, as measured by increased platelet aggregation. Compared to TA muscles in the WT, resting β1β2M-KO muscles displayed significantly higher levels of CD41-positive areas overlaying PECAM-positive cells, a measure of platelet aggregation (Fig. 4A). TUNEL staining was then undertaken to ascertain whether the increase in platelet aggregation was associated with an increase in the number of apoptotic nuclei within resting β1β2M-KO muscle. These stains revealed that β1β2M-KO TA muscles exhibited significantly more apoptotic nuclei than WT muscles (Fig. 4B, C); a finding consistent with the myopathic state of the muscle. However, many apoptotic nuclei were also noted outside of the myofiber proper (i.e., in the interstitial space) consistent with an increased number of apoptotic capillaries. To determine whether these interstitial apoptotic nuclei were capillaries, we undertook an alkaline phosphatase stain and quantified capillary density. As seen in Fig. 4D, a significant reduction in capillary density of almost 50% was measured in TA muscles of β1β2M-KO mice compared to WT.
Figure 4.

Increased platelet aggregation and apoptosis in β1β2M-KO TA muscles. A) A significant increase in platelet aggregation, as measured by the ratio of CD41-positive to PECAM-positive, was observed in β1β2M-KO (M-KO) TA muscles compared to WT. Right panel is a representative immunofluorescent stain of WT and β1β2M-KO muscle cross sections with PECAM (green) labeling all capillaries and CD41 (red) labeling platelet aggregates. B) Representative images of TUNEL staining in WT and β1β2M-KO TA muscles. TUNEL-positive areas (red, left panels) were overlaid with DAPI (blue, right panels) to ascertain the number of apoptotic nuclei (yellow arrows, apoptotic interstitial nuclei; yellow arrows with asterisk inside, apoptotic myonuclei). Insets highlight TUNEL-positive, DAPI-positive nuclei that are within the myofiber (m) or outside of the myofiber (e), presumably capillaries. C) Quantification of the TUNEL-positive nuclei within the TA reveals a significant increase in apoptotic nuclei within the muscle (as would be expected in myopathic muscles) and an even more robust difference in TUNEL-positive nuclei that were outside of the muscle fiber (nonmyonuclei) in the β1β2M-KO TA. D) Alkaline phosphatase stain in WT and β1β2M-KO TA muscles. Left panel: quantification of capillary density as percentage of WT values demonstrates significantly lower capillary density in TA muscles of β1β2M-KO mice (∼45%) compared to WT. Right panel: representative images. Data are expressed as means ± se; n = 4–8. Scale bars = 50 μm. *P < 0.05 vs. WT.
Soleus muscles of β1β2M-KO mice exhibit a more modest reduction in capillary density and do not present a myopathic phenotype
As our data support impairment in nNOSμ-mediated vasodilation as the basis of the myopathic phenotype, we were interested to determine whether postural muscles, which exhibit tonic contractile activity, allowing for contraction-mediated mechanisms to promote vasodilation/blood flow (i.e., skeletal muscle pump; refs. 13, 14), would also display a myopathy. In contrast to the observed myopathic features in the nonpostural (TA) muscles, β1β2M-KO soleus muscles were phenotypically similar to WT soleus muscles (Fig. 5A), with an absence of the myopathic characteristics noted above (e.g., necrotic fibers, central-nucleated myofibers, split fibers). Quantification of TUNEL-positive nuclei supported the absence of myopathy and no difference in apoptotic nuclei (both myonuclei and nonmyonuclear) were observed in the resting β1β2M-KO soleus muscles (Fig. 5B).
Figure 5.

β1β2M-KO soleus muscles display no myopathy and only modest reductions in capillary density. A) There is an absence of myopathic traits in soleus (SOL) muscles of β1β2M-KO (M-KO) mice, as can be seen in these H&E stains. B) Quantification of the TUNEL-positive nuclei within the soleus muscle demonstrated no difference in either muscle or nonmuscle nuclei that are TUNEL-positive. C) Representative alkaline phosphatase stains (left panel) and quantification of capillary density in soleus muscles of WT and β1β2M-KO mice. Reduction in capillary density is much less (only ∼20%) in soleus compared to TA muscles of β1β2M-KO mice. Data are expressed as means ± se; n = 3–4. Scale bars = 50 μm. *P < 0.05.
Although no myopathy or difference in apoptotic nuclei was noted in the soleus muscles of β1β2M-KO mice, a 20% reduction in capillary density, compared to WT soleus, was observed (P<0.05; Fig. 5C). Given a lack of difference in CD41-positive capillaries (not shown) and apoptotic nuclei observed between β1β2M-KO and WT soleus muscles (Fig. 5B), we hypothesize that the reduced capillary density is likely the result of AMPK's role in regulating basal skeletal muscle capillarization through VEGF (15).
DISCUSSION
Our analysis of β1β2M-KO skeletal muscles has uncovered a significant myopathic phenotype within nonpostural muscles (e.g., TA), but not in tonically active, postural (soleus) muscles. In contrast to a mitochondrial-based etiology to the muscle pathology as was anticipated on the basis of the role of AMPK in metabolic regulation, our results indicate reductions in AMPK-nNOSμ signaling as the underlying cause. Reductions in AMPK-mediated phosphorylation of nNOSμ are observed in β1β2M-KO muscles concomitant with significant increases in platelet aggregation (metric of thrombus formation and blocked capillaries). We propose that the resultant impairment in blood flow leads to injury/death to downstream capillaries and myofibers; a hypothesis supported by significant increases in apoptosis of both muscle and capillary nuclei and significant reductions in total muscle capillary area. Taken together, the present study sheds new light on the central role of skeletal muscle AMPK in paracrine signaling to the surrounding capillary bed. This signaling between muscle and the capillary bed would provide an intricate cascade to allow resting skeletal muscle fibers to dynamically regulate their own perfusion in response to energy demand.
Given the significant role of AMPK in skeletal muscle metabolism and mitochondrial biogenesis (1), a mitochondrial basis to the muscle pathology was initially hypothesized. However, our histochemical analysis did not reveal any hallmark signs of mitochondrial myopathies, such as mitochondrial proliferation, ragged blue fibers, or COX−/SDH+ fibers (8). While these results were contrary to our original hypothesis, they are consistent with the lack of dramatic changes in mitochondrial function and content observed in AMPK-KO mouse models (4, 16–18).
Mice with a skeletal- and cardiac-specific KO of LKB1, an upstream activator of AMPK, also display reduced muscle mitochondrial marker enzyme expression and significant exercise intolerance (19), akin to that observed in β1β2M-KO mice (4). Although these LKB1-KO mice were noted to be outwardly normal at younger ages (<20 wk of age), no investigations into the muscle morphology were presented. However, with increasing age (>30 wk of age), a notable myopathy developed resulting in severely restricted hind-limb function (20). In contrast, young β1β2M-KO mice do not display an overt ambulatory phenotype (4), but despite no gross abnormalities in ambulatory activity, β1β2M-KO mice display hallmark characteristics of muscle pathology, including decreased myofiber size, irregular shaped muscle fibers, swollen and opaque fibers, necrotic fibers, centrally nucleated myofibers, and a consistent presence of split fibers. An investigation into older β1β2M-KO mice would be of interest to ascertain whether limb function is impaired similar to that of muscle-specific LKB1-KO mice. It would also be of interest to assess the AMPK-nNOSμ relationship in LKB1-deficient mice, though it should be noted that the LKB1 mice also present with congestive heart failure so discerning between the skeletal muscle defects and indirect effects of heart failure would be more complicated.
In addition to a role for AMPK in metabolism, previous reports have defined other roles for AMPK, including the regulation of nNOS (21). The nNOSμ splice variant is preferentially localized to the dystrophin-associated complex of the muscle sarcolemma, via its binding to α-syntrophin, and is found in greater abundance in muscles composed of more glycolytic fiber types, such as the TA, plantaris, and gastrocnemius (22–25). When a mitochondrial origin to the myopathy was ruled out, we speculated that the underlying cause of the myopathy in β1β2M-KO mice may be mediated through reductions in nNOSμ activity. As hypothesized, a significant decrease in dystrophin-associated nNOSμ phosphorylation at the AMPK phosphorylation site (Ser1446) was noted in resting β1β2M-KO muscle. This was further supported by the finding that Ser1446 phosphorylation of nNOSμ in WT muscles and C2C12 myotubes could be rapidly induced with the AMPK activator, AICAR, a finding that was not reproduced in β1β2M-KO muscle. In addition, nitric oxide production was increased in C2C12 myotubes incubated with the AMPK activator, AICAR. While a decreased nNOS protein content has been observed in AMPK-kinase dead (AMPK-KD) mice (21), to our knowledge no reports of muscle myopathy have been reported.
In healthy skeletal muscle, it has been known for some time that nNOS-generated nitric oxide modulates contractile force (23) and exercise-induced glucose uptake, with NOS inhibitors selectively blunting exercise-induced, but not insulin-stimulated, glucose transport (26). Following these reports, the diverse roles of dystrophin-associated nNOS (nNOSμ) in regulating other aspects of skeletal muscle health has been the subject of more intense investigations, as alterations to nNOSμ localization and/or content have been reported to be involved in the pathophysiology of various myopathic states, including Duchenne muscular dystrophy (and in mdx mice), Becker muscular dystrophy, calveolinopathy, sarcoglycanopathies, and even aging (27–31). Mechanistically, nNOSμ-mediated nitric oxide production will attenuate α-adrenergic vasoconstriction, thereby promoting rapid, localized vasodilation. In the absence of this system, functional muscle ischemia occurs (32). Additionally, nitric oxide has also been shown to decrease leukocyte adherence (33) and down-regulate platelet aggregation and adherence, thereby reducing thrombus formation (34). While it was assumed that endothelial NOS (eNOS) was the primary source of this nitric oxide, Tymvious et al. (35) recently reported that platelet aggregation was regulated by sources of nitric oxide other than eNOS, and from sources outside the vascular endothelium. Thus, the findings of the present study (increased platelet aggregation, increased apoptosis, loss of capillary density, and presence of myopathy) are consistent with the relationship of AMPK and nNOSμ, and this conclusion is supported by the ability of the AMPK to phosphorylate nNOSμ and significantly increase nitric oxide production within minutes (Fig. 3). In nNOSμ-deficient mice, impaired functional capacity has also been reported along with evidence of reduced muscle health (decreased fiber areas and muscle mass; refs. 36, 37). Though myopathic changes were not specifically investigated in those studies, a report from Percival and colleagues (38) suggests that a significant myopathy was only noted in mice lacking both nNOSμ and the splice variant nNOSβ. It is not known whether AMPK regulates nNOSβ, but since this splice variant retains the corresponding C-terminal AMPK phosphorylation site (nNOSβ, Ser1182), this Golgi-associated nNOS splice variant would be expected to be a substrate.
The resting β1β2M-KO TA muscles displayed hallmarks of myopathy, such as decreased fiber cross-sectional area and the presence of opaque and necrotic fibers. There was also an order of magnitude increase in the presence of centrally located nuclei, indicative of increased muscle repair/turnover. These observations were consistent with measures in other nonpostural muscles (gastrocnemius; Supplemental Fig. S1). Interestingly, the postural soleus muscles and the tonically active diaphragm muscle (Supplemental Fig. S1) did not show signs of a myopathy. The reasons underlying this are likely due to the fact that postural muscles have ∼3 times greater resting perfusion (14) and more homogeneous blood flow through the capillaries (39). Also, their tonic activity would allow for an AMPK-nNOS-independent means of regulating resting blood flow (23). Further support for this dichotomy in nNOS function between muscle groups comes from Copp et al. (40), who demonstrate that inhibition of nNOS in rats reduced skeletal muscle blood flow and vascular conductance during intense exercise, predominantly in fast-twitch muscle fibers. Planitzer et al. (41) have also reported that soleus has lower nNOS expression than other faster muscles and, thus, has other compensatory mechanisms to maintain resting muscle blood flow (muscle pump; ref. 42). The maintenance of blood flow would prevent the apoptosis noted above which we hypothesize leads to the observed myopathy in the TA but not the soleus.
Although the relationship between AMPK and VEGF has been well defined (15), we would propose that a reduction in VEGF expression plays a secondary role in the pathology observed. We based this hypothesis on our observations that there is an approximate 20% decrease in capillary density in the soleus muscles of β1β2M-KO mice (likely consequence of the reduced basal angiogenesis driven by the AMPK-VEGF relationship) but no measurable apoptosis or myopathy. However, the more dramatic reduction in capillary density in β1β2M-KO TA muscles (∼45% reduction) would be a consequence of reduced basal angiogenesis (as seen in soleus) and continual capillary dropout resulting from the impaired nNOS capillary signaling. Nearly 90% of capillaries support blood flow at rest although velocity of red blood cells is heterogeneous, particularly in more glycolytic muscles (39). The absence of skeletal muscle AMPK prevents the activation of sarcolemmal nNOSμ in response to changing metabolic demands of individual muscle fibers and thus no increase in red blood cell velocity and oxygen delivery. This then causes periods of ischemia in the skeletal muscle microvasculature, resulting in cell death and a reduced capacity for AMPK-VEGF angiogenesis thereafter.
While it has been previously shown that nNOSμ is involved in matching blood supply to the metabolic demands of the active muscle (38, 40), the present findings demonstrate for the first time a critical relationship between AMPK and nNOSμ in resting skeletal muscle. This relationship would allow the energy-sensing enzyme of skeletal muscle to readily regulate perfusion in resting nonpostural muscles at times of energy need; findings that support a novel model to help explain the heterogeneity of vascular perfusion that is observed in resting skeletal muscle. In the absence of AMPK, an impaired vasodilatory capacity and increased platelet aggregation ensues, resulting in functional ischemia and downstream apoptosis. The outcome is a significant muscle pathology, including a loss of capillary content, impairments that would contribute to the exercise intolerance and increased fatigability previously observed in these β1β2M-KO mice (4).
Supplementary Material
Acknowledgments
The 10F5 antibody developed by C. Lucas was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the U.S. National Institute of Child Health and Human Development and maintained by the Department of Biology, University of Iowa (Iowa City, IA, USA).
These studies were supported by grants and fellowships from the Natural Science and Engineering Research Council of Canada (T.J.H., G.R.S.) and the Canadian Institutes of Health Research (CIHR; T.J.H., G.R.S.), the Canadian Foundation for Innovation (T.J.H., G.R.S.), the Australian Research Council (B.E.K.), and the Australian National Health and Medical Research Council (B.E.K., G.R.S.). This work was supported, in part, by the Victorian government's Operational Infrastructure Support program (B.E.K.). G.R.S. holds a Canada Research Chair in Metabolism and Obesity.
The authors also acknowledge the contributions of the Michael DeGroote Fellowship (M.M.T.), the Ontario Graduate Scholarship program (M.P.K.), and the Canadian Institutes of Health Research Banting and Best Doctoral Fellowship program (D.M.D.).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- β1β2M-KO
- muscle-specific AMPK β1β2 double-knockout
- AICAR
- 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- AMPK
- AMP-activated protein kinase
- COX
- cytochrome oxidase
- DAPI
- 4,6-diamidino-2-phenylindole
- EDL
- extensor digitorum longus
- H&E
- hematoxylin and eosin
- KO
- knockout
- L-NMMA
- L-NG-monomethyl arginine citrate
- PECAM
- platelet endothial cell adhesion molecule
- nNOS
- neuronal nitric oxide synthase
- nNOSμ
- muscle-specific neuronal nitric oxide synthase
- SDH
- succinate dehydrogenase
- TA
- tibialis anterior
- TUNEL
- terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling
- WT
- wild type
REFERENCES
- 1. Steinberg G. R., Kemp B. E. (2009) AMPK in health and disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 2. Dzamko N., van Denderen B. J., Hevener A. L., Jorgensen S. B., Honeyman J., Galic S., Chen Z. P., Watt M. J., Campbell D. J., Steinberg G. R., Kemp B. E. (2010) AMPK β1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J. Biol. Chem. 285, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinberg G. R., O'Neill H. M., Dzamko N. L., Galic S., Naim T., Koopman R., Jorgensen S. B., Honeyman J., Hewitt K., Chen Z. P., Schertzer J. D., Scott J. W., Koentgen F., Lynch G. S., Watt M. J., van Denderen B. J., Campbell D. J., Kemp B. E. (2010) Whole body deletion of AMP-activated protein kinase β2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem. 285, 37198–37209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Neill H. M., Maarbjerg S. J., Crane J. D., Jeppesen J., Jorgensen S. B., Schertzer J. D., Shyroka O., Kiens B., van Denderen B. J., Tarnopolsky M. A., Kemp B. E., Richter E. A., Steinberg G. R. (2011) AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. U. S. A. 108, 16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jorgensen S. B., Nielsen J. N., Birk J. B., Olsen G. S., Viollet B., Andreelli F., Schjerling P., Vaulont S., Hardie D. G., Hansen B. F., Richter E. A., Wojtaszewski J. F. (2004) The α2–5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 53, 3074–3081 [DOI] [PubMed] [Google Scholar]
- 6. Mounier R., Lantier L., Leclerc J., Sotiropoulos A., Pende M., Daegelen D., Sakamoto K., Foretz M., Viollet B. (2009) Important role for AMPK α1 in limiting skeletal muscle cell hypertrophy. FASEB J. 23, 2264–2273 [DOI] [PubMed] [Google Scholar]
- 7. Lantier L., Mounier R., Leclerc J., Pende M., Foretz M., Viollet B. (2010) Coordinated maintenance of muscle cell size control by AMP-activated protein kinase. FASEB J. 24, 3555–3561 [DOI] [PubMed] [Google Scholar]
- 8. Taylor R. W., Schaefer A. M., Barron M. J., McFarland R., Turnbull D. M. (2004) The diagnosis of mitochondrial muscle disease. Neuromuscul. Disord. 14, 237–245 [DOI] [PubMed] [Google Scholar]
- 9. Shortreed K. E., Krause M. P., Huang J. H., Dhanani D., Moradi J., Ceddia R. B., Hawke T. J. (2009) Muscle-specific adaptations, impaired oxidative capacity and maintenance of contractile function characterize diet-induced obese mouse skeletal muscle. PLoS One 4, e7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baudouin-Brignole F., Bayle J., Goguel A., Philip P. J. (1997) PMA induces platelet activation of specific antigens (CD62/CD63) in GpIIb-IIIa deficient platelets from Glanzmann's thrombasthenia. Platelets 8, 391–395 [DOI] [PubMed] [Google Scholar]
- 11. Dubowitz V. (2007) Commentary from the editor. Neuromuscul. Disord. 17, 1–5 [DOI] [PubMed] [Google Scholar]
- 12. Sewry C. A. (2008) Pathological defects in congenital myopathies. J. Muscle Res. Cell Motil. 29, 231–238 [DOI] [PubMed] [Google Scholar]
- 13. Folkow B., Halicka H. D. (1968) A comparison between “red” and “white” muscle with respect to blood supply, capillary surface area and oxygen uptake during rest and exercise. Microvasc. Res. 1, 1–14 [Google Scholar]
- 14. McDonagh P. F., Gore R. W. (1982) A comparison of capillary hydraulic conductivities in postural and locomotor muscle. Am. J. Physiol. 243, H491–H497 [DOI] [PubMed] [Google Scholar]
- 15. Zwetsloot K. A., Westerkamp L. M., Holmes B. F., Gavin T. P. (2008) AMPK regulates basal skeletal muscle capillarization and VEGF expression, but is not necessary for the angiogenic response to exercise. J. Physiol. 586, 6021–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jorgensen S. B., Jensen T. E., Richter E. A. (2007) Role of AMPK in skeletal muscle gene adaptation in relation to exercise. Appl. Physiol. Nutr. Metab. 32, 904–911 [DOI] [PubMed] [Google Scholar]
- 17. Klein D. K., Pilegaard H., Treebak J. T., Jensen T. E., Viollet B., Schjerling P., Wojtaszewski J. F. (2007) Lack of AMPK α2 enhances pyruvate dehydrogenase activity during exercise. Am. J. Physiol. Endocrinol.Metab. 293, E1242–E1249 [DOI] [PubMed] [Google Scholar]
- 18. Zong H., Ren J. M., Young L. H., Pypaert M., Mu J., Birnbaum M. J., Shulman G. I. (2002) AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. U. S. A. 99, 15983–15987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomson D. M., Porter B. B., Tall J. H., Kim H. J., Barrow J. R., Winder W. W. (2007) Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am. J. Physiol. Endocrinol.Metab. 292, E196–E202 [DOI] [PubMed] [Google Scholar]
- 20. Thomson D. M., Hancock C. R., Evanson B. G., Kenney S. G., Malan B. B., Mongillo A. D., Brown J. D., Hepworth S., Fillmore N., Parcell A. C., Kooyman D. L., Winder W. W. (2010) Skeletal muscle dysfunction in muscle-specific LKB1 knockout mice. J. Appl. Physiol. 108, 1775–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee-Young R. S., Griffee S. R., Lynes S. E., Bracy D. P., Ayala J. E., McGuinness O. P., Wasserman D. H. (2009) Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J. Biol. Chem. 284, 23925–23934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Z. P., McConell G. K., Michell B. J., Snow R. J., Canny B. J., Kemp B. E. (2000) AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am. J. Physiol. Endocrinol. Metab. 279, E1202–E1206 [DOI] [PubMed] [Google Scholar]
- 23. Kobzik L., Reid M. B., Bredt D. S., Stamler J. S. (1994) Nitric oxide in skeletal muscle. Nature 372, 546–548 [DOI] [PubMed] [Google Scholar]
- 24. Shearer J., Fueger P. T., Vorndick B., Bracy D. P., Rottman J. N., Clanton J. A., Wasserman D. H. (2004) AMP kinase-induced skeletal muscle glucose but not long-chain fatty acid uptake is dependent on nitric oxide. Diabetes 53, 1429–1435 [DOI] [PubMed] [Google Scholar]
- 25. Stephens T. J., Chen Z. P., Canny B. J., Michell B. J., Kemp B. E., McConell G. K. (2002) Progressive increase in human skeletal muscle AMPK α2 activity and ACC phosphorylation during exercise. Am. J. Physiol. Endocrinol. Metab. 282, E688–E694 [DOI] [PubMed] [Google Scholar]
- 26. Roberts C. K., Barnard R. J., Scheck S. H., Balon T. W. (1997) Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am. J. Physiol. 273, E220–E225 [DOI] [PubMed] [Google Scholar]
- 27. Asai A., Sahani N., Kaneki M., Ouchi Y., Martyn J. A., Yasuhara S. E. (2007) Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS One 2, e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brenman J. E., Chao D. S., Xia H., Aldape K., Bredt D. S. (1995) Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752 [DOI] [PubMed] [Google Scholar]
- 29. Chao D. S., Gorospe J. R., Brenman J. E., Rafael J. A., Peters M. F., Froehner S. C., Hoffman E. P., Chamberlain J. S., Bredt D. S. (1996) Selective loss of sarcolemmal nitric oxide synthase in Becker muscular dystrophy. J. Exp. Med. 184, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crosbie R. H., Dovico S. A., Flanagan J. D., Chamberlain J. S., Ownby C. L., Campbell K. P. (2002) Characterization of aquaporin-4 in muscle and muscular dystrophy. FASEB J. 16, 943–949 [DOI] [PubMed] [Google Scholar]
- 31. Herrmann R., Straub V., Blank M., Kutzick C., Franke N., Jacob E. N., Lenard H. G., Kroger S., Voit T. (2000) Dissociation of the dystroglycan complex in caveolin-3-deficient limb girdle muscular dystrophy. Hum.Mol. Genet. 9, 2335–2340 [DOI] [PubMed] [Google Scholar]
- 32. Sander M., Chavoshan B., Harris S. A., Iannaccone S. T., Stull J. T., Thomas G. D., Victor R. G. (2000) Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A. 97, 13818–13823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lefer D. J., Jones S. P., Girod W. G., Baines A., Grisham M. B., Cockrell A. S., Huang P. L., Scalia R. (1999) Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 276, H1943–H1950 [DOI] [PubMed] [Google Scholar]
- 34. Tousoulis D., Kampoli A. M., Tentolouris C., Papageorgiou N., Stefanadis C. (2012) The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 10, 4–18 [DOI] [PubMed] [Google Scholar]
- 35. Tymvios C., Moore C., Jones S., Solomon A., Sanz-Rosa D., Emerson M. (2009) Platelet aggregation responses are critically regulated in vivo by endogenous nitric oxide but not by endothelial nitric oxide synthase. Br. J. Pharmacol. 158, 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kobayashi Y. M., Rader E. P., Crawford R. W., Iyengar N. K., Thedens D. R., Faulkner J. A., Parikh S. V., Weiss R. M., Chamberlain J. S., Moore S. A., Campbell K. P. (2008) Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 456, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Percival J. M., Anderson K. N., Gregorevic P., Chamberlain J. S., Froehner S. C. (2008) Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS One 3, e3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Percival J. M., Anderson K. N., Huang P., Adams M. E., Froehner S. C. (2010) Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J. Clin. Invest. 120, 816–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hudlicka O. (2011) Microcirculation in skeletal muscle. Muscles Ligaments Tendons J. 1, 3–11 [PMC free article] [PubMed] [Google Scholar]
- 40. Copp S. W., Holdsworth C. T., Ferguson S. K., Hirai D. M., Poole D. C., Musch T. I. (2013) Muscle fibre-type dependence of neuronal nitric oxide synthase-mediated vascular control in the rat during high speed treadmill running. J. Physiol. 591, 2885–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Planitzer G., Miethke A., Baum O. (2001) Nitric oxide synthase-1 is enriched in fast-twitch oxidative myofibers. Cell Tissue Res. 306, 325–333 [DOI] [PubMed] [Google Scholar]
- 42. Miller J. D., Pegelow D. F., Jacques A. J., Dempsey J. A. (2005) Skeletal muscle pump versus respiratory muscle pump: modulation of venous return from the locomotor limb in humans. J. Physiol. 563, 925–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.