

Abstract
The FERM domain containing protein Kindlin-3 has been recognized as a major regulator of integrin function in hematopoietic cells, but its role in neoplasia is totally unknown. We have examined the relationship between Kindlin-3 and breast cancer in mouse models and human tissues. Human breast tumors showed a ∼7-fold elevation in Kindlin-3 mRNA compared with nonneoplastic tissue by quantitative polymerase chain reaction. Kindlin-3 overexpression in a breast cancer cell line increased primary tumor growth and lung metastasis by 2.5- and 3-fold, respectively, when implanted into mice compared with cells expressing vector alone. Mechanistically, the Kindlin-3-overexpressing cells displayed a 2.2-fold increase in vascular endothelial growth factor (VEGF) secretion and enhanced β1 integrin activation. Increased VEGF secretion resulted from enhanced production of Twist, a transcription factor that promotes tumor angiogenesis. Knockdown of Twist diminished VEGF production, and knockdown of β1 integrins diminished Twist and VEGF production by Kindlin-3-overexpressing cells, while nontargeting small interfering RNA had no effect on expression of these gene products. Thus, Kindlin-3 influences breast cancer progression by influencing the crosstalk between β1 integrins and Twist to increase VEGF production. This signaling cascade enhances breast cancer cell invasion and tumor angiogenesis and metastasis.—Sossey-Alaoui, K., Pluskota, E., Davuluri, G., Bialkowska, K., Das, M., Szpak, D., Lindner, D. J., Downs-Kelly, E., Thompson, C. L., Plow, E. F. Kindlin-3 enhances breast cancer progression and metastasis by activating Twist-mediated angiogenesis.
Keywords: integrins, VEGF, VEGFR2, epithelial-to-mesenchymal-transition, tumor macrophages
A well-established characteristic that distinguishes transformed from nontransformed cells is their enhanced adhesive and migratory properties. The integrins are a large family of heterodimeric cell-surface receptors that have been extensively linked to the adhesive phenotype of neoplastic cells (1). Not only integrins themselves but also many of their intracellular binding partners, such as focal adhesion kinase (FAK), integrin-linked kinase (ILK), and Talin, which organize with integrins into adhesive structures, are altered in transformed cells (2–4). Recently, considerable evidence has emerged to implicate members of the kindlin family of FERM domain proteins as integrin binding partners and as key regulators of the adhesive functions mediated by integrins (5–7). Kindlins influence integrin activation, changes in their affinity or avidity for extracellular ligands (8–11). Specific and severe pathologies have been ascribed to deficiencies of each kindlin family member in both mice and humans; Kindlin-1 deficiency leads to skin and intestinal disorders (12, 13); Kindlin-2 deficiency is embryonically lethal in mice (14, 15); and Kindlin-3-deficient mice and humans display bleeding, susceptibility to infections and osteopetrosis (16, 17). Altered levels of Kindlin-1 (FERMT1) have been associated with the pathology of pancreatic and lung cancers (18, 19), and Kindlin-2 (FERMT2) levels are increased in lung and gastric cancers (19, 20). To date, the association of Kindlin-3 (FERMT3) levels with malignancy has not been considered as this homologue was believed to be restricted to cells of hematopoietic origin (21, 22). This notion was dispelled by the demonstration that Kindlin-3 was not only present but also contributes to the function of endothelial cells (23).
In examining endothelial cells within tumor tissue by immunofluorescence, we were surprised to note that tumor cells themselves were also reactive with Kindlin-3 antibodies (23). Knowing that a hallmark of transformed cells is their enhanced invasion and migration and knowing that Kindlin-3 can regulate the adhesive functions of αvβ3 and α5β1, 2 integrins with well-established connections to tumor metastasis (24, 25), we screened several different human tumor cell lines for Kindlin-3 mRNA by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR). Kindlin-3 was not only present but also was more highly expressed in more aggressive tumor lines. With these provocative correlations, we undertook a systematic study of the interrelationship between Kindlin-3 and tumor metastasis in the context of breast cancer (BC). Our data lead us to conclude that Kindlin-3 is a promoter of BC in humans and in mouse models. A mechanism underlying this unanticipated function of Kindlin-3 is delineated by showing that Kindlin-3 controls tumor angiogenesis.
MATERIALS AND METHODS
Patient selection
This study was conducted after approval from the Institutional Review Board at University Hospitals Case Medical Center (Cleveland, OH, USA), which waived the need for additional consent of participants, since there was no interaction with patients involved in the present study, and all samples and clinical data on specimens were provided in a deidentified form.
Fifty-eight pairs of formalin-fixed-paraffin-embedded (FFPE) human primary breast tumors/normal adjacent mammary tissue (Supplemental Table S1) were obtained from the paraffin archives of University Hospitals Case Medical Center. Molecular classification of the primary BC was achieved by immunohistochemistry (IHC) for estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) biomarkers. This cohort was used to quantify Kindlin-1, Kindlin-2, and Kindlin-3 mRNA expression levels as described below. Sections from each paraffin block were analyzed by hematoxylin and eosin (H&E) staining and evaluated by macrodissection to select cores that contained maximal amounts of cancer tissue.
Histopathological analysis
In addition to the 58 paired samples used for mRNA analysis, 12 tumor/normal breast sections representing 4 early-stage, 4 late-stage, and 4 metastatic breast tumors were obtained from the same paraffin archives and used to assess Kindlin-3 and von Willebrand factor (vWF) levels by IHC. Tumor specimens were reviewed and scored by a single pathologist (E.D.-K.). Cytosolic Kindlin-3 expression was scored as follows: 0 for negative, 1 for weak, 2 for moderate, and 3 for strong. The percentage of overall positively stained cells in each tumor was recorded. The product of the intensity score and percentage of positively stained cells is the Kindlin-3 score (Kindlin-3 score = intensity × percentage of positively stained cells; refs. 26, 27). Kindlin-3 expression was reported both for tumor and adjacent benign ductal epithelium. Adjacent benign epithelium was assessed to determine if there was any difference in the level, pattern, or intensity of Kindlin-3 expression between tumor and normal ductal epithelium. Similar procedures were followed to score vWF staining in the same tumor blocks.
Immunostaining
Tumor and lung tissues were collected after 4 or 8 wk and snap-frozen in optimal cutting temperature (OCT) medium (Sakura Finetek, Torrance, CA, USA), and 8-μm sections were prepared. Blood vessels were visualized with biotinylated rat anti-mouse CD31 (BD Biosciences, San Jose, CA, USA) followed by streptavidin-Alexa Fluor 568 conjugate or with rabbit anti-vWF (Dako, Carpinteria, CA, USA). Macrophages were detected with rat anti-F4/80 (eBioscience, San Diego, CA, USA). Tumor sections were also stained with the following antibodies: rabbit anti-fibronectin (Millipore, Temecula, CA, USA), rabbit anti-vimentin (Cell Signaling Technology, Danvers, MA, USA), rabbit anti-N-cadherin (Thermo Scientific, Rockford, IL, USA), and rabbit anti-P-vascular endothelial growth factor receptor 2 (VEGFR2; pYpY 1054/1059; Invitrogen, Camarillo, CA, USA) followed by Alexa 488 or Alexa 568-conjugated goat anti-rabbit IgG. Lung sections were stained with hematoxylin (Vector Laboratories, Burlingame, CA, USA) and eosin (Fisher Scientific Co., Kalamazoo, MI, USA) according to the manufacturer's instructions. Paraffin-embedded sections of human BC were stained with rabbit anti-kindlin-3 (F5276) developed by our group (23) and rabbit anti-vWF followed by rabbit IgG Vectastain Elite ABC-Peroxidase Kit (Vector Laboratories). Stained sections were analyzed using fluorescent or bright-field imaging microscopy (Leica, Wetzlar, Germany) and ImagePro Plus Capture and Analysis software (Media Cybernetics, Rockville, MD, USA). Vascular and F4/80-positive areas were quantified in 10–15 independent fields/section using Image Pro-Plus software as described previously (28).
Real-time qRT-PCR
Total RNA was extracted from cancer cell lines or tumor tissue using TRIzol reagent (Invitrogen), following to the manufacturer's instructions. cDNA was generated and used as a template for qRT-PCR performed as described previously (26, 29). qRT-PCR was performed using the respective gene-specific primers (SABiosciences, Valencia, CA, USA) and the RT2 SYBR Green/Fluorescein qPCR Master Mix (SABiosciences) following the manufacturer's instructions. qPCR was performed on the Bio-Rad iCycler PCR system (Bio-Rad, Berkeley, CA, USA) where the reaction mixtures were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The cycle threshold (Ct) values were calculated with SDS 1.4 software (Bio-Rad). The expression levels of each transcript were normalized using the 2−ΔΔCt method (30, 31) relative to GAPDH. The ΔCt was calculated by subtracting the Ct values of GAPDH from the Ct values of the transcript of interest. The ΔΔCt was then calculated by subtracting ΔCt of the matching normal human breast tissue from the ΔCt of cancer tissues or the ΔCt of MCF10A cell line for the established cancer cell lines. Fold change in the gene was calculated according to the equation 2−ΔΔCt.
Cell migration, invasion, and proliferation assays
Cell migration was assessed by wounding confluent cultures with a micropipette tip and immediately placing them in complete medium. Bright-field images were obtained immediately after wounding and after 16 h. The extent of wound closure was quantified by measuring the wound areas obtained from ≥6 different fields using ImageJ 1.34s (U.S. National Institutes of Health, Bethesda, MD, USA). For invasion assays, modified Boyden chambers were coated with Matrigel (1:10 dilution; BD Biosciences) and used to compare the invasiveness of MDA-MB-231 cells expressing enhanced green fluorescent protein (EGFP) alone to that expressing EGFP-Kindlin-3 and to compare the invasiveness of BT549 transfected with a nontargeting small interfering RNA (siRNA) to that transfected Kindlin-3-siRNA in response to 10% serum as described previously (27, 29, 32). Alterations in MDA-MB-231 and BT549 cell proliferation were determined by counting and comparing the number of viable cells over a period of 5 d after seeding the same number of cells on d 1.
Cell culture and transfections
The human MDA-MB-231 and BT549 BC cell lines and the nonmalignant mammary MCF10A cell line were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured as per ATTC instructions. Growth and morphology of the cells were routinely monitored, and key features were consistent with prior descriptions of the line. Derivative clones of the MDA-MB-231 cells with stable expression of EGFP and EGFP-Kindlin-3 fusion proteins were established by nucleofection of EGFPC2 plasmids expressing either EGFP alone or EGFP-Kindlin-3 fusion constructs (17). Stable and continuous expression of the EGFP derivatives was maintained by growing cells in the presence of neomycin (1 mg/ml), and enrichment for the GFP-expressing cells was established by fluorescence-activated cell sorting (FACS).
siRNA gene knockdown
The On-Target plus SMARTpool siRNA L-01677-01 against Kindlin-3 was from Thermo Scientific (Waltham, MA, USA); siRNAs targeting β1-integrin (sc-35674) and Twist (sc-38604) were from Santa Cruz Biotechnology (Dallas, TX, USA). Nontargeting siRNA siGenome control pool (D-001206-14-05) was from Thermo Scientific. Transient transfections were performed according to the manufacturer's instructions as described previously (29, 33).
Twist nuclear localization analysis
MDA-MB-231 cells expressing EGFP alone or EGFP-Kindlin-3 were seeded on 20 μg/ml fibronectin for 2 h, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked in horse serum, and stained with Twist antibodies for ∼18 h. Antigen-antibody complexes were detected by staining with Alexa 568-coupled secondary antibodies (Life Technologies) for 1 h. EGFP-expressing cells were visualized for EGFP fluorescence and actin with phalloidin conjugated to Alexa 633 (Life Technologies). DAPI-containing ProLong Gold Antifade reagent (Life Technologies) was used to mount the slides and to stain the nucleus. Stained cells were visualized with a ×63 1.4 oil objective using a Leica TCS-NT laser scanning confocal microscope (Imaging Core, Cleveland Clinic). Laser intensities were adjusted to eliminate crosstalk between channels, and images were collected using Leica Confocal Software (version 2.5 Build 1227). For the quantitative analysis of Twist localization, 309 GFP-expressing and 480 Kindlin 3-expressing cells were scored.
Tube formation assay
Human umbilical vein endothelial cell (HUVEC)-based tube formation assays were performed as described previously (23). Multiwell culture plates (12 wells/plate) were coated with 500 μl of growth factor-reduced Matrigel (BD Biosciences) and incubated at 37°C for 30 min. HUVECs were seeded at a density of 2.5 × 105 cells/well on the polymerized Matrigel, treated as specified, and incubated at 37°C for 18 h. Bright-field images were obtained immediately after seeding and again after 18 h. Tube formation was analyzed and quantified using ImageJ 1.34s.
Vascular endothelial growth factor (VEGF) ELISA assay
VEGF secreted in the conditioned medium was quantified using the ELISA development kit and the polyclonal rabbit anti-human VEGF from PeproTech (Rocky Hill, NJ, USA), following the manufacturer's instructions. In brief, 100 μl of capture antibody was transferred to a 96-well ELISA plate and incubated overnight at room temperature. Each well was washed 3 times and blocked by adding 300 μl of phosphate-buffered saline (PBS) containing 0.05% Tween 20, 5% sucrose, and 0.05% NaN3 for a minimum of 1 h. Following 3 washes, 100 μl conditioned medium/well was added, and the ELISA plate was incubated for 2 h at room temperature. Subsequently, 100 μl of streptavidin horseradish peroxidase was added to each well, and the plate was incubated for 30 min at room temperature. The plates were then washed, 10 μl of ABTS substrate (Sigma-Aldrich) was added, and optical density was measured after ∼20 min at 450 nm.
RESULTS
Kindlin-3 expression levels are increased in human BC
Several studies have reported the involvement of Kindlin-1 and Kindlin-2 in different human cancers (18–20), but the association of Kindlin-3 with the cancer etiology in general and human BC in particular has not been previously considered. To begin, we analyzed the expression levels of Kindlin-3 mRNA in a cohort of 58 paired human breast tumor/normal specimens. The tumors represented different stages, pathologies, and subtypes (Supplemental Table S1). By qRT-PCR, Kindlin-3 mRNA expression levels were elevated in >70% of tumors compared with their paired normal tissues (Fig. 1A). The average increase in Kindlin-3 expression levels was >7-fold in tumor vs. adjacent normal breast tissue (Fig. 1A and Supplemental Table S1). In 9 tumors (>17%), Kindlin-3 levels were elevated >10-fold (Fig. 1A and Supplemental Table S1). Furthermore, immunohistochemical analyses showed that Kindlin-3 immunoreactivity increased in late-stage and metastatic tumors compared with early-stage tumors (Fig. 1B). Kindlin-3 expression levels did not show a significant change (up-regulation or down-regulation) in the normal tissue adjacent to the tumors, with an average fold change of 1.1 ± 0.4 when normalized to Kindlin-3 expression levels in the nonmalignant breast cell line MCF10A (Supplemental Fig. S1A). We also used qRT-PCR analysis to assess Kindlin-1 and Kindlin-2 expression levels in this same BC cohort. We found no significant changes in the expression levels of Kindlin-1 (average expression levels of 1.18±0.56; Supplemental Fig. S1B). Kindlin-2, on the other hand, was down-regulated in most of the tumors (average expression levels of 0.68±0.57; Supplemental Fig. S1C). Similar trends in the expression profiles of the kindlins were observed in the MDA-MB-231 BC cell line (Supplemental Fig. S1D).
Figure 1.
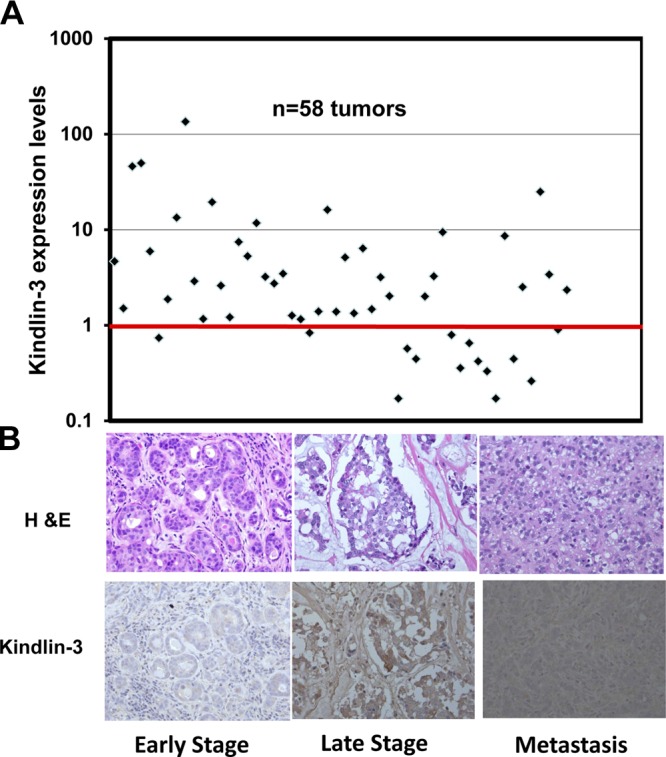
Kindlin-3 expression levels are increased in human BC tumors. A) Real-time qRT-PCR of Kindlin-3 transcripts in 58 BC tumors. Each data point represents the average fold change of Kindlin-3, from 3 independent PCRs, in 1 BC tumor compared with the adjacent normal mammary tissue. GAPDH was used as an internal control for normalization. Points under the red bar represent tumors in which Kindlin-3 is down-regulated; points above the red bar represent tumors in which Kindlin-3 is up-regulated. B) Immunohistochemical staining of BC human tumor sections with H&E (top panels) and Kindlin-3 antibody (bottom panels). Intensity of brown staining correlates with Kindlin-3 expression levels; low in early stage BC and high in metastatic tumors. Four tumors from early stage, 4 tumors from late stage, and 4 metastatic tumors were stained; 1 representative section from each stage is shown.
Kindlin-3 enhances migration and invasion of BC cells in vitro
In screening a series of BC cell lines by RT-PCR, we noted that MDA-MB-231 cells, a well-established human triple-negative BC (TNBC) cell line, had abundant albeit relatively low Kindlin-3 mRNA expression compared with other BC lines (BT549, Supplemental Fig. S2, and T47D, not shown). In addition, the expression profile of the kindlins in MDA-MB-231 cells was similar to that in tumors; i.e., neutral Kindlin-1, down-regulated Kindlin-2 and up-regulated Kindlin-3 (Supplemental Fig. S1D). Since the MDA-MB-231 line is widely used in BC progression and metastasis studies, we focused on this cell line to manipulate Kindlin-3 expression. We generated MDA-MB-231-derived clones with stable expression of EGFP-alone or EGFP-Kindlin-3 fusion protein (Kindlin-3). Stable expression of EGFP-alone or Kindlin-3 was monitored periodically by flow cytometry (Fig. 2A) and Western blots (Fig. 2B). A major characteristic of cancer cells is their motility and invasive properties, and we used wound closure and Boyden chamber invasion assays to assess the effects of Kindlin-3 on these cellular responses. Overexpression of Kindlin-3 in MDA-MB-231 cells resulted in a significant increase of migration in the wound closure assay; whereas the Kindlin-3-overexpressing cells almost completely closed the wounded area after 16 h, the cells expressing EGFP alone failed to do so under the same conditions (Fig. 2C, D). In the invasion assay, significantly more Kindlin-3-overexpressing cells traversed the Matrigel-coated inserts and invaded the lower chamber as compared with the cells expressing EGFP alone (Fig. 2E, F). Thus, Kindlin-3 confers enhanced migratory and invasive advantages to these BC cells. The enhanced migration and invasion of the Kindlin-3-overexpressing MDA-MB-231 cells were not due to increased cell proliferation. No difference in cell proliferation between the EGFP-expressing and the Kindlin-3-overexpressing MDA-MB-231 cells was detected (Supplemental Fig. S3). On the other hand, siRNA knockdown of Kindlin-3 in BT549 cells resulted in significant inhibition of migration (∼70% of the wound remained open in the Kindlin-3 knockdown cells compared with 40% in cells transfected with the nontargeting si-RNA, P<0.001; Supplemental Fig. S4). Similarly, knockdown of Kindlin-3 inhibited BT549 invasion (>3-fold decrease, P<0.005; Supplemental Fig. S5).
Figure 2.

Kindlin-3 enhances migration and invasion of BC cells in vitro. A) Flow cytometry of untransfected (red histogram), EGFP-expressing (gray histogram), and EGFP-Kindlin-3-overexpressing (solid black histogram) MDA-MB-231 cells. B) Immunoblots of total cell lysates from MDA-MB-231 cells expressing EGFP alone or EGFP-Kindlin-3, probed with anti-Kindlin-3 (top panel), anti-GFP (middle panel), or anti-β-actin as a loading control (bottom panel). C) Representative micrographs of EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells that were induced to migrate into denuded wounds over a 16 h period. D) Size of the remaining open wound after 16 h was measured from 12 different wounds and plotted as the percentage of wound at time 0. E) Representative micrographs of EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells that were induced to invade through a Matrigel-coated membrane. F) Invading cells were counted from 6 different fields and plotted as an average of the number of invading cells per field. All data are representative of 3 independent experiments or are means ± se (n=3; P<0.05, Student's t test).
Kindlin-3 enhances growth and metastasis of BC cells in in vivo
To extend the effects of Kindlin-3 overexpression to an in vivo setting, we performed spontaneous metastasis and lung colonization assays in mice. In the spontaneous metastasis assay, the MDA-MB-231 cells expressing EGFP alone or Kindlin-3 were implanted in the mammary fat pads of SCID mice, and tumor growth was assessed over 8 wk, after which the primary tumors were excised, and the mice were followed for an additional 5 wk to assess metastasis to the lungs. We found that Kindlin-3 overexpression enhanced the growth of primary tumors and metastasis to lungs (Fig. 3). While every mouse in the EGFP-alone and the Kindlin-3 groups developed tumors (100% tumor incidence) after a ∼4-wk latency period, the tumor burden was significantly higher (P<0.05) in the mice implanted with the Kindlin-3-overexpressing cells than those implanted with cells expressing EGFP alone (Fig. 3A). Metastasis to the lungs was also significantly higher (P<0.001) in the mice injected with Kindlin-3-overexpressing cells compared with the cells expressing EGFP alone, as determined by the number and size of metastatic foci quantified in the lungs (Fig. 3B, C). Thus, Kindlin-3 not only enhances the rate of tumor growth at the primary site in vivo but also increases the formation of distant metastases. These increases were not a result of an activation of tumor cell proliferation by Kindlin-3 overexpression, as the number of viable cells between the EGFP- and Kindlin-3-expressing cells was similar over extended growth periods.
Figure 3.

Kindlin-3 enhances primary tumor growth and metastasis of BC in vivo in mouse models. Effect of Kindlin-3 on primary tumor growth (A) and spontaneous lung metastasis (B, C) and in lung colonization model (D–F). A) Tumor growth curves for EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells. Data points represent means ± sd of tumor volume, showing a significant difference (P<0.05) between the EGFP- and Kindlin-3-expressing cells. Mice (SCID) were monitored for 70 d after implantation of cancer cells in their mammary fat pads, after which all were analyzed for lung metastases. B) H&E staining of lung sections of mice implanted with the EGFP-expressing MDA-MB-231 cells (GFP) and mice implanted with Kindlin-3-overexpressing MDA-MB-231 cells. Metastases are delineated by dark lines. C) Quantification of lung surface metastasis nodules from the groups of mice injected with the EGFP-expressing and EGFP-Kindlin-3-overexpressing cells and plotted as an average of the number of metastatic nodules per lung lobe (P<0.001, Student's t test). D) Representative confocal micrographs of lung sections and their corresponding H&E staining from mice injected with EGFP-expressing and Kindlin-3-overexpresing MDA-MB-231 cells. Each green area represents a lung metastasis and is delineated by a dark line in the H&E staining micrographs. E) Quantification of lung metastasis from the 2 groups of mice described in D. F) Genomic PCR analysis of DNA from lungs of mice injected with the EGFP-expressing or Kindlin-3-overexpressing cells. Human-specific ERVK6A gene can be amplified in human DNA (231 and H-Ctrl) but not in mouse DNA (M-Ctrl). Presence of the human-specific ERVK6A DNA sequences in the mouse tissues (GFP and K3) indicates the presence of human cells in these tissues. Intensity of the PCR product of the ERVK6A gene correlates with the level of metastasis. Mouse Wave3 gene is used as a control for integrity of the mouse DNA in each sample. N, number of mice per group.
For the lung colonization assay, the 2 MDA-MB-231 cell cultures were injected into the tail veins of SCID mice. Metastasis to the lungs was surveyed by H&E staining and immunofluorescence of lung sections. Overexpression of Kindlin-3 resulted in a significant increase in pulmonary metastasis (Fig. 3D). By quantifying the lung surface tumor nodules, the Kindlin-3-overexpressing cells produced larger and numerous tumors in the lungs, whereas the cells expressing EGFP alone produced smaller and fewer (3 times fewer, P<0.001) metastases (Fig. 3E). In addition, we also surveyed metastasis to the lungs using genomic DNA-based PCR with primers specific to the human ERVK6 DNA sequence (27). A strong PCR product was detected in the lungs from the mouse injected with Kindlin-3-overexpressing cells, while the PCR signal obtained from lungs of mice injected with the cells expressing EGFP alone was much weaker (Fig. 3F). By both methods of quantitation, Kindlin-3-overexpressing cells resulted in a 3-fold increase in the tumors that metastasized to the lung (Fig. 3F).
Kindlin-3 enhances tumor angiogenesis by increasing VEGF-A expression
Angiogenesis is critical for tumor growth at primary and metastatic sites; hence, we determined whether Kindlin-3 affected tumor microvasculature. IHC using antibody to CD31, an endothelial cell-specific marker, revealed a significant increase (3-fold, P=0.001) in the number and size, as well as a dismorphic architecture of blood vessels in tumors from Kindlin-3-overexpressing compared with EGFP-expressing cells (Fig. 4A, B). As an additional measure of tumor angiogenesis, CD31 mRNA expression levels were assessed by qRT-PCR and were found to be significantly higher (7-fold, P=0.035) in the tumors derived from the Kindlin-3-overexpressing cells (Fig. 4C). These observations from the mouse models were further confirmed in human primary BC specimens. Staining for vWF, another blood vessel marker, was elevated in late-stage compared with early-stage tumors (Fig. 4D).
Figure 4.

Kindlin-3 enhances tumor angiogenesis by increasing VEGF-A expression. A) Representative immunofluorescence confocal micrographs of sections of mammary fat pad tumors from mice implanted with EGFP-expressing (GFP) and Kindlin-3-overexpressing MDA-MB-231 cells stained with anti CD31 (red) to detect tumor-associated blood vessels. Cell nuclei were counterstained with DAPI (blue). B) Quantification of angiogenesis from the EGFP and Kindlin-3 groups, as determined by the average blood vessel area per tumor section (n=number of mice per group). C) qRT-PCR of the CD31 transcripts from the mammary fat pad tumors described in A. D) Representative confocal micrographs of human BC tumor sections stained with a vWF antibody and their corresponding H&E staining. Intensity of the brown staining correlates with expression of vWF protein; low in early stage and high in late stage BC tumors. E) qRT-PCR of the VEGF-A transcripts from the mammary fat pad tumors described in A. F) Representative immunofluorescence confocal micrographs of sections of mammary fat pad tumors described in A, stained for the phosphorylated form of VEGFR2 (green fluorescence). Cell nuclei were counterstained with DAPI. G) Immunoblots of protein lysates from mammary fat pad tumors from mice implanted with EGFP-expressing (GFP-T1 to -T3) and Kindlin-3-overexpressing (K3-T1 to -T3) MDA-MB-231 cells with anti-phospho-VEGFR2. β-Actin antibody is a loading control.
We next investigated whether the increased angiogenesis induced by Kindlin-3 was driven by VEGF. Using qRT-PCR, we found that VEGF-A levels were significantly elevated (∼3-fold, P<0.05) in tumors derived from the mice implanted with Kindlin-3-overexpressing cells compared with those derived from cells expressing EGFP alone (Fig. 4E). We then assessed phosphorylation status of VEGFR2, the major receptor for VEGF, using both IHC (Fig. 4F) and immunoblotting (Fig. 4G). Phosphorylation of VEGFR2, indicative of receptor activation, was elevated in the Kindlin-3-derived tumors, compared with their EGFP-alone-derived controls (Fig. 4F). Densitometry of the immunoblots (Fig. 4G) showed a 3-fold increase of p-VEGFR2 in the Kindlin-3-derived tumors.
Blockade of secreted VEGF-A from cancer cells inhibits endothelial angiogenesis
To test whether Kindlin-3 enhances tumor angiogenesis by stimulating VEGF production, we performed an ELISA to quantify VEGF-A levels in the conditioned media of Kindlin-3-overexpressing and EGFP-expressing cells. Secreted levels of VEGF-A were elevated in the Kindlin-3-overexpressing cells (2-fold, P<0.05) compared with the cells expressing EGFP alone (Fig. 5A). Conversely, knockdown of Kindlin-3 in BT549 cells resulted in a 5-fold decrease of secreted VEGF-A compared with the nontargeting-siRNA-(NT-siRNA)-transfected cells (P<0.001, Supplemental Fig. S6). We next tested whether the conditioned medium of Kindlin-3-overexpressing cells would stimulate angiogenesis to a greater extent than the conditioned medium from cells expressing EGFP alone in an in vitro angiogenesis assay. Early-passage HUVECs were seeded onto growth factor-reduced Matrigel with or without VEGF-A supplementation. Over time, the HUVECs given VEGF-A organized in tube-like networks. The HUVECs that were supplemented with the conditioned medium derived from the Kindlin-3-overexpressing cells formed closed and regularly shaped tube-like structures in the absence of added VEGF-A, resembling those formed with addition of VEGF-A. In contrast, HUVECs supplemented with the conditioned medium derived from the cells expressing EGFP alone formed fewer tubes and, in most cases, failed to fully close, resembling those formed in serum-free medium (Fig. 5B, C). These results are consistent with our findings that the Kindlin-3-overexpressing cells secrete higher VEGF-A levels (Fig. 5A). In addition, preincubation of HUVECs plus Kindlin-3-conditioned medium with a soluble VEGFR inhibitor (sFLT) completely inhibited tube formation by the HUVECs (Fig. 5D), consistent with VEGF being the major angiogenic factor contributed by the conditioned medium.
Figure 5.

Blockade of secreted VEGF-A from cancer cells inhibits angiogenesis. A) Quantification of secreted VEGF-A in the conditioned media of EGFP-expressing (GFP) and Kindlin-3-overexpressing (K3) MDA-MB-231 cells using ELISA; n = number of repeats. B) Bright-field microscopy of tube-formation structures of HUVECs treated with serum-free medium (SFM), SFM supplemented with VEGF-A (10 ng/ml), or conditioned media from EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells. C) Quantification of tube formation. Number of closed tubes was counted in 12 different fields and plotted as the average ± se number of closed tubes per field. Each assay was repeated ≥3 times. *P < 0.05, **P < 0.05; Student's t test. D) Bright-field micrographs of tube structures of HUVECs treated with SFM supplemented with VEGF-A (10 ng/ml) or conditioned media from EGFP- and Kindlin-3-expressing MDA-MB-231 cells, in the absence (top panels) or the presence (bottom panels) of sFLT, a soluble VEGFR inhibitor.
Kindlin-3 modulates cancer angiogenesis through regulation of Twist expression and macrophage recruitment
The transcription factor Twist has been shown to be a master regulator of cancer metastasis by promoting tumor angiogenesis (34). Accordingly, we tested differences in the Twist levels, as well as its related transcription factors Slug and Snail, in Kindlin-3-overexpressing cells and their derived tumors compared with cells expressing EGFP alone and their derived tumors by Western blot. While a slight increase was observed in Slug and Snail levels, Twist expression was readily detected in the Kindlin-3-overexpressing cells and their derived tumors, while little or no Twist could be detected in the cells expressing EGFP alone and their derived tumors (Fig. 6A). In addition, a slight decrease, albeit not significant, was observed for Snail in the tumors derived from the Kindlin-3-overexpressing cells (Fig. 6A). Based on densitometry, the levels of Twist were ≥10 times higher in the Kindlin-3-derived tumors than in the EGFP-alone-derived counterparts. Furthermore, by immunocytochemistry, we showed a significant increase of Twist localization to the nucleus of cancer cells expressing Kindlin-3, which is an indication of increased potential for activation of Twist target genes (Fig. 6B, C).
Figure 6.

Kindlin-3 modulates angiogenesis through the regulation of Twist expression and macrophage recruitment. A) Immunoblotting with the indicated antibodies of protein lysates from EGFP-expressing (GFP) and Kindlin-3-overexpressing (K3) MDA-MB-231 cells and their respective mammary fat pad-derived tumors (GFP-T1 to -T3 and K3-T1 to -T3, respectively). B) Representative immunofluorescence confocal microscopy of EGFP-expressing (top panels) and Kindlin-3-overexpressing (bottom panels) MDA-MB-231 cells stained with Twist antibody. Cell nuclei were counterstained with DAPI. C) Quantification of Twist nuclear localization in the EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells, plotted as the average percentage of cells with nuclear Twist staining per field. (n=number of fields per experiment from ≥3 repeats). D) Immunoblotting of protein lysates from the Kindlin-3-overexpressing MDA-MB-231 cells with anti-Twist. Cells were first transfected with either an NT-siRNA (NT-si) or a Twist-targeting siRNA (si-Twist). Anti-β-actin serves as a loading control. E) Quantification of secreted VEGF-A into the conditioned medium of Kindlin-3-overexpressing MDA-MB-231 cells using ELISA after transfection with either NT-si or si-Twist; n = number of repeats. F) Representative immunofluorescence confocal micrographs of sections of mammary fat pad tumors derived from EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells, stained with anti-F4/80 macrophage specific antibody (green fluorescence). Cell nuclei were counterstained with DAPI. G) Quantification of macrophage infiltration in the tumors described in F, plotted as the average of the percentage of F4/80-positive area per field; n = number of fields.
To further probe the role of Kindlin-3 in the Twist-VEGF-angiogenesis axis, we assessed the effect of loss of Twist expression on VEGF-A secretion. Knockdown of Twist in the Kindlin-3-overexpressing cells (Fig. 6D) decreased VEGF-A secretion by 3-fold (P<0.05), compared with the levels of VEGF-A secreted by the same cells transfected with the control NT-siRNA (Fig. 6E). In support of this finding, we found that siRNA knockdown of Kindlin-3 in BT549 also resulted in inhibition of Twist expression (Supplemental Fig. S2) and subsequent inhibition of VEGF-A secretion (Supplemental Fig. S6). A recent study has also shown that Twist induces tumor angiogenesis via recruitment of the tumor-associated macrophages (TAMs; ref. 35). Notably, immunostaining of tumor sections for a macrophage-specific marker, F4/80, detected a significant enrichment (4-fold, P=0.01) in TAMs in the Kindlin-3-derived tumors (Fig. 6F, G), where higher levels of Twist were also detected (Fig. 6A) compared with the EGFP-alone-derived tumors.
Kindlin-3-Twist-VEGF-angiogenesis axis is integrin dependent
Kindlin-3 is an established modulator of the integrin activation. We found that both the total surface expression (measured with monoclonal antibody HB1.1) and the activation of β1-integrin (measured with HUTS-4, an antibody specific for activated β1 integrins; ref. 36) increased on the Kindlin-3-overexpressing vs. EGFP-expressing cells (Fig. 7A, B), as measured by flow cytometry. Next, we sought to determine whether manipulation of β1-integrin levels in the Kindlin-3-overexpressing cells would affect VEGF-A secretion. We used siRNA to specifically knock down β1-integrin expression in the Kindlin-3-overexpressing cells and assessed the effects on β1-integrin activation levels with HUTS-4. As expected, loss of β1-integrin expression (Fig. 7C) resulted in a significant decrease in the activation levels of cell surface β1-integrin in the Kindlin-3-expressing cells when compared with the same cells transfected with NT-siRNA (Fig. 7D). More important, loss of β1-integrin expression and activation in the Kindlin-3-overexpressing cells also resulted in a significant decrease (3-fold, P<0.05) in the levels of secreted VEGF-A (Fig. 7E), thereby supporting the association between VEGF-A production by Kindlin-3 and β1-integrin and placing the β1-integrin ⇔ Kindlin-3 interplay upstream of this signaling pathway.
Figure 7.

Kindlin-3-Twist-VEGF-angiogenesis signaling axis is integrin dependent. A) Quantification of cell surface β1-integrin activation in the EGFP-expressing (GFP) and Kindlin-3-overexpressing (K3) MDA-MB-231 cells, as measured by HUTS4 antibody binding, normalized to total β1-integrin expression levels and plotted as a fold change to the EGFP-expressing cells; n = number of repeats. B) Quantification of cell surface β1-integrin expression levels from the cells described in A; n = number of repeats. C) Immunoblotting with the indicated antibodies of protein lysates from the Kindlin-3-overexpressing MDA-MB-231 cells. Cells were first transfected with either an NT-siRNA (NT-si) or with a β1-integrin-targeting siRNA (si-β1). Anti-β-actin is a loading control. D) Quantification of cell surface β1-integrin activation levels in the EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells transfected with either NT-si or β1-si. β1-Integrin activation levels were measured by HUTS4 antibody binding, normalized to total β1-integrin expression levels, and plotted as a fold change in the EGFP-expressing cells transfected with the NT-siRNA. *P < 0.05, Student's t test. E) Quantification of secreted VEGF-A in the conditioned medium of Kindlin-3-overexpressing MDA-MB-231 cells using ELISA after transfection with NT-si or si-β1; n = number of repeats. F) Immunoblotting with the indicated antibodies of protein lysates from the Kindlin-3-overexpressing MDA-MB-231 cells that were transfected with either an NT-siRNA (NT), a Kindlin-3 siRNA (si-K3), si-β1, or a Twist siRNA (si-Twist). G) Quantification of secreted VEGF-A in the conditioned media of cells described in F using ELISA; n = number of repeats. *P < 0.05, **P < 0.05, ***P < 0.05; Student's t test.
To further confirm the involvement of the individual components of this signaling axis leading to increased VEGF-A production and of tumor angiogenesis, we used siRNAs to specifically inhibit expression of each gene separately in the Kindlin-3-overexpressing cells and assessed changes in secreted VEGF-A. As shown in Fig. 7F, each siRNA was very effective in knocking down the expression of its respective gene target, i.e., Kindlin-3, β1-integrin, and Twist, without affecting expression of the other 2 genes (Fig. 7F). Furthermore, the control NT-siRNA had no effect on expression of any of the 3 genes, confirming the specificity of the siRNAs used (Fig. 7F). When the conditioned media from cells of individual knockdown experiments were analyzed for VEGF-A levels, we found that loss of expression of Kindlin-3, β1-integrin, or Twist individually resulted in 3-, 2-, and 6-fold reduction, respectively, in VEGF secretion (Fig. 7G).
Kindlin-3-mediated regulation of Twist activates the epithelial-to-mesenchymal transition program
One of the hallmarks of neoplastic transformation is the epithelial-to-mesenchymal transition (EMT), whereby polarized cancer cells acquire an apolar fibroblastoid-like phenotype and become highly motile. EMT programs are also accompanied by up-regulation of fibroblastoid markers (e.g., N-cadherin and fibronectin). Based on our findings that Kindlin-3 enhances tumor progression and metastasis of BC, a process that also requires EMT, we examined whether Kindlin-3 expression modulates EMT markers. Western blots of cell and tumor lysates showed that expression levels of both N-cadherin and fibronectin were markedly elevated (4- and 10-fold, respectively) in the tumors derived from mice implanted with the Kindlin-3-overexpressing cells compared with the cells expressing EGFP alone (Fig. 8A). This finding was confirmed by immunostaining of tumor sections, where increased fibronectin deposition (Fig. 8B) and N-cadherin expression (Fig. 8C) was observed in tumors derived from the Kindlin-3-overexpressing cells compared with the tumors derived from the cells expressing EGFP alone. The observation that overexpression of Kindlin-3 resulted in the activation of these EMT markers in the derived tumors while having no significant effect in the cells grown in the tissue culture supports the involvement of the tumor microenvironment in the Kindlin-3-mediated activation of the EMT program.
Figure 8.

Kindlin-3-mediated regulation of Twist activates the EMT program in BC. A) Immunoblotting with the indicated antibodies of protein lysates from the EGFP-expressing (GFP) and Kindlin-3-overexpressing (K3) MDA-MB-231 cells and their respective mammary fat pad-derived tumors (GFP-T1 to -T3 and K3-T1 to -T3, respectively). Anti-β-actin antibody serves as a loading control. B, C) Representative immunofluorescence confocal micrographs of sections of mammary fat pad tumors derived from EGFP-expressing and Kindlin-3-overexpressing MDA-MB-231 cells, stained with anti-fibronectin (B) and anti-N-cadherin (C) (green fluorescence). Cell nuclei were counterstained with DAPI. D) Model describing how the β1-integrin-Kindlin-3 interplay modulates the molecular signaling pathways that regulate BC progression and metastasis.
DISCUSSION
Metastasis remains the major cause of death in patients with BC. Moreover, disseminated BC cells can escape clinical detection by remaining dormant for years before reemerging as incurable secondary tumors that are resistant to standard-of-care chemotherapeutic agents. Our findings now establish Kindlin-3 as a promoter of BC progression and metastasis as supported by the following findings: Kindlin-3 is overexpressed in the majority of human BCs; Kindlin-3 stimulates BC migration and invasion in vitro and tumor progression and metastasis in mice in vivo; Kindlin-3 induces tumor angiogenesis through the activation of VEGF secretion and macrophage recruitment, downstream of Twist; and the integrin-Kindlin-3 interface encourages Twist-mediated activation of tumor angiogenesis and induction of the EMT program. Collectively, our findings identify Kindlin-3 as a novel and key mediator of metastatic progression in BC by stimulating tumor angiogenesis via the activation of the Twist-VEGF signaling axis and by activating the EMT, both of which are critical for cancer progression and metastasis. A model depicting the interrelationships that we have uncovered is depicted in Fig. 8D. The hallmarks of cancer delineated by Hanahan and Weinberg (37) include activation of invasion and metastasis, induction of angiogenesis, sustainability of proliferative signaling, evasion from growth suppressors, resistance to cell death, and gain of replicative immortality (37). Our data show that Kindlin-3 modulates at least 3 of the 6 hallmarks of cancer; i.e., activation of cancer invasion and metastasis and induction of tumor angiogenesis. Strongly supporting the involvement of Kindlin-3 in BC pathology, mining of the Oncomine database (http://www.oncomine.com) indicated that Kindlin-3 was in the top 3% of gene products elevated in BC (P<10−12). Also of note, in the Oncomine database, Kindlin-2 is down-regulated and Kindlin-1 is unaffected in BC, although these other kindlins have been reported to be elevated in other cancers (18–20). Thus, our panel of 58 BC tumors appears to be representative with respect to kindlin expression levels.
Shuttling of Twist between the cytoplasm and the nucleus is regulated through post-translational modifications and was also shown to be modulated by integrin-mediated interaction with both the extracellular matrix and intracellular cytoskeleton proteins (38). Our results show that the Kindlin-3-mediated regulation of angiogenesis, which requires nuclear localization of Twist, is influenced by integrin activation, since inhibition of integrin expression and activation severely dampened VEGF-A secretion and consequent angiogenesis. Overexpression of Twist, a phenomenon that we found to be dependent on increased expression of Kindlin-3, has been found to confer resistance to doxorubicin and paclitaxel, 2 chemotherapeutic drugs commonly used for BC treatment (39, 40). Therefore, targeting Kindlin-3, the upstream activator of Twist, in a therapeutic setting may prove beneficial for patients with BC.
Supplementary Material
Acknowledgments
The authors thank Brian McCue for technical assistance.
This work was supported in part by National Heart, Lung, and Blood Institute (U.S. National Institutes of Health) grants P01-HL-073311 and R01-HL-096062 and Case Comprehensive Cancer Center grant P30-CA-43703.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- Ab
- antibody
- BC
- breast cancer
- EGFP
- enhanced green fluorescent protein
- EMT
- epithelial-to-mesenchymal transition
- ER
- estrogen receptor
- H&E
- hematoxylin and eosin
- HER2
- human epidermal growth factor receptor 2
- HUVEC
- human umbilical vein endothelial cell
- IHC
- immunohistochemistry
- NT-siRNA
- nontargeting small interfering RNA
- PR
- progesterone receptor
- PCR
- polymerase chain reaction
- qRT-PCR
- quantitative reverse transcription polymerase chain reaction
- siRNA
- small interfering RNA
- VEGF
- vascular endothelial growth factor
- VEGFR2
- vascular endothelial growth factor receptor 2
- vWF
- vonWillebrand factor
REFERENCES
- 1. Ganguly K. K., Pal S., Moulik S., Chatterjee A. (2013) Integrins and metastasis. Cell Adh. Migr. 7, 251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cance W. G., Kurenova E., Marlowe T., Golubovskaya V. (2013) Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal. 6, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desiniotis A., Kyprianou N. (2011) Significance of talin in cancer progression and metastasis. Int. Rev. Cell. Mol. Biol. 289, 117–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shibue T., Brooks M. W., Weinberg R. A. (2013) An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 24, 481–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malinin N. L., Plow E. F., Byzova T. V. (2010) Kindlins in FERM adhesion. Blood 115, 4011–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plow E. F., Qin J., Byzova T. (2009) Kindling the flame of integrin activation and function with kindlins. Curr. Opin. Hematol. 16, 323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ye F., Petrich B. G. (2010) Kindlin: helper, co-activator, or booster of talin in integrin activation? Curr. Opin. Hematol. 18, 356–360 [DOI] [PubMed] [Google Scholar]
- 8. Larjava H., Plow E. F., Wu C. (2008) Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 9, 1203–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Legate K. R., Fassler R. (2009) Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J. Cell Sci. 122, 187–198 [DOI] [PubMed] [Google Scholar]
- 10. Moser M., Legate K. R., Zent R., Fassler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 11. Brahme N. N., Calderwood D. A. (2012) Cell adhesion: a FERM grasp of the tail sorts out integrins. Curr. Biol. 22, R692–R694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ussar S., Moser M., Widmaier M., Rognoni E., Harrer C., Genzel-Boroviczeny O., Fassler R. (2008) Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 4, e1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siegel D. H., Ashton G. H., Penagos H. G., Lee J. V., Feiler H. S., Wilhelmsen K. C., South A. P., Smith F. J., Prescott A. R., Wessagowit V., Oyama N., Akiyama M., Al Aboud D., Al Aboud K., Al Githami A., Al Hawsawi K., Al Ismaily A., Al-Suwaid R., Atherton D. J., Caputo R., Fine J. D., Frieden I. J., Fuchs E., Haber R. M., Harada T., Kitajima Y., Mallory S. B., Ogawa H., Sahin S., Shimizu H., Suga Y., Tadini G., Tsuchiya K., Wiebe C. B., Wojnarowska F., Zaghloul A. B., Hamada T., Mallipeddi R., Eady R. A., McLean W. H., McGrath J. A., Epstein E. H. (2003) Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am. J. Hum. Genet. 73, 174–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montanez E., Ussar S., Schifferer M., Bosl M., Zent R., Moser M., Fassler R. (2008) Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 22, 1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dowling J. J., Gibbs E., Russell M., Goldman D., Minarcik J., Golden J. A., Feldman E. L. (2008) Kindlin-2 is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ. Res. 102, 423–431 [DOI] [PubMed] [Google Scholar]
- 16. Svensson L., Howarth K., McDowall A., Patzak I., Evans R., Ussar S., Moser M., Metin A., Fried M., Tomlinson I., Hogg N. (2009) Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malinin N. L., Zhang L., Choi J., Ciocea A., Razorenova O., Ma Y. Q., Podrez E. A., Tosi M., Lennon D. P., Caplan A. I., Shurin S. B., Plow E. F., Byzova T. V. (2009) A point mutation in KINDLIN3 ablates activation of three integrin subamilies in humans. Nat. Med. 15, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mahawithitwong P., Ohuchida K., Ikenaga N., Fujita H., Zhao M., Kozono S., Shindo K., Ohtsuka T., Aishima S., Mizumoto K., Tanaka M. (2013) Kindlin-1 expression is involved in migration and invasion of pancreatic cancer. Int. J. Oncol. 42, 1360–1366 [DOI] [PubMed] [Google Scholar]
- 19. Zhan J., Zhu X., Guo Y., Wang Y., Wang Y., Qiang G., Niu M., Hu J., Du J., Li Z., Cui J., Ma B., Fang W., Zhang H. (2012) Opposite role of Kindlin-1 and Kindlin-2 in lung cancers. PLoS One 7, e50313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen Z., Ye Y., Dong L., Vainionpaa S., Mustonen H., Puolakkainen P., Wang S. (2012) Kindlin-2: a novel adhesion protein related to tumor invasion, lymph node metastasis, and patient outcome in gastric cancer. Am. J. Surg. 203, 222–229 [DOI] [PubMed] [Google Scholar]
- 21. Moser M., Nieswandt B., Ussar S., Pozgajova M., Fassler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 22. Moser M., Bauer M., Schmid S., Ruppert R., Schmidt S., Sixt M., Wang H. V., Sperandio M., Fassler R. (2009) Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305 [DOI] [PubMed] [Google Scholar]
- 23. Bialkowska K., Ma Y. Q., Bledzka K., Sossey-Alaoui K., Izem L., Zhang X., Malinin N., Qin J., Byzova T., Plow E. F. (2010) The integrin co-activator Kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J. Biol. Chem. 285, 18640–18649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varner J. A., Cheresh D. A. (1996) Tumor angiogenesis and the role of vascular cell integrin alphavbeta3. Important Adv. Oncol. 1996, 69–87 [PubMed] [Google Scholar]
- 25. Juliano R. L. (1993) The role of beta 1 integrins in tumors. Semin. Cancer Biol. 4, 277–283 [PubMed] [Google Scholar]
- 26. Kulkarni S., Augoff K., Rivera L., McCue B., Khoury T., Groman A., Zhang L., Tian L., Sossey-Alaoui K. (2012) Increased expression levels of WAVE3 are associated with the progression and metastasis of triple negative breast cancer. PLoS One 7, e42895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sossey-Alaoui K., Safina A., Li X., Vaughan M. M., Hicks D. G., Bakin A. V., Cowell J. K. (2007) Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am. J. Pathol. 170, 2112–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pluskota E., Dowling J. J., Gordon N., Golden J. A., Szpak D., West X. Z., Nestor C., Ma Y. Q., Bialkowska K., Byzova T., Plow E. F. (2011) The integrin coactivator kindlin-2 plays a critical role in angiogenesis in mice and zebrafish. Blood 117, 4978–4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sossey-Alaoui K., Bialkowska K., Plow E. F. (2009) The miR200 family of microRNAs regulates WAVE3-dependent cancer cell invasion. J. Biol. Chem. 284, 33019–33029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the [2−delta delta C(T)] method. Methods 25, 402–488 [DOI] [PubMed] [Google Scholar]
- 31. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 32. Sossey-Alaoui K., Ranalli T. A., Li X., Bakin A. V., Cowell J. K. (2005) WAVE3 promotes cell motility and invasion through the regulation of MMP-1, MMP-3, and MMP-9 expression. Exp. Cell Res. 308, 135–145 [DOI] [PubMed] [Google Scholar]
- 33. Sossey-Alaoui K., Downs-Kelly E., Das M., Izem L., Tubbs R., Plow E. F. (2011) WAVE3, an actin remodeling protein, is regulated by the metastasis suppressor microRNA, miR-31, during the invasion-metastasis cascade. Int. J. Cancer 129, 1331–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mironchik Y., Winnard P. T., Jr., Vesuna F., Kato Y., Wildes F., Pathak A. P., Kominsky S., Artemov D., Bhujwalla Z., Van D. P., Burger H., Glackin C., Raman V. (2005) Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 65, 10801–10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Low-Marchelli J. M., Ardi V. C., Vizcarra E. A., van R. N., Quigley J. P., Yang J. (2013) Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. 73, 662–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luque A., Gomez M., Puzon W., Takada Y., Sanchez-Madrid F., Cabanas C. (1996) Activated conformations of very late activation integrins detected by a group of antibodies (HUTS) specific for a novel regulatory region (355–425) of the common beta 1 chain. J. Biol. Chem. 271, 11067–11075 [DOI] [PubMed] [Google Scholar]
- 37. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 38. Alexander N. R., Tran N. L., Rekapally H., Summers C. E., Glackin C., Heimark R. L. (2006) N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Res. 66, 3365–3369 [DOI] [PubMed] [Google Scholar]
- 39. Cheng G. Z., Chan J., Wang Q., Zhang W., Sun C. D., Wang L. H. (2007) Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 67, 1979–1987 [DOI] [PubMed] [Google Scholar]
- 40. Li Q. Q., Xu J. D., Wang W. J., Cao X. X., Chen Q., Tang F., Chen Z. Q., Liu X. P., Xu Z. D. (2009) Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 15, 2657–26565 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.