

Abstract
Small intestine luminal nutrient sensing may be crucial for modulating physiological functions. However, its mechanism of action is incompletely understood. We used a model of enteral nutrient deprivation, or total parenteral nutrition (TPN), resulting in intestinal mucosal atrophy and decreased epithelial barrier function (EBF). We examined how a single amino acid, glutamate (GLM), modulates intestinal epithelial cell (IEC) growth and EBF. Controls were chow-fed mice, T1 receptor-3 (T1R3)-knockout (KO) mice, and treatment with the metabotropic glutamate receptor (mGluR)-5 antagonist MTEP. TPN significantly changed the amount of T1Rs, GLM receptors, and transporters, and GLM prevented these changes. GLM significantly prevented TPN-associated intestinal atrophy (2.5-fold increase in IEC proliferation) and was dependent on up-regulation of the protein kinase pAkt, but independent of T1R3 and mGluR5 signaling. GLM led to a loss of EBF with TPN (60% increase in FITC-dextran permeability, 40% decline in transepithelial resistance); via T1R3, it protected EBF, whereas mGluR5 was associated with EBF loss. GLM led to a decline in circulating glucagon-like peptide 2 (GLP-2) during TPN. The decline was regulated by T1R3 and mGluR5, suggesting a novel negative regulator pathway for IEC proliferation not previously described. Loss of luminal nutrients with TPN administration may widely affect intestinal taste sensing. GLM has previously unrecognized actions on IEC growth and EBF. Restoring luminal sensing via GLM could be a strategy for patients on TPN.—Xiao, W., Feng, Y., Holst, J. J., Hartmann, B., Yang, H., Teitelbaum, D. H. Glutamate prevents intestinal atrophy via luminal nutrient sensing in a mouse model of total parenteral nutrition.
Keywords: proliferation, epithelial barrier function, taste receptor, metabotropic glutamate receptor
The gastrointestinal (GI) mucosa responds to a vast array of signals originating in the lumen, including nutrient and nonnutrient stimuli. Subsequently, these signals modulate physiological functions, including digestion, absorption, secretion, and protection from hazardous intraluminal contents (1). In recent years, there has been growing interest in luminal signaling pathways in the GI tract and in elucidating the possible roles and the underlying mechanisms of gut luminal sensing in various physiological and pathologic conditions. Recent studies have demonstrated that G-protein-coupled receptors (GPCRs), typically taste receptors (TRs) via G-protein subunits, play a key role in luminal molecule recognition and initiate luminal signaling pathways by activation of enteroendocrine cells and nerve fibers (2, 3). This signaling has been shown to integrate luminal sensing processes at different levels and thereby to modulate physiological responses in the GI tract, as well as to activate mucosal defense mechanisms (4). Moreover, evidence is accumulating that altering the content of the GI lumen may lead to disease states ranging from feeding disorders to inflammation (5). However, despite the clinical importance, effective strategies for preventing inappropriate luminal sensing responses from various pathologic stimuli are still needed. Total parenteral nutrition (TPN), or the intravenous feeding of patients, is used in clinical patient care almost 400,000×/yr in the United States (6). However, there is a substantial body of evidence that TPN can lead to complications, including the development of mucosal atrophy and loss of intestinal epithelial barrier function (EBF), both of which may contribute to an associated increase in clinical infections and loss of immune reactivity (7). However, the underlying mechanisms contributing to TPN-associated intestinal atrophy and EBF dysfunction are not fully understood. Because TPN is accompanied with a loss of nutrients within the GI lumen, it presents as a very powerful tool for understanding the mechanisms that guide luminal sensing of a single nutrient. By this method, a single nutrient can be examined in isolation without the interference of nutrients that would confound the results. In addition, the influence that this single nutrient may have on the TPN-induced intestinal mucosal atrophy and EBF dysfunction allows for a very precise way to track such physiological changes. In this study, we tested whether it is possible to prevent development of this TPN-associated GI physiological phenotype through restoring luminal sensing signaling with a specific tastant.
Glutamate (GLM) is a basic taste stimulant that mediates the umami taste (8). It has been shown to be closely involved in luminal chemosensing in duodenal mucosa, via multiple GPCRs, including the T1 receptor (T1R) TR family, namely, the T1R1/T1R3 heterodimer, and the metabotropic GLM receptors (mGluRs) (9–11). Luminal GLM perfusion significantly enhances duodenal mucosal defense mechanisms against injury due to acid exposure (11). GLM also has the capability of stimulating the proliferation of several kinds of cells outside the GI tract (12). Thus, we hypothesized that GLM would drive intestinal epithelial cell (IEC) proliferation and prevent TPN-associated mucosal atrophy through modulating luminal sensing signaling in the GI mucosa.
This study was conducted to assess the possible alteration of luminal sensing signaling in the small intestine during TPN administration, with or without oral GLM supplementation. The present results demonstrate the novel finding that GLM can drive IEC proliferation and prevent TPN-associated atrophy. Interestingly, our study also demonstrated a dose-dependent modulation of EBF action and identified unique signaling pathways, by which GLM can influence EBF in TPN-fed mice (hereafter referred to as TPN mice). Therefore, modulation of luminal sensing via GLM signaling affects not only IEC proliferation, but also EBF in TPN mice. In the present work, we identified a differential action of GLM via T1R3 that promoted EBF, whereas mGluR5 signaling led to EBF loss. The study also showed that GLM's promotion of IEC proliferation was associated with a decline in circulating glucagon-like peptide 2 (GLP-2), suggesting a novel negative regulator mechanism of IEC proliferation. Although an optimal dose remains to be determined, the current work suggests a potentially important role for GLM in protecting patients receiving TPN against the associated intestinal atrophy and in minimizing TPN′s negative effects on EBF.
MATERIALS AND METHODS
Animals
C57BL/6 male, specific pathogen-free 10- to 11-wk-old mice (Jackson Laboratory, Bar Harbor, ME, USA) were maintained under temperature-, humidity-, and light-controlled conditions. Body weight- and age-matched adult male T1R3-knockout (T1R3KO; B6;129-Tas1r3tm1Csz/J) mice with a C57BL/6 background (Jackson Laboratory) were also studied. The mice were initially fed standard mouse chow and water ad libitum and allowed to acclimate for 1 wk before surgery. During the administration of intravenous solutions, the mice were housed in metabolic cages to prevent coprophagia. The study protocol was approved by the University of Michigan Committee on the Use and Care of Animals (approval 7703).
Operative procedures and TPN delivery
Cannulation and administration of TPN was identical with that previously described (13, 14). TPN mice received intravenous TPN at 4.8 ml/d. Nitrogen and energy delivery was matched between groups (isonitrogenous/isocaloric). This TPN solution, and the administration rate, allows for adequate caloric delivery, matching the amount that the enterally fed mice received (14, 15). The TPN + GLM group received monosodium glutamate (MSG; Ajinomoto Co. Inc., Tokyo, Japan) at 75 mM diluted in drinking water (5 ml/d), starting the day of cannulation. The TPN group that was not given GLM received a reduction in the crystalline amino acids to match the nitrogen delivery in the TPN + GLM (75 mM) group. The sham-treatment group consisted of enterally fed mice given intravenous saline at the same rate as the infusions in the TPN group (5 ml water/d).
3-((2-Methyl-4-thiazolyl)ethynyl)pyridine (MTEP; R&D Systems, Minneapolis, MN, USA) was used to block mGluR5's activity and was diluted with saline and administered intraperitoneally via the indwelling catheter at 30 mg/kg daily, beginning on the day of cannulation. In the respective control group, MTEP was replaced with saline.
Mice were killed with CO2 7 d after cannulation. Body weight was recorded on the day of cannulation and the 7th day after cannulation. The amount of water taken by the mice was measured daily to ensure that the mice took the appropriate amount of GLM-treated water. Mice were euthanized before the end of the 7 d period if their health deteriorated to a level found to be unacceptable for their well-being and were not included in the final data analysis. All experiments were performed with ≥6 mice/group.
RNA isolation and quantitative RT-PCR
RNA extraction from mucosal scrapings was performed according to published procedures (16, 17). Quantitative PCR was measured with a Rotor-Gene 6000 (Corbett Life Science, Sydney, NSW, Australia), and β-actin was used as the internal control for normalization. For the initial screening of candidate TRs, GLM receptors, and transporters, a customized RT2 Profiler PCR Array was used (SABioscience; Qiagen, Frederick, MD, USA).
Western immunoblot analysis
The isolation of IECs and Western blot analysis were performed (18) with antibodies (Abs) including rabbit anti-T1R3 (1:200; ab-74732; Abcam, Cambridge, UK), rabbit-anti-α-gustducin (Gα; 1:400; sc-395; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-mGluR2/3 (1:250; ab-6438; Abcam), rabbit anti-mGluR4 (1:400; ab-6438; Abcam), rabbit anti-mGluR5 (1:300; ab-53090; Abcam), rabbit anti-EAAT2 (1:400; SC-15317; Santa Cruz Biotechnology), rabbit anti-EAAT3 (1:400; SC-25658; Santa Cruz Biotechnology), rabbit anti-zona occludens 1 (ZO-1; 1:250; 617300; Life Technologies, Grand Island, NY, USA), mouse anti-occludin (1:200; 331500; Life Technologies), rabbit anti-phospho-Akt (pAkt)-1/2/3 (Ser473 and Thr308; 1:200; sc-25658; Santa Cruz Biotechnology), rabbit anti-Akt (pan; C67E7; 1:400; 4691; Cell Signaling Technology, Beverly, MA, USA), mouse anti-proliferating cell nuclear antigen (PCNA; 1:3000; 2586, Cell Signaling Technology), and mouse anti-β-actin (1:1000; sc-130300; Santa Cruz Biotechnology). The secondary Ab was the horseradish peroxidase conjugate of either goat anti-mouse or goat anti-rabbit IgG (1:2000; Santa Cruz Biotechnology).
Immunofluorescence (IF) microscopy
Sections (10 μm) of jejunum were embedded and frozen in optimal cutting temperature (OCT) compound (Tissue-Tek; Sakura Finetek, Tokyo, Japan). IF staining was performed (16) with concentrations of the primary Abs as follows: 1:3000 PCNA, 1:50 5-bromo-2-deoxy uridine (BrdU), 1:250 ZO-1, and 1:150 occludin.
IEC proliferation assessment
IEC proliferation was assessed by IF staining of the markers PCNA and BrdU. For BrdU detection, all mice received BrdU (000103; BrdU labeling reagent; Life Technologies) at 50 mg/kg via i.p. injection 2 h before death (19). An index of the crypt cell proliferation rate was expressed as the ratio of the number of PCNA- or BrdU-positive cells per crypt to the total number of crypt cells. The total number of proliferating cells per crypt was defined as the mean of proliferating cells in 10 crypts for each mouse. Immunoblot density of PCNA was also used as a measure of the IEC proliferation rate.
Intestinal morphology assessment
Villus height and crypt depth were measured in ≥10 well-oriented, full-length crypt-villus units per specimen and averaged. Data were analyzed with commercially available digital image analysis software (NIS-Elements, AR 3.0; Nikon, Melville, NY, USA).
Ussing chamber and permeability studies
Experiments were performed in modified Ussing chambers (Physiological Instruments, San Diego, CA, USA) on jejunal segments (20). The transmembrane resistance (TER) was determined according to Ohm's law. The permeability of the jejunum was also assessed by measuring the mucosal-to-serosal permeation of FITC-dextran (FD4; average molecular weight, 3000–5000; Sigma-Aldrich, St. Louis, MO, USA; ref. 21). Briefly, 150 μl FITC-dextran (50 mg/ml, diluted with fresh Krebs buffer) was added to the mucosal compartment after a 30 min equilibration period. Then, a 1 ml sample was removed from the serosal compartment after 60 min, kept in −80°C for subsequent fluorescence quantitative analysis, and replaced by the same volume of fresh Krebs buffer. Fluorescence was measured with a Synergy 2 multimode microplate reader (BioTek Instruments, Winooski, VT, USA) at an excitation wavelength of 492 nm and an emission wavelength of 515 nm. Permeability was expressed as the percentage of mucosal-to-serosal clearance of FITC-dextran.
Plasma GLP-2 measurements
Plasma GLP-2 was measured by radioimmunoassay with an N-terminal specific antiserum that measures only GLP-2 with an intact N terminus (22). DPP4 inhibitor (2 μl; EMD Millipore, Billerica, MA, USA) was added in the collected blood samples to avoid GLP-2 degradation.
Statistical analysis
Data are expressed as means ± sd. A preliminary power analysis determined that a minimum of 6 subjects/study group would be needed on the basis of previous variance data from proliferation assays, our primary outcome measure. Paired t tests were used for comparing 2 groups. Significance was defined as P < 0.05.
RESULTS
TPN induces altered mRNA expression of T1Rs, GLM receptors, and transporters in the jejunum, and oral GLM supplementation prevents these TPN-associated changes
It is presently unknown how luminal sensing molecules are affected by enteral nutrient deprivation (i.e., TPN). Therefore, a specially designed mRNA array was created to study the gene expression profile, including TRs, GI receptors [GLM receptors (GLMRs)], and transporters involved in gut luminal sensing signaling in TPN mice. From this screening, 13 genes were found to be significantly altered by TPN. As shown in Table 1, compared with levels in the sham-treatment (enterally fed) group, TPN led to significant changes in the level of several T1Rs, GLMRs, and selected nutrient transporters. Surprisingly, of the 3 T1Rs, T1R1 and T1R3, which form a heterodimeric complex for effective umami taste sensing, both showed an upregulation of mRNA expression to various degrees during TPN administration. In contrast, T1R2 expression, which mediates sweet taste sensing, displayed a significant decrease. TPN also induced a significant decline in the mRNA expression of mGluR5 and a significant upregulation of mGluR7; glutamate receptor, ionotropic, kainate 5 (GRIK5); and GluR1. In addition, several transporters, including excitatory amino acid transporter 2 and 3 (EAAT2 and EAAT3) and sodium glucose cotransporter 1 (SGLT-1), displayed a significant downregulation during TPN treatment (Table 1). However, supplementation with oral GLM induced an upregulation of mRNA level in most T1Rs, GLMRs, and transporters to various degrees, suggesting that GLM significantly stimulates luminal sensing signaling in the small intestine of TPN mice.
Table 1.
mRNA alteration of luminal sensing molecules, including T1Rs, GLM receptors, and transporters in the jejunal mucosa of TPN mice, with or without GLM supplementation (75 mM)
| Molecule | Sham | TPN | TPN + GLM |
|---|---|---|---|
| T1R1 | 0.76 ± 0.34 | 2.37 ± 0.53* | 2.89 ± 0.81* |
| T1R2 | 3.72 ± 1.09 | 1.34 ± 0.14* | 5.21 ± 2.52# |
| T1R3 | 4.20 ± 1.48 | 6.49 ± 2.37 | 14.78 ± 1.50*,# |
| mGluR3 | 1.25 ± 0.70 | 1.16 ± 0.40 | 5.56 ± 1.51*,# |
| mGluR4 | 2.74 ± 0.87 | 2.97 ± 1.15 | 5.66 ± 0.64*,# |
| mGluR5 | 3.30 ± 1.02 | 1.29 ± 0.75* | 4.48 ± 1.07*,# |
| mGluR7 | 0.98 ± 0.18 | 2.56 ± 0.84* | 4.65 ± 1.67*,# |
| GluR1 | 1.98 ± 1.55 | 3.93 ± 1.99* | 7.29 ± 1.99*,# |
| GRIK5 | 0.73 ± 1.99 | 1.99 ± 0.79* | 1.38 ± 0.28* |
| EAAT2 | 1.47 ± 0.40 | 0.20 ± 0.04* | 0.61 ± 0.16*,# |
| EAAT3 | 3.54 ± 1.30 | 1.61 ± 0.55* | 4.39 ± 1.74# |
| VGLUT3 | 2.14 ± 0.56 | 5.02 ± 2.21* | 11.25 ± 1.62*,# |
| SGLT1 | 1.09 ± 0.35 | 0.66 ± 0.24* | 1.45 ± 0.72# |
Values are means ± sd.
P < 0.05 vs. sham-treatment group;
P < 0.05 vs. TPN group.
Next, IF staining of 3 T1Rs in the present study was identified in the cytoplasm of endocrine-like solitary cells dispersed throughout the villus epithelium, with rare cells identified in the jejunal glands having the configuration of enteroendocrine cells (Fig. 1A). This observation suggests that enteroendocrine cells play an important role in the intestinal TR signaling pathway, and the distribution of these T1Rs was similar to that previously reported (23). The localization of each of the T1Rs was similar between the sham-treatment, TPN, and TPN + GLM groups. No differences were noted in staining intensity or number of T1R+ cells; however, the fluorescence intensity of these cells and the overall number were quite low, making quantitative differentiation difficult. Consistent with the result of PCR analyses, T1R3 protein expression analysis showed a 1.6-fold increase in the TPN group and a 2.4-fold rise in the TPN+GLM mice, compared with expression in the sham group. Unfortunately, we were unable to obtain ideal immunoblot results of T1R1 and T1R2 because of the presence of a very high background and relatively weak signal of the target blots. A similar problem has been identified by several other groups; these researchers found that the poor quality of the currently available antibodies and the low level of TRs contribute to the difficulty of quantitative analysis of T1R expression (24–26).
Figure 1.

Influence of TPN administration and GLM supplementation on the amount of luminal sensing molecules of isolated IECs at the protein level. A) IF staining was performed on jejunal mucosa from TPN mice with anti-T1Rs and anti-Gα Abs and counterstained with DAPI nuclear stain. Note the similar localization of 3 T1Rs and Gα proteins in the cytoplasm of the endocrine-like solitary cells of the jejunal villi. B, C) Protein expression of T1R3, mGluRs, transporters, and Gα in sham-treatment mice compared with TPN and TPN + GLM (75 mM) mice. B) Representative immunoblots of isolated small-bowel IECs from each group. C) Mean protein level for each group. For representative images and immunoblots, a minimum of 3 experiments and images were used for each selection. Scale bars = 50 μm. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
Of note, immunoblot results for mGluRs varied compared with mRNA expression. As shown in Fig. 1B, the expression of mGluR2/3, mGluR4, and mGluR5 was down-regulated during TPN administration. As well, the expression level of mGluR2/3 and mGluR4 proteins declined even further with GLM supplementation, whereas mGluR5 showed a significant up-regulation. This result suggests that different mGluRs play distinct roles in GLM-mediated actions in modulating luminal sensing. In addition, the expression of the GLM transporters EAAT2 and EAAT3 showed a significant decrease in the TPN mice and an increase in the TPN + GLM group at the protein level, similar to the mRNA results.
It is well recognized that all T1Rs and mGluRs belong to GPCRs, with transmembrane domains to facilitate taste sensing (8, 27). These GPCRs activate G proteins, most notably α-gustducin (Gα), to transduce taste signaling to downstream signaling molecules (3). In the current study, IF staining results demonstrated a similar localization of Gα protein in the IECs, as seen with T1R staining (Fig. 1A), suggesting a possible colocalization with T1Rs in enteroendocrine cells, as described in a previous study (24). However, we failed to obtain ideal colocalization images of Gα with T1Rs. Immunoblot analysis showed that Gα protein expression in the TPN group significantly dropped (to ∼30% of that in sham-treated mice), whereas GLM supplementation completely prevented this reduction (Fig. 1B, C). This result suggests that TPN simultaneously influences the expression of GPCRs and the G protein Gα and affects the downstream taste signaling cascades, whereas GLM prevents the loss of intestinal luminal sensing via stimulation of umami TRs and reactivation of Gα transduction.
GLM stimulated IEC proliferation in TPN mice and prevented TPN-induced intestinal atrophy
As shown in Fig. 2A, after TPN administration, both jejunal villus length and crypt depth decreased to ∼40 and ∼70% of those in the sham-treatment group, respectively, whereas GLM supplementation significantly increased villus height to 62% of the sham-treatment group, suggesting that oral GLM supplementation results in significant prevention of the intestinal atrophy associated with TPN administration.
Figure 2.

GLM supplementation prevents the intestinal atrophy induced by TPN. A) Hematoxylin and eosin staining was performed on jejunum from sham-treatment, TPN, and TPN + GLM mice (×100). Villi length and crypt depth of the jejunal mucosa in the TPN mice were significantly shortened, compared with the sham-treatment group, whereas GLM supplementation (75 mM) markedly increased jejunal villi length in the TPN mice. Left panels: representative images of 3 groups. Right panels: changes in the jejunal villi and crypts of each study group. B) IF staining was performed on jejunal mucosa from the sham-treatment, TPN, and TPN + GLM groups with anti-PCNA and anti-BrdU Abs and counterstained with DAPI. Left panels: representative images of 3 groups. Right panels: PCNA- or BrdU-positive rate in the intestinal mucosal crypt of each group. Note the significant decrease in PCNA- and BrdU-positive rates in the TPN group compared with that in the sham-treatment group, whereas GLM supplementation (75 mM) significantly increased the PCNA- and BrdU-positive rates in the TPN mice. C) Level of PCNA at the mRNA and protein levels. Left panel: mRNA level of PCNA from isolated mucosal scrapings of each group. Right panel: protein level of PCNA from isolated IECs of each group. Representative immunoblots of isolated small-bowel IECs are given from each group. When compared to that in the sham-treatment group, TPN administration significantly decreased the expression of jejunal PCNA at the mRNA and protein levels, whereas GLM supplementation (75 mM) significantly prevented the down-regulation of PCNA during TPN administration. Scale bars = 50 μm. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
To determine the mechanisms that led to GLM's preventing TPN-associated atrophy, we next examined IEC proliferation rates. IF staining demonstrated that the PCNA-positive rate in intestinal mucosal crypts of the TPN mice dropped to ∼37% of that in the sham-treatment group; however, proliferation rates rose to 89% of sham-treated controls when supplemented with GLM (Fig. 2B). This result was confirmed with BrdU staining, with similar changes in IEC proliferation rates between these methods of measurement. To further confirm these changes in proliferation, we performed both PCR and immunoblot analysis of PCNA expression, and the results were similar to those seen with IF staining (Fig. 2C). Thus, GLM supplementation led to a significant prevention of TPN-associated loss of IEC proliferation. It should be noted that there was no significant change in body weight throughout the TPN period between the TPN and TPN + GLM groups (data not shown). In addition, the water intake of the TPN mice supplemented with GLM did not obviously differ from that of the nonsupplemented TPN mice (data not shown).
It is known that TPN-induced intestinal mucosal atrophy is also due to an increase in epithelial cell (EC) apoptosis (17). Therefore, an assessment of the effect that GLM might have on IEC apoptosis rates was performed with active caspase-3 staining. However, GLM showed no obvious effect on apoptosis, and there was no significant difference in apoptosis IEC-positive rates between the TPN and TPN + GLM groups (Supplemental Fig. S1). This result was further confirmed by immunoblot analysis of apoptosis-associated protein Bcl-2 and Bax. As well, the mRNA levels of caspase-3 were not significantly different between the TPN and TPN + GLM groups (0.030±0.001 vs. 0.04±0.002, respectively).
GLM restored pAkt expression, which was down-regulated during TPN
Our laboratory has recently reported a significant loss of pAkt signaling with TPN administration and the key role of pAkt signaling in modulating EC proliferation in the TPN mice (19). Consistent with this, the present work showed a significant decrease in jejunal pAkt expression in the TPN mice compared with that in the sham-treatment group (Fig. 3). Interestingly, GLM supplementation fully prevented the decline in pAkt expression, with amounts returning to sham-treatment levels. This result suggests that pAkt signaling plays an important role in the proproliferation mechanism of GLM on IECs in this TPN model.
Figure 3.
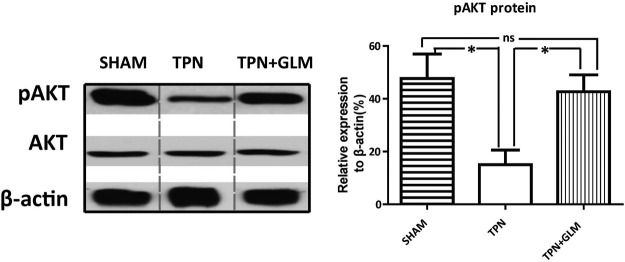
TPN administration induces a significant decrease in pAkt expressed by IECs, which is reversed with GLM supplementation. Representative immunoblots of isolated small-bowel IECs are given from each group. Compared with sham-treatment, TPN administration significantly decreased expression of jejunal pAkt protein, whereas GLM supplementation (75 mM) significantly prevented the down-regulation of pAkt during TPN administration. No difference in Akt level was observed among the 3 groups. A minimum of 3 experiments were used for each selection. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
GLM supplementation influenced EBF dysfunction induced by TPN administration
Besides intestinal mucosal atrophy, EBF dysfunction is another important physiological manifestation associated with TPN administration and may contribute to the increasing rates of systemic infections and septicemia (28). Interestingly, GLM supplementation at 75 mM in the TPN mice led to a significant (1.6-fold) increase in FITC-dextran permeability and a 0.6-fold decrease in TER value in jejunal mucosa, when compared the same measures in the nonsupplemented TPN group (Fig. 4A, B). Of note, this change in TER and FITC-dextran permeability was highly dose dependent, with the higher concentration (75 mM) of GLM resulting in a loss of EBF, whereas 25 mM had no obvious effect on EBF (Supplemental Fig. S2). In addition, administering GLM to the enterally fed mice (sham-treatment group) had no effect on either TER or dextran permeability. It should be noted that there was no obvious difference in survival between the GLM and non-GLM TPN groups (survival in both was ∼75%; data not shown), suggesting that GLM supplementation had little to no adverse influence on the outcome of the TPN mice.
Figure 4.

GLM supplementation affects EBF in TPN mice. A, B) FITC-dextran permeability (A) and Ussing chamber results (B) in each group. Note that TPN administration significantly induced an increase in FITC-dextran permeability and a decline in TER of jejunal mucosa, compared to that in the sham-treatment group, whereas GLM supplementation at 75 mM intensified the changes induced by TPN treatment. C) Representative images of IF staining for the TJ proteins occludin and ZO-1 in the villus region, counterstained with DAPI. The abnormality of the TJ network, shown as a marked disconnection of the sharp, mesh-like grid, was demonstrated in the jejunal villi from the TPN mice, compared to that in the sham-treatment group, whereas GLM exposure at 75 mM intensified the abnormality. D) Protein expression of occludin and ZO-1 from isolated IECs in each group. TPN administration significantly decreased the expression of both occludin and ZO-1 proteins, compared to that in the sham-treatment group, whereas GLM supplementation (75 mM) caused a lower protein level of occludin but not of ZO-1 in the TPN mice. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
IF staining with the tight junction (TJ) proteins occludin and ZO-1 was then used to further address the integrity of EBF in the TPN mice. Staining in the TPN mice displayed a marked disconnection of the sharp, mesh-like grid, as well as a loss of the colocalization of ZO-1 and occludin that was seen in the sham mice (Fig. 4C). Administration of GLM (75 mM) to the TPN mice actually augmented the loss of colocalization and led to a greater reduction of occludin along the jejunal villi (Fig. 4C). Immunoblot analysis of the TJ proteins also demonstrated a significant reduction of ZO-1 and occludin protein expression in the TPN group, compared with that in the sham mice. GLM supplementation led to an even lower level of occludin protein expression but did not affect ZO-1 protein expression, compared with that in the nonsupplemented TPN group (Fig. 4D).
Loss of T1R3 signaling does not influence the proproliferative effect of GLM, but promotes further loss of EBF in TPN mice
As a common key component of the heterodimeric umami TRs and sweet TRs, T1R3 plays a pivotal role in intestinal luminal sensing (8). In light of the significant alteration of T1R3 expression under TPN and GLM treatments, T1R3KO mice were used to clarify the role of T1R3 in the proproliferation and barrier-modulating mechanism of GLM in the TPN mice. Although we anticipated a blockade of GLM action in the T1R3KO mice, no significant differences between them and the wild-type (WT) mice were observed in the measurement of jejunal villus length, crypt depth, and IEC proliferation, when measured with PCNA or BrdU staining in the TPN mice, with or without GLM treatment. This finding was further confirmed with immunoblot analysis of PCNA protein (Fig. 5A–C). As well, removal of T1R3 did not influence the up-regulation of pAkt protein expression induced by GLM treatment in the TPN mice. These results suggest that GLM-mediated restoration of IEC proliferation and pAkt up-regulation in TPN mice is independent of T1R3.
Figure 5.

T1R3KO promotes intestinal epithelial barrier dysfunction in TPN mice, but has no obvious influence on the proproliferative effect of GLM in TPN mice. A–C) Jejunal villi and crypt measurements (A), PCNA- and BrdU-positive rate in the intestinal mucosal crypt (B), and levels of PCNA and pAkt in each group (C), determined by immunoanalysis of isolated small-bowel IECs. No significant difference was observed in these measurements between the T1R3KO and WT TPN mice, with or without GLM supplementation. D, E) Ussing chamber (D) and FITC-dextran permeability (E) results of each group. T1R3KO TPN mice showed significantly lower TER values (D) and higher FITC-dextran concentrations (E) than did the WT TPN mice, with or without GLM supplementation. F) Protein expression of occludin and ZO-1 from isolated IECs in each group. G) Representative images of IF staining for occludin and ZO-1 in the villus region, counterstained with DAPI. A significant decrease in occludin protein expression was induced in T1R3KO TPN mice, as compared with that in WT TPN mice, with or without GLM supplementation. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
In contrast, the following results provide evidence of a possible EBF-protective role for the T1R3 TR. T1R3KO TPN mice showed significantly reduced TER and increased FITC-dextran permeability compared with WT TPN mice. Accordingly, the T1R3KO TPN mice given GLM showed even further loss of TER and an increase in permeability compared with that in the WT mice (Fig. 5D, E), whereas no change in EBF was detected in the chow-fed T1R3KO mice. Use of T1R3KO mice also resulted in a significant down-regulation of the occludin protein in TPN mice, with or without GLM supplementation, compared with that in WT TPN mice; however, there was no effect on ZO-1 expression (Fig. 5C). This finding was further confirmed with IF staining of TJ proteins (Fig. 5D), which displayed an obviously weakened and interrupted occludin linear immunoreactivity along the villi. Taken together, these data support the possibility that T1R3 serves an important role in maintaining homeostasis of the EBF, potentially by maintaining an integrated TJ configuration. It should be noted that the simple removal of T1R3 is insufficient to alter EBF, as barrier function was not affected in sham-treatment (enterally fed) T1R3KO mice (data not shown).
mGluR5 antagonist MTEP has no obvious influence on the proproliferative effect of GLM on TPN mice, but significantly prevents EBF loss in TPN mice with GLM supplementation
It has been shown that some mGluRs are involved in the umami taste-sensing mechanism (27, 29). Of these mGluRs in our PCR screening profile, mGluR5 was the only one that displayed an obvious down-regulation in the TPN group, which was reversed after GLM supplementation. This finding suggests that mGluR5 is involved in the regulatory mechanism of GLM's action in TPN mice. Most important, evidence is accumulating that mGluR5 plays an important role in the modulation of cell proliferation (30, 31). Hence, the classic mGluR5 antagonist MTEP was used in the present study to explore the role of mGluR5 in the GLM mechanism in TPN mice. However, neither histologic analysis nor proliferation assessment with PCNA or BrdU staining demonstrated any influence of MTEP on the proproliferative effect of GLM in the WT or T1R3KO TPN mice, suggesting that the proliferation modulation of GLM is independent of mGluR5 or T1R3, either alone or in concert (Fig. 6A–C).
Figure 6.

mGluR5 antagonist MTEP significantly prevents intestinal epithelial barrier dysfunction, but has no obvious influence on the proproliferative effect of GLM in TPN mice. A–C) Intestinal villi and crypt measurements (A), PCNA- and BrdU-positive rate in the intestinal mucosal crypt (B), and levels of PCNA and pAkt (C). No significant differences in any of the measurements were observed with MTEP exposure in the WT or T1R3KO TPN + GLM (75 mM) mice. D) Ussing chamber and FITC-dextran permeability results. MTEP exposure significantly blocked the decline in TER and the increase in the FITC permeability rate induced by TPN + GLM treatments (75 mM), when compared with saline treatment in WT or T1R3KO mice. E) Protein expression of occludin and ZO-1 of isolated small-bowel IECs, determined by immunoanalysis. F) Representative images of IF staining of the TJ proteins occludin and ZO-1 in the villus region, with DAPI counterstaining. MTEP exposure markedly prevented the decline in occludin protein expression induced by TPN + GLM (75 mM) treatment when compared with saline treatment in WT or T1R3KO mice. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
Given the influence of GLM on EBF in TPN mice, assessment of EBF with MTEP treatment was performed to determine whether mGluR5 is involved in this process. TER and permeability results demonstrated that MTEP treatment induced a 1.8-fold increase in TER and a 2.9-fold decrease in FITC-dextran permeability in TPN mice given GLM supplementation, compared with those receiving saline treatment (Fig. 6D). This result suggests that mGluR5 plays an important role in GLM-mediated EBF dysfunction in TPN mice. This same inhibition of EBF dysfunction by MTEP was also seen in the T1R3KO TPN mice (Fig. 6D), further supporting the importance of the mGluR5 pathway in the GLM-mediated loss of EBF. Coincident with these physiological changes was a significant (P<0.05) preservation of occludin protein levels in MTEP-treated TPN + GLM mice (Fig. 6E). IF staining results showed that MTEP treatment markedly attenuated the TJ abnormalities at the jejunal villi in WT or T1R3KO TPN mice with GLM supplementation (Fig. 6C), including prevention of the loss of these junctional proteins along the interepithelial space and of the internalization of occludin. It should be noted that, as mentioned above, T1R3 removal intensified barrier dysfunction in TPN + GLM mice, whereas it appears that blocking mGluR5 activity with MTEP neutralized the negative effect of T1R3KO in GLM-treated TPN mice. This finding suggests a differential action of GLM on different receptors, where GLM appears to support EBF via T1R3 and to degrade EBF via mGluR5.
GLM supplementation decreases plasma GLP-2 levels of TPN mice via both T1R3 and mGluR5
Our previous work demonstrated that TPN results in a significant loss of plasma GLP-2 and a down-regulation of GLP-2 receptors (7). As some GLP-2 receptors are on mucosal neuroendocrine cells, we next asked whether GLM supplementation could stimulate luminal sensing and drive an increase in GLP-2. The supplementation of enterally fed (sham-treatment) mice with GLM led to an unexpected dose-dependent decline in plasma GLP-2 levels (Fig. 7A). GLM at the higher concentrations (75 and 150 mM) resulted in a significant decrease in GLP-2, whereas 25 mM had no obvious effect on plasma GLP-2 concentration in the sham-treatment mice. Fig. 7B shows that TPN administration led to a significant decline in GLP-2 levels, whereas GLM supplementation at 75 mM further decreased the plasma GLP-2 concentration to ∼40% of that in the TPN group. Most strikingly, blockade of T1R3 signaling and mGluR5 both increased GLP-2 levels in the TPN + GLM group in an additive fashion. This result suggests a novel mechanism whereby the IEC proliferative effect of GLM is negatively regulated by down-regulation of the expression of GLP-2, potentially via enteroendocrine cells (T1R3 receptors) as well as other epithelial cell populations (mGLuR5).
Figure 7.
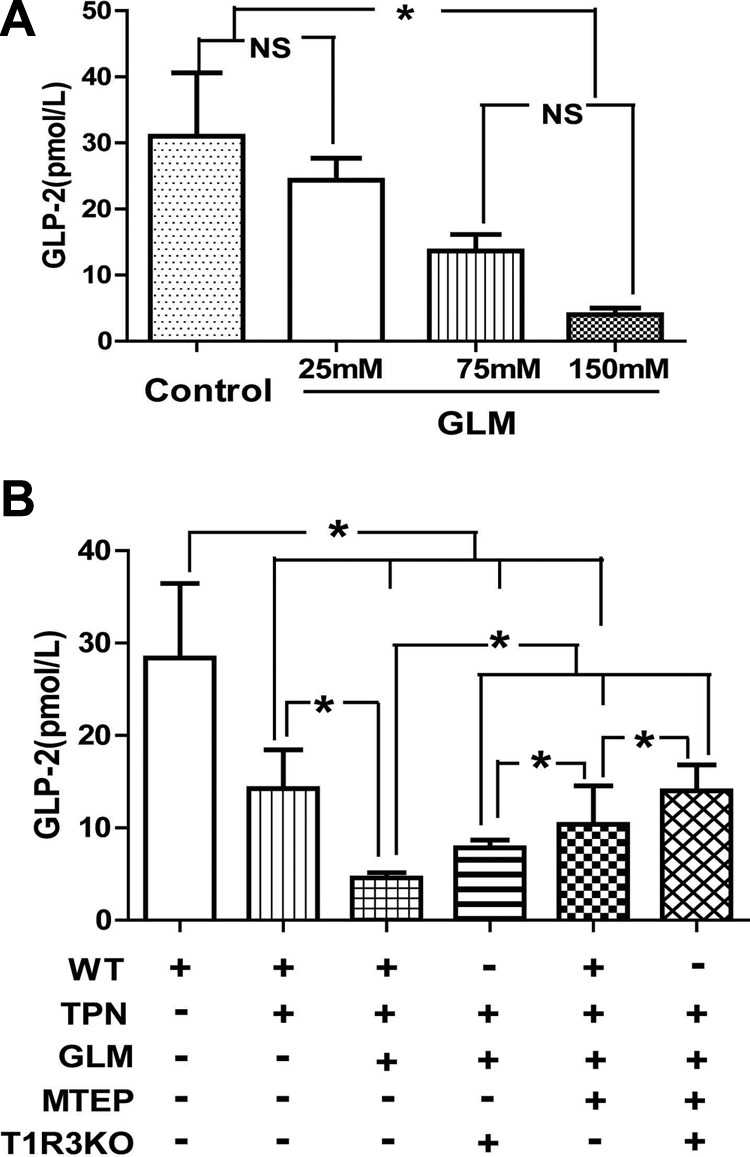
GLM supplementation decreased plasma GLP-2 levels in TPN mice via both T1R3 and mGluR5. A) Supplementation of enterally fed (sham-treatment) mice with GLM led to a dose-dependent decline in plasma GLP-2 levels. GLM at the higher concentration (75 and 150 mM) resulted in a significant decrease in GLP-2, whereas 25 mM had no obvious effect on plasma GLP-2 concentration in sham-treatment mice. B) TPN administration led to a significant decline in GLP-2 levels, and GLM supplementation at 75 mM further decreased plasma GLP-2 concentrations in TPN mice. Block of T1R3 signaling and mGluR5 both increased GLP-2 levels in the TPN+GLM group in an additive fashion. Values are means ± sd (error bars). ns, not significantly different. *P < 0.05.
To better understand the mechanisms of these changes, we looked at a time course of proglucagon mRNA expression in the mucosa of the ileum and colon, the 2 sites of proglucagon production, which is post-translationally cleaved to GLP-2. Supplemental Fig. S4 shows that proglucagon levels were similar to those in enterally fed mice at 3 d of TPN, but significantly declined by 1 wk. Interestingly, no differences in proglucagon levels were seen between the TPN and TPN + GLM groups, suggesting that a post-transcriptional change is responsible for the decline in GLP-2 circulating levels. Alternatively, the differences may also be due to changes in the degradation of GLP-2 by endogenous peptidases.
DISCUSSION
In the present study, TPN administration led to significant alterations in a series of luminal sensing-associated molecules, including T1Rs, GLMRs, and transporters in the jejunal mucosa, whereas GLM supplementation at 75 mM significantly influenced the alterations associated with TPN. Notably, GLM supplementation in our TPN model significantly prevented TPN-associated intestinal mucosal atrophy via a stimulatory effect on IEC proliferation. GLM administration at 75 mM also led to a partial loss of EBF that was confined to our TPN mouse study group. Further exploration of the mechanisms guiding this effect suggests that two GPCRs, T1R3 and mGluR5, may be involved in the mechanisms by which GLM modulates EBF, whereby GLM stimulation of T1R3 appears to promote EBF, and GLM, acting on mGluR5, may have a negative effect on barrier function. However, GLM stimulation of these receptors failed to explain the positive effect on epithelial proliferation in the TPN mice. Another finding was that GLM supplementation further decreased plasma GLP-2 levels in the TPN mice via both T1R3 and mGluR5 signaling, suggesting that the proproliferative effect of GLM on the TPN mice is independent of GLP-2 release. Collectively, these results demonstrated for the first time that loss of gut luminal nutrient sensing contributes to TPN-associated intestinal atrophy. Modulation of intestinal luminal sensing pathways through GLM supplementation may not only drive IEC proliferation but also may affect EBF of TPN mice. Clearly, further work is necessary to understand what mechanisms drive this robust increase in IEC proliferation and to determine the optimal dose of GLM for preventing intestinal atrophy without promoting epithelial barrier dysfunction during TPN administration.
Luminal nutrient sensing and TPN
Sensing of luminal contents is important in initiating the appropriate response of digestion and absorption of nutrients and can also neutralize and help remove drugs, toxins, and microorganisms and play an important role in host defense mechanisms (32–34). It has been shown that the taste-sensing system plays a critical role in luminal sensing in the GI tract, especially for luminal nutrient signals (1, 5, 35, 36). The interaction between luminal nutrients and TRs through G-coupled proteins initiates various downstream pathways and exerts different effects on the body that depend on the combination of 3 T1Rs (4, 25, 35, 37–39).
TPN administration is associated with numerous complications, ranging from an increase in infections to a loss of immune reactivity, most likely owing to mucosal atrophy (7). Although the underlying mechanisms of TPN-associated complications are still not fully established, it is clear that a major contributing factor is enteral nutrient deprivation (28, 40). Indeed, the present study supports our hypothesis that TPN administration leads to a significant alteration in mRNA expression of T1Rs, GLMRs, and transporters in the jejunal mucosa, some of which were further confirmed with immunoblot analysis. This result strongly suggests that the loss of luminal nutrients during TPN administration widely affects nutrient taste sensing, nutrient transport, and energy metabolism, but also could be a factor in TPN-associated complications.
Of the T1Rs, T1R1 and T1R3 form a heterodimer that responds to umami taste. These TRs were up-regulated during TPN administration, whereas T1R2 moved in the opposite direction (reduction). As well, the G-protein subunit Gα demonstrated a significant down-regulation during TPN. Recent work has revealed that intestinal T1Rs and other taste-signaling molecules are under dynamic metabolic and luminal control for feedback of GI function in response to luminal nutrients (23). For example, intestinal T1R3 and Gα expression has been shown to be up-regulated in germ-free mice, indicating that intestinal T1Rs may be adaptively regulated by the absence of microbiota and driven by a compensatory mechanism promoting the proximal intestine to detect and absorb more nutrients (26). Our laboratory also confirmed the possible connection between the alteration of luminal microbiome and TPN-associated intestinal atrophy (7). Thus, it is likely that removal of food intake with TPN administration results in reduced taste, which may trigger certain adaptive machinery to modulate the level of T1Rs and Gα, in order to detect the decreased nutrient signals. Interestingly, a similar adaptive response of T1R2/T1R3 was observed in reactive astrocytes under ischemia stimulation, which is believed to maintain the glucose supply to these stressed neurons (41).
GLM supplementation prevents the loss of TPN-associated intestinal atrophy by promoting the proliferation of IECs
GLM, as a nonessential amino acid, is well-known to be responsible for umami taste (27). Akiba et al. (1, 9–11) showed in an elegant study that luminal GLM perfusion activates duodenal mucosal defense mechanisms via umami sensing. Therefore, one goal of the present work was to investigate whether GLM supplementation could similarly have benefit in preventing TPN-associated complications.
Indeed, in our study, GLM supplementation stimulated the expression of most of the nutrient-sensing molecules. Notably, the expression of all GPCRs, including T1Rs and mGluRs, as well as the G protein Gα, was up-regulated in the TPN mice after GLM treatment, indicating that GLM could reactivate GPCR signaling pathways that are negatively influenced by TPN. In addition, GLM supplementation reversed the decline in expression of SGLT-1, EAAT2, and EAAT3, again suggesting that GLM can greatly improve the transportation of luminal nutrients across the epithelium that is inhibited by TPN (42).
Our previous work (43, 44) demonstrated that TPN administration can induce intestinal mucosal atrophy, which may be one of the fundamental events that contribute to TPN-associated clinical complications. In the present study, we showed that GLM supplementation could significantly prevent intestinal mucosal atrophy in the TPN mice via promotion of IEC proliferation. In accordance, GLM has been shown to be involved in the mucosal healing process and may well mediate this action by IECs that use GLM as an energy source for cell proliferation, as suggested in previous studies (45, 46). Thus, GLM supplementation in the TPN model may provide an important energy source that drives this proliferation (47).
pAkt is an important molecule that plays a central role in regulating IEC proliferation (48). Our previous work also showed that TPN leads to decreased pAkt, which contributes to intestinal atrophy, whereas pAkt activation leads to a significant prevention of mucosal atrophy (19). In the present work, GLM led to a significant increase in pAkt expression, which was otherwise down-regulated in the TPN group. This result suggests that pAkt signaling plays an important role in the GLM proproliferation mechanism (19). In addition, it is possible that GLM via conversion into glutamine exerts protective effects on IECs, which itself has been shown to have strong benefits in maintaining IEC proliferation (49, 50).
Of note, because the nutrient was delivered orally in the present study, we cannot rule out the possibility that GLM's effects derive from sense of taste via activation of umami receptors in the tongue, which can also indirectly regulate multiple intestinal physiological functions via the gut–brain axis (51). Therefore, in subsequent experiments, luminal GLM perfusion by gavaging may be a better way to observe the direct effect of GLM on the intestinal mucosa.
GLM supplementation affects EBF in TPN mice
A loss of EBF during TPN administration is also believed to contribute to the increased rate of systemic infections and septicemia (28). Surprisingly, it appears that GLM supplementation at 75 mM affected EBF in the TPN mice, as manifested by increased permeability and even worse disarrangement and down-regulation of the occludin protein. It should be noted that no obvious change in EBF was observed in lower doses of GLM (25 mM) in the TPN mice as well at 75 mM of GLM in the chow-fed mice, suggesting that the influence of GLM on EBF in TPN mice is highly dose dependent, as reported in other works (12, 52).
To date, the exact role of GLM in modulating EBF has yet to be defined by the limited available data. For example, Vermeulen et al. (53) showed that exposure to GLM significantly reduces the phorbol-12,13-dibutyrate-induced hyperpermeability of IECs. As well, blocking the conversion from glutamine to GLM has been shown to inhibit the barrier-protective effect of glutamine (49, 53). A recent study may help us to illustrate the influence of GLM on EBF of the TPN mice. Znalesniak et al. (54) demonstrated that IEC restitution induced by epidermal growth factor leads to a significant down-regulation of the cell-cell contact-associated genes, including TJ-associated proteins, which is thought to severely affect EBF. Thus, it is likely that the rapid proliferation and migration of IECs along the crypt–villus axis in the GLM-treated TPN group led to this loss of barrier function. More investigation is needed to further determine the dose–effect relationship of GLM in both in vitro and in vivo tests, to clarify the exact role of GLM in EBF, and to precisely determine the optimal dose of GLM in preventing TPN-associated intestinal atrophy and avoid negative effects on the EBF.
Possible role of T1R3 and mGluR5 in GLM's effects on IEC proliferation and EBF
As the common component of the T1R1/T1R3 and T1R2/T1R3 heterodimers, T1R3 is generally thought to play a pivotal role in the luminal sensing machinery (8). Loss of T1R3 (via transgenic KOs) led to total loss of preference for both umami stimuli and artificial sweeteners and had only minimal response to sugars (55). Quite unexpectedly, the use of T1R3KO mice failed to influence the effect of GLM on IEC proliferation, but promoted a further weakening of EBF in the TPN mice, with or without GLM supplementation. Thus, the effect of GLM on IEC proliferation in the TPN mice appears not to be T1R3 dependant. It is possible that other mechanisms drive this proliferative action. Emerging evidence suggests that besides T1R3, there are other receptors responsible for the umami taste. Yasumatsu et al. (27) recently reported that multiple receptors and transduction pathways, as well as multiple types of GLM-sensitive gustatory nerve fibers, are involved in umami taste sensing. Moreover, T1R3KO in different models caused only a partial reduction in the capability of detecting and discriminating MSG and sucrose (56). Notably, in our study, the loss of T1R3 exacerbated EBF in TPN mice, suggesting that T1R3 is involved in maintaining EBF. The significant up-regulation of T1R3 in TPN or TPN + GLM mice may represent a dynamic protective role of T1R3 in maintaining barrier integrity against external stimulus. In addition, no obvious change in EBF was observed in T1R3KO normal or sham mice, indicating that T1R3 may not be a pivotal factor in EBF regulation.
mGluR5 is another type of GPCR, belonging to the group I metabotropic GLM receptors (57). Evidence is accumulating that mGluR5 plays an important role in the modulation of cell proliferation (30, 31, 58). However, it appears that mGluR5 is not involved in the mechanism by which GLM increases IEC proliferation in TPN mice. Furthermore, MTEP failed to block the proproliferative effect of GLM in T1R3KO TPN mice, suggesting that the proliferative action of GLM is independent of mGluR5 or T1R3, either alone or in concert. Interestingly, mGluR5 is involved in the effect of GLM on EBF in TPN mice, as the mGluR5 antagonist MTEP significantly alleviated the reduced EBF of TPN mice with GLM supplementation. It appears that T1R3 and mGluR5 have distinct functions in EBF regulation: stimulation of T1R3 may promote barrier function, whereas mGluR5 may participate in GLM-induced EBF dysfunction in TPN mice (summarized in Fig. 8).
Figure 8.

Schematic view of the possible mechanisms by which GLM modulates intestinal epithelial proliferation and alters EBF in mice receiving TPN. TPN administration leads to significant alterations in a series of luminal sensing–associated molecules, including T1Rs, mGluRs, Gα, and transporters in the intestinal mucosa. This disturbance of luminal sensing machinery may contribute to the development of mucosal atrophy and loss of intestinal EBF during TPN treatment. Luminal GLM supplementation stimulates the expression of GPCRs, such as T1R3, mGluR5, and the G-protein subunit Gα, thereby reactivating the luminal sensing signaling pathways that are down-regulated by TPN. Subsequently, GLM promotes IEC proliferation in TPN mice through activating the pAkt/mTOR signaling pathway, which may be initiated via some presently poorly characterized TRs localized on small-bowel enteroendocrine cells (19, 62). In addition, GLM supplementation leads to a decline in circulating plasma GLP-2 levels in TPN mice, suggesting that the proproliferative effect of GLM on TPN mice is independent of GLP-2 release. As well, by inhibiting the expression of the TJ protein occludin in IECs, GLM may lead to a partial loss of EBF in TPN mice. Of note, T1R3 and mGluR5 appear to have distinct functions in EBF regulation by GLM, whereby stimulation of T1R3 by GLM promotes barrier function, whereas mGluR5 may participate in GLM-induced epithelial barrier dysfunction in TPN mice. In addition, GLM could improve the transportation of luminal nutrients across the epithelium, which is normally down-regulated with TPN, by stimulating the expression of multiple transporters such as EAAT2, EAAT3, and SGLT-1.
GLP-2 is not involved in the proproliferative effect of GLM on TPN mice
GLP-2 is a nutrient-dependent, proglucagon-derived, intestinotrophic hormone that has been shown to support intestinal epithelial growth during TPN administration or in patients with short bowel syndrome (59, 60). A recent report showed that GLM increases GLP-2 release from the GI tract, which is well recognized to stimulate IEC proliferation (61). Accordingly, we hypothesized that the increase in GLP-2 release is part of the mechanism that accounts for GLM's profound action in driving IEC proliferation. Surprisingly, supplementation with GLM led to a dose-dependent decline in plasma GLP-2 levels in either enterally fed or TPN mice, whereas blockade of T1R3 signaling and mGluR5 both increased GLP-2 levels in an additive fashion, suggesting that both receptors sense intraluminal GLM, leading to a decline in GLP-2 in a negative-regulatory fashion. It is possible that this is a counterregulatory control mechanism that prevents an otherwise excessive increase in IEC proliferation induced by GLM. As noted in the Results section, TPN administration over 3 d to 1 wk led to a significant decline in proglucagon expression in the ileum and colon, the two sources of proglucagon, the peptide from which GLP-2 is derived. This decline occurred in the TPN and TPN+GLM groups. We were surprised that the mRNA levels did not correlate at 1 wk with the decline in blood levels of GLP-2. A variety of mechanisms may contribute to such differences, including changes in the post-transcriptional expression of GLP-2 or changes in the degradation of GLP-2 by endogenous peptidases. Further work is needed to improve our understanding of these unique findings.
There are some shortcomings to the study, in that the complete control of electrolyte delivery in the GLM group may not have been perfectly matched to that in the TPN group without GLM, but the disparity was thought to be slight and did not account for the substantial changes detected in this study. Taken together, evidence from the present study shows for the first time the relationship between intestinal luminal nutrient-sensing mechanisms and TPN-associated complications. GLM supplementation at 75 mM significantly prevents TPN-associated intestinal atrophy. Two GPCRs, T1R3 and mGluR5, were not involved in the proproliferation mechanism of GLM in IECs, but played distinct and divergent roles in modulating the EBF of TPN mice. GLM supplementation further decreased plasma GLP-2 levels in TPN mice via both T1R3 and mGluR5, suggesting that the proproliferative effect of GLM on the TPN mice may be independent of GLP-2 release. More research is needed to understand the important role of luminal sensing in IEC proliferation and EBF modulation, as well as to determine the optimal dose of GLM to prevent intestinal atrophy without promoting epithelial barrier dysfunction under TPN administration. The use of GLM may be beneficial in preventing the development of clinical TPN-associated atrophy.
Supplementary Material
Acknowledgments
The authors thank P. J. Browner and Drs. R. Sueyoshi, M. W. Ralls, and R.S. Herman for assistance with mouse care, sample collection, and the Ussing chamber experiment, and H. K. Yoon for technical assistance.
This work was supported by Ajinomoto Co. Inc. (Tokyo, Japan) and U.S. National Institutes of Health grant 2R01AI-44076-14.
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- Ab
- antibody
- BrdU
- 5-bromo-2-deoxy uridine
- EAAT
- excitatory amino acid transporter
- EBF
- epithelial barrier function
- EC
- epithelial cell
- Gα
- α-gustducin
- GI
- gastrointestinal
- GLM
- glutamate
- GLP-2
- glucagon-like peptide-2
- GLMR
- glutamate receptor
- GPCR
- G-protein coupled receptor
- GRIK5
- glutamate receptor, ionotropic, kainate 5
- KO
- knockout
- IEC
- intestinal epithelial cell
- IF
- immunofluorescence
- mGluR
- metabotropic glutamate receptor
- MSG
- monosodium glutamate
- MTEP
- 3-((2-methyl-4-thiazolyl)ethynyl)pyridine
- pAkt
- phospho-Akt
- PCNA
- proliferating cell nuclear antigen
- SGLT-1
- sodium glucose cotransporter 1
- T1R
- T1 receptor
- TER
- transmembrane resistance
- TJ
- tight junction
- TR
- taste receptor
- TPN
- total parenteral nutrition
- WT
- wild-type
- ZO-1
- zona occludens 1
REFERENCES
- 1. Akiba Y., Kaunitz J. D. (2009) Luminal chemosensing and upper gastrointestinal mucosal defenses. Am. J. Clin. Nutr. 90, 826S–831S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yasumatsu K., Horio N., Murata Y., Shirosaki S., Ohkuri T., Yoshida R., Ninomiya Y. (2009) Multiple receptors underlie glutamate taste responses in mice. Am. J. Clin. Nutr. 90, 747S–752S [DOI] [PubMed] [Google Scholar]
- 3. Finger T. E., Kinnamon S. C. (2011) Taste isn't just for taste buds anymore. F1000 Biol. Rep. 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sternini C. (2007) Taste receptors in the gastrointestinal tract: IV, functional implications of bitter taste receptors in gastrointestinal chemosensing. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G457–G461 [DOI] [PubMed] [Google Scholar]
- 5. Negri R., Morini G., Greco L. (2011) From the tongue to the gut. J. Pediatr. Gastroenterol. Nutr. 53, 601–605 [DOI] [PubMed] [Google Scholar]
- 6. Agency for Healthcare Research and Quality (2010) Data on PN and EN Use. Healthcare Cost and Utilization Project, National Center for Health Statistics, Washington, DC [Google Scholar]
- 7. Feng Y., Ralls M. W., Xiao W., Miyasaka E., Herman R. S., Teitelbaum D. H. (2012) Loss of enteral nutrition in a mouse model results in intestinal epithelial barrier dysfunction. Ann. N. Y. Acad. Sci. 1258, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li X. (2009) T1R receptors mediate mammalian sweet and umami taste. Am. J. Clin. Nutr. 90, 733S–737S [DOI] [PubMed] [Google Scholar]
- 9. Akiba Y., Kaunitz J. D. (2011) Duodenal chemosensing and mucosal defenses. Digestion. 83(Suppl. 1), 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akiba Y., Kaunitz J. D. (2011) Luminal chemosensing in the duodenal mucosa. Acta Physiol. (Oxf.) 201, 77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akiba Y., Watanabe C., Mizumori M., Kaunitz J. D. (2009) Luminal l-glutamate enhances duodenal mucosal defense mechanisms via multiple glutamate receptors in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G781–G791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schunemann D. P., Grivicich I., Regner A., Leal L. F., de Araújo D. R., Jotz G. P., Fedrigo C. A., Simon D., da Rocha A. B. (2010) Glutamate promotes cell growth by EGFR signaling on U-87MG human glioblastoma cell line. Pathol. Oncol. Res. 16, 285–293 [DOI] [PubMed] [Google Scholar]
- 13. Yang H., Gumucio D. L., Teitelbaum D. H. (2008) Intestinal specific overexpression of interleukin-7 attenuates the alternation of intestinal intraepithelial lymphocytes after total parenteral nutrition administration. Ann. Surg. 248, 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang C., Feng Y., Yang H., Koga H., Teitelbaum D. H. (2009) The bone morphogenetic protein signaling pathway is upregulated in a mouse model of total parenteral nutrition. J. Nutr. 139, 1315–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun X., Spencer A. U., Yang H., Haxhija E. Q., Teitelbaum D. H. (2006) Impact of caloric intake on parenteral nutrition-associated intestinal morphology and mucosal barrier function. JPEN J. Parenter. Enteral Nutr. 30, 474–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng Y., Sun X., Yang H., Teitelbaum D. H. (2009) Dissociation of E-cadherin and beta-catenin in a mouse model of total parenteral nutrition: a mechanism for the loss of epithelial cell proliferation and villus atrophy. J. Physiol. 587, 641–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang H., Fan Y., Teitelbaum D. H. (2003) Intraepithelial lymphocyte-derived interferon-gamma evokes enterocyte apoptosis with parenteral nutrition in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G629–G637 [DOI] [PubMed] [Google Scholar]
- 18. Grossmann J., Maxson J. M., Whitacre C. M., Orosz D. E, Berger N. A., Fiocchi C., Levine A. D. (1998) New isolation technique to study apoptosis in human intestinal epithelial cells. Am. J. Pathol. 153, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng Y., McDunn J. E., Teitelbaum D. H. (2010) Decreased phospho-Akt signaling in a mouse model of total parenteral nutrition: a potential mechanism for the development of intestinal mucosal atrophy. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G833–G841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun X., Yang H., Nose K., Nose S., Haxhija E. Q., Koga H., Feng Y., Teitelbaum D. H. (2008) Decline in intestinal mucosal IL-10 expression and decreased intestinal barrier function in a mouse model of total parenteral nutrition. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G139–G147 [DOI] [PubMed] [Google Scholar]
- 21. Krug S. M., Amasheh S., Richter J. F., Milatz S., Günzel D., Westphal J. K., Huber O., Schulzke J. D., Fromm M. (2009) Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol. Biol. Cell 20, 3713–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nelson D. W., Murali S. G., Liu X., Koopmann M. C., Holst J. J., Ney D. M. (2008) Insulin-like growth factor I and glucagon-like peptide-2 responses to fasting followed by controlled or ad libitum refeeding in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1175–R1184 [DOI] [PubMed] [Google Scholar]
- 23. Young R. L., Sutherland K., Pezos N., Brierley S. M., Horowitz M., Rayner C. K., Blackshaw L. A. (2009) Expression of taste molecules in the upper gastrointestinal tract in humans with and without type 2 diabetes. Gut 58, 337–346 [DOI] [PubMed] [Google Scholar]
- 24. Mace O. J., Affleck J., Patel N., Kellett G. L. (2007) Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J. Physiol. 582, 379–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mace O. J., Lister N., Morgan E., Shepherd E., Affleck J., Helliwell P., Bronk J. R., Kellett G. L., Meredith D., Boyd R., Pieri M., Bailey P. D., Pettcrew R., Foley D. (2009) An energy supply network of nutrient absorption coordinated by calcium and T1R taste receptors in rat small intestine. J. Physiol. 587, 195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swartz T. D., Duca F. A., de Wouters T., Sakar Y., Covasa M. (2012) Up-regulation of intestinal type 1 taste receptor 3 and sodium glucose luminal transporter-1 expression and increased sucrose intake in mice lacking gut microbiota. Br. J. Nutr. 107, 621–630 [DOI] [PubMed] [Google Scholar]
- 27. Yasumatsu K., Ogiwara Y., Takai S., Yoshida R., Iwatsuki K., Torii K., Margolskee R. F., Ninomiya Y. (2012) Umami taste in mice uses multiple receptors and transduction pathways. J. Physiol. 590, 1155–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang H., Feng Y., Sun X., Teitelbaum D. H. (2009) Enteral versus parenteral nutrition: effect on intestinal barrier function. Ann. N. Y. Acad. Sci. 1165, 338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kinnamon S. C. (2009) Umami taste transduction mechanisms. Am J. Clin. Nutr. 90, 753S–755S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao L., Jiao Q., Yang P., Chen X., Zhang J., Zhao B., Zheng P., Liu Y. (2011) Metabotropic glutamate receptor 5 promotes proliferation of human neural stem/progenitor cells with activation of mitogen-activated protein kinases signaling pathway in vitro. Neuroscience 29, 185–194 [DOI] [PubMed] [Google Scholar]
- 31. Nochi R., Kato T., Kaneko J., Itou Y., Kuribayashi H., Fukuda S., Terazono Y., Matani A., Kanatani S., Nakajima K., Hisatsune T. (2012) Involvement of metabotropic glutamate receptor 5 signaling in activity-related proliferation of adult hippocampal neural stem cells. Eur. J. Neurosci. 36, 2273–2283 [DOI] [PubMed] [Google Scholar]
- 32. Janssen S., Laermans J., Verhulst P. J., Thijs T., Tack J., Depoortere I. (2011) Bitter taste receptors and alpha-gustducin regulate the secretion of ghrelin with functional effects on food intake and gastric emptying. Proc. Natl. Acad. Sci. U. S. A. 108, 2094–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nguyen C. A., Akiba Y., Kaunitz J. D. (2012) Recent advances in gut nutrient chemosensing. Curr. Med. Chem. 19, 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaji I., Karaki S., Fukami Y., Terasaki M., Kuwahara A. (2009) Secretory effects of a luminal bitter tastant and expressions of bitter taste receptors, T2Rs, in the human and rat. large intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G971–G981 [DOI] [PubMed] [Google Scholar]
- 35. Dockray G. J. (2003) Luminal sensing in the gut: an overview. J Physiol Pharmacol. 54, 9–17 [PubMed] [Google Scholar]
- 36. Bezencon C., le Coutre J., Damak S. (2007) Taste-signaling proteins are coexpressed in solitary intestinal epithelial cells. Chem. Senses 32, 41–49 [DOI] [PubMed] [Google Scholar]
- 37. Conigrave A. D., Brown E. M. (2006) Taste receptors in the gastrointestinal tract: II, l-amino acid sensing by calcium-sensing receptors: implications for GI physiology. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G753–G761 [DOI] [PubMed] [Google Scholar]
- 38. Rozengurt E. (2006) Taste receptors in the gastrointestinal tract: I, Bitter taste receptors and alpha-gustducin in the mammalian gut. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G171–G177 [DOI] [PubMed] [Google Scholar]
- 39. Mayer E. (2011) Gut feelings: the emerging biology of gut-brain communication. Nat. Rev. Neurosci. 12, 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wildhaber B. E., Yang H., Spencer A. U., Drongowski R. A., Teitelbaum D. H. (2005) Lack of enteral nutrition: effects on the intestinal immune system. J. Surg. Res. 123, 8–16 [DOI] [PubMed] [Google Scholar]
- 41. Shin Y. J., Park J. H., Choi J. S., Chun M. H., Moon Y. W., Lee M. Y. (2010) Enhanced expression of the sweet taste receptors and alpha-gustducin in reactive astrocytes of the rat hippocampus following ischemic injury. Neurochem. Res. 35, 1628–1634 [DOI] [PubMed] [Google Scholar]
- 42. Zhang J., Yin Y., Shu X. G., Li T., Li F., Tan B., Wu Z., Wu G. (2013) Oral administration of MSG increases expression of glutamate receptors and transporters in the gastrointestinal tract of young piglets. Amino Acids 45, 1169–1177 [DOI] [PubMed] [Google Scholar]
- 43. Wildhaber B. E., Lynn K. N., Yang H., Teitelbaum D. H. (2002) Total parenteral nutrition-induced apoptosis in mouse intestinal epithelium: regulation by the Bcl-2 protein family. Pediatr. Surg. Int. 18, 570–575 [DOI] [PubMed] [Google Scholar]
- 44. Wildhaber B. E., Yang H., Teitelbaum D. H. (2003) Total parenteral nutrition-induced apoptosis in mouse intestinal epithelium: modulation by keratinocyte growth factor. J. Surg. Res. 112, 144–151 [DOI] [PubMed] [Google Scholar]
- 45. Reeds P. J., Burrin D. G., Stoll B., Jahoor F. (2002) Intestinal glutamate metabolism. J. Nutr. 2000;130: 978S–982S [DOI] [PubMed] [Google Scholar]
- 46. Amagase K., Ochi A., Kojo A., Mizunoe A., Taue M., Kinoshita N., Nakamura E., Takeuchi K. (2012) New therapeutic strategy for amino acid medicine: prophylactic and healing promoting effect of monosodium glutamate against NSAID-induced enteropathy. J. Pharmacol. Sci. 118, 131–137 [DOI] [PubMed] [Google Scholar]
- 47. Reeds P. J., Burrin D. G., Stoll B., Jahoor F. (2000) Intestinal glutamate metabolism. J. Nutr. 130: 978S–982S [DOI] [PubMed] [Google Scholar]
- 48. Sheng H., Shao J., Townsend C. M. J., Evers B. M. (2003) Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 52, 1472–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nose K., Yang H., Sun X., Nose S., Koga H., Feng Y., Miyasaka E., Teitelbaum D. H. (2010) Glutamine prevents total parenteral nutrition-associated changes to intraepithelial lymphocyte phenotype and function: a potential mechanism for the preservation of epithelial barrier function. J. Interferon Cytokine Res. 30, 67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Newsholme P., Procopio J., Lima M. M., Pithon-Curi T. C., Curi R. (2003) Glutamine and glutamate: their central role in cell metabolism and function. Cell Biochem. Funct. 21, 1–9 [DOI] [PubMed] [Google Scholar]
- 51. Kondoh T., Mallick H. N., Torii K. (2009) Activation of the gut-brain axis by dietary glutamate and physiologic significance in energy homeostasis. Am. J. Clin. Nutr. 90, 832S–837S [DOI] [PubMed] [Google Scholar]
- 52. Sharp C. D., Hines I., Houghton J., Warren A., Jackson T. H., 4th, Jawahar A., Nanda A., Elrod J. W., Long A., Chi A., Minagar A., Alexander J. S. (2003) Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am. J. Physiol. Heart Circ. Physiol. 285, H2592–H2598 [DOI] [PubMed] [Google Scholar]
- 53. Vermeulen M. A., de Jong J., Vaessen M. J., van Leeuwen P. A, Houdijk A. P. (2011) Glutamate reduces experimental intestinal hyperpermeability and facilitates glutamine support of gut integrity. World J. Gastroenterol. 17, 1569–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Znalesniak E. B., Hoffmann W. (2010) Modulation of cell-cell contacts during intestinal restitution in vitro and effects of epidermal growth factor (EGF). Cell Physiol. Biochem. 25, 533–542 [DOI] [PubMed] [Google Scholar]
- 55. Zhao G. Q., Zhang Y., Hoon M. A., Chandrashekar J., Erlenbach I., Ryba N. J., Zuker C. S. (2003) The receptors for mammalian sweet and umami taste. Cell 115, 255–266 [DOI] [PubMed] [Google Scholar]
- 56. Delay E. R., Hernandez N. P., Bromley K., Margolskee R. F. (2006) Sucrose and monosodium glutamate taste thresholds and discrimination ability of T1R3 knockout mice. Chem. Senses 31, 351–357 [DOI] [PubMed] [Google Scholar]
- 57. Niswender C. M., Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kubera C., Hernandez A. L., Heng V., Bordey A. (2012) Transient mGlu5R inhibition enhances the survival of granule cell precursors in the neonatal cerebellum. Neuroscience 219, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu X., Nelson D. W., Holst J. J., Ney D. M. (2006) Synergistic effect of supplemental enteral nutrients and exogenous glucagon-like peptide 2 on intestinal adaptation in a rat model of short bowel syndrome. Am. J. Clin. Nutr. 84, 1142–1150 [DOI] [PubMed] [Google Scholar]
- 60. Chance W. T., Sheriff S., Dayal R., Friend L. A., Thomas I., Balasubramaniam A. (2006) The role of polyamines in glucagon-like peptide-2 prevention of TPN-induced gut hypoplasia. Peptides 27, 883–892 [DOI] [PubMed] [Google Scholar]
- 61. Wang J. H., Inoue T., Higashiyama M., Guth P. H., Engel E., Kaunitz J. D., Akiba Y. (2011) Umami receptor activation increases duodenal bicarbonate secretion via glucagon-like peptide-2 release in rats. J. Pharmacol. Exp. Ther. 339, 464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wauson E. M., Zaganjor E., Lee A. Y, Guerra M. L., Ghosh A. B., Bookout A. L., Chambers C. P., Jivan A., McGlynn K., Hutchison M. R., Deberardinis R. J., Cobb M. H. (2012) The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol. Cell 47, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.