

Abstract
Duchenne muscular dystrophy in boys progresses rapidly to severe impairment of muscle function and death in the second or third decade of life. Current supportive therapy with corticosteroids results in a modest increase in strength as a consequence of a general reduction in inflammation, albeit with potential untoward long-term side effects and ultimate failure of the agent to maintain strength. Here, we demonstrate that alternative approaches that rescue defective autophagy in mdx mice, a model of Duchenne muscular dystrophy, with the use of rapamycin-loaded nanoparticles induce a reproducible increase in both skeletal muscle strength and cardiac contractile performance that is not achievable with conventional oral rapamycin, even in pharmacological doses. This increase in physical performance occurs in both young and adult mice, and, surprisingly, even in aged wild-type mice, which sets the stage for consideration of systemic therapies to facilitate improved cell function by autophagic disposal of toxic byproducts of cell death and regeneration.—Bibee, K. P., Cheng, Y.-J., Ching, J. K., Marsh, J. N., Li, A. J., Keeling, R. M., Connolly, A. M., Golumbek, P. T., Myerson, J. W., Hu, G., Chen, J., Shannon, W. D., Lanza, G. M., Weihl, C. C., Wickline, S. A. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function.
Keywords: perfluorocarbon, Duchenne, cardiomyopathy, nanomedicine, drug delivery
Duchenne muscular dystrophy (DMD) is a fatal X-linked muscle degenerative disorder that affects 1:3500 males and is caused by mutations in the dystrophin gene, leading to complete absence of cell membrane-stabilizing dystrophin proteins (1). Progressive weakness leading to early demise results from widespread cellular injury, as muscle cell membranes undergo repeated mechanical tearing and disruption. This form of muscular dystrophy now is treated symptomatically with corticosteroids, resulting in a modest increase in strength, albeit with expected and unavoidable side effects (2–5). Exon-skipping gene therapy to correct the basic defect also is showing promise, and larger clinical trials are now under way, yet the cardiac phenotype has proven difficult to modify, and pursuit of combination therapies that might be steroid sparing remains an active area of research (6–9).
With regard to newer pharmaceutical strategies, a recent promising drugable target is autophagy, which is a cellular process responsible for recycling reusable products of metabolism and/or efficient elimination of toxic byproducts (10). Indeed, rapamycin, a known inducer of autophagy, was reported by Eghtesad et al. (11) in 6-wk-old mdx mice to improve diaphragm muscle histopathology; and protein restriction has been tested by De Palma et al. to activate autophagy in 16-wk-old mdx leading to enhanced grip strength (12). Recent work by Pauly et al. (13) has demonstrated that autophagy induction in the diaphragm of 6-wk-old mdx animals by daily intraperitoneal injections of an AMP-activated protein kinase agonist also promotes clearance of defective mitochondria and improves diaphragm strength. However, the potential benefit of enhancing autophagy in multiple muscle groups with the use of clinically practicable therapeutic agents that might elicit rapid and repeatable responses in both global physical performance and cardiac functionality in vivo over a broad range of ages (e.g., ≥18 mo) has not been examined.
Rapamycin, an immune-suppressing macrolide typically used to prevent organ transplant rejection and as an anti-inflammatory agent to prevent angioplasty restenosis (14–16), is known to induce autophagy, thereby inciting a cell survival program that enables useful recycling of amino acids via nonspecific degradation of long-lived proteins and dysfunctional organelles (17). By binding to the mammalian target of rapamycin complex 1 (mTORC1), rapamycin blocks proproliferative, antiapoptotic signaling (18). Paradoxically, blocking mTORC1 with rapamycin might be expected to exert deleterious effects on protein synthesis, muscle fiber regeneration, and cell growth in mdx mice, all of which might be expected to maintain strength. However, if autophagy is fundamentally defective in DMD/mdx and can be shown to be related to weakness and cardiac dysfunction and if a method for effective delivery of clinically approved doses of rapamycin to muscle tissues that might enhance autophagy and improve endurance can be designed, then a compelling mechanistic link between disturbed autophagy and progressive deterioration of global physical strength and contractile performance in vivo would be suggested.
Nanoparticle (NP) therapeutics for treating cancer and other pathologies are now undergoing clinical trials in myriad drug combinations (19), but heretofore have not been evaluated in neuromuscular diseases. The potential advantage conferred by such agents is their high local accumulation at sites of disease in concert with the use of lower serum levels of free drug that would limit unwanted off-target side effects, as has been shown for Doxil and Abraxane in cancer, for example. Accordingly, we compared both an oral preparation of rapamycin and an i.v. rapamycin-loaded nanoparticle (RNP) formulation for ability to improve a clinically significant global performance factor related to endurance and grip strength through the use of validated methods that are translationally relevant and highly correlated with mortality in the mdx model (20). Furthermore, we measured autophagic responses to RNPs in skeletal muscle tissues for correlation with grip strength metrics. Finally, we delineated the effects of these agents on cardiac function in aged mdx and wild-type mice.
MATERIALS AND METHODS
Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified.
NP formulation
The nontargeted perfluorocarbon (PFC) NP emulsions without or with rapamycin (at a concentration of 0.275 mol%) were prepared as described previously (21). In general, the sizes of the PFC NPs range between ∼160 and 240 nm (22, 23). As an example, batch MS1-92b exhibited a diameter of 184.7 nm, polydispersity of 0.120, and ζ potential of −15.31 mV. We note from prior experience that drug loading has little effect on the size but may affect ζ potential, depending on the exact composition and charge of the loaded moiety (24, 25). An additional, dual-fluorophore emulsion was formulated to verify delivery of both NPs and rapamycin to skeletal muscle. The emulsion lipid moiety incorporated rhodamine B (Avanti Polar Lipids, Alabaster, AL, USA), and the rapamycin was conjugated with a near-infrared (IR) fluorescent dye (Cy7.5; Lumiprobe Corporation, Hallandale Beach, FL) before formulation into the NPs. PFC NPs are exceedingly stable in blood, as they were originally developed as a blood substitute (26). The stability of rapamycin incorporated into NPs has been reported previously (21). Drug release kinetics against an infinite sink of releasing medium in vitro verified retention of 97% of NP rapamycin over 3 d.
Animal studies
Strength studies
Male mdx mice (strain C57BL/10ScSn-Dmdmdx/J) and age-matched controls (strain C57BL/10SnJ) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All mice were housed in the same animal facility and cared for under approval by Washington University's animal study committee. Starting at age 14 wk, mice were injected 2×/wk for 4 wk with plain NPs (n=18 for mdx, n=4 for control) or NPs loaded with rapamycin (n=16 for mdx, n=5 for wild-type). Dosage was 1 ml emulsion/kg body mass, injected via lateral tail vein, resulting in ∼0.002 mg rapamycin/animal/treatment. An additional set of mdx (n=8) mice was housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility until they were 17 mo of age, at which point they received 4 wk of RNP treatment as described above.
Separate groups were utilized for oral rapamycin therapy. For these animals, rapamycin was solubilized in grain alcohol and was delivered in a mixture of dextrose and water 2×/wk for 4 wk via micropipette gavage, typically 40–50 μl/mouse. There were 3 treatment groups: mdx mice given rapamycin (n=6), control mice given rapamycin (n=6), and mdx mice given placebo (n=6).
Treatment effectiveness was determined by measuring forelimb grip strength within 1 wk before beginning each treatment regimen, and subsequently within 1 wk after treatment completion. Strength testing was performed in blinded fashion by an experienced technician as described previously by our group (20) and by others who have used this standardized test of physical fitness and endurance. Strength testing was performed using a grip meter attached to a force transducer (E-DF 002 Digital Force Gauge; Chatillon Force Measurement Systems, Largo, FL, USA). The tests relied on the animal's instinct to grab hold as it was pulled backward. Each mouse was grasped by the tail and encouraged to grab a trapeze bar attached to the force transducer using its forelimbs. The mouse was then pulled away, and the peak force was recorded on a digital display. This procedure was repeated 5 times in a row for each animal. The 3 highest readings were averaged and normalized by the animal's weight to give the strength score.
RNP drug holiday studies
To determine whether the effects of RNPs on grip strength were reproducible in individual animals, a cohort (n=7 for mdx/no drug, n=8 for mdx/rapamycin, n=4 for wild-type/no drug, and n=5 for wild-type/rapamycin) of the originally treated 14-wk-old mice was maintained off any drug therapy over the span of 18–34 wk of age to allow washout of the RNP effect. The same 1-mo RNP treatment and grip strength testing regimen was then repeated for 1 mo starting at age 34 wk.
Cardiac studies
A cohort of 17-mo-old mdx mice received either i.v. RNPs (n=9) or no treatment (n=7) via 8 tail vein injections (1.5 ml/kg/dose), 2×/wk over 4 wk to deliver the same cumulative amount of rapamycin as for the other studies above. Wild-type mice of the same age were used as controls (n=6 for RNPs, n=8 for no treatment). The mice were lightly anesthetized for cardiac ultrasonography with isoflurane and placed supine for examination with a 16-MHz array (Spark; Ardent Sound, Inc., Mesa, AZ, USA). Prior to imaging, the thorax of each animal was shaved and depilated. Animal body temperature was maintained with a heated water blanket and thermistor-controlled heating lamp. Heated ultrasound coupling gel was liberally applied to the shaved area. Waveforms were sampled at the system's clock frequency (66.67 MHz) using a 12-bit digitizer (GaGe Compuscope 12400, DynamicSignals LLC, Lockport, IL, USA). Cardiac data (200 frames) were acquired for each view at a frame rate of 152 Hz, with each frame consisting of 128 A-lines (2048 sampled points/line). Each loop thus consisted of between 8 and 12 heart cycles. Radio frequency (RF) data were converted to scaled image data using custom plugins written for the open-source image processing software ImageJ (W. S. Rasband, U.S. National Institutes of Health, Bethesda, MD, USA; http://imagej.nih.gov/ij/). Backscattered ultrasound energy was mapped to grayscale by taking the log of the sum of the squares of the digitized voltage values in a center-weighted moving window. The resulting image loops were scaled to a resolution of 0.05 mm/pixel. Left ventricular ejection fraction (LVEF) was computed using ImageJ by manually tracing the ventricular endocardium border at systole and diastole in the short-axis (SAX) view for each beat and recording the area region of interest (ROI). Ventricular length was computed from the long-axis (LAX) view by determining the length of a line manually drawn from the apex endocardium to the aortic valve plane. Left ventricular volume at systole (or diastole) was estimated by computing the volume of an ellipsoid having a circular cross section equal to the mean systolic (or diastolic) ROI area and length equal to the mean systolic (or diastolic) measured ventricular length. LVEF was then computed as the ratio of the difference in volume of this ellipsoid between diastole and systole, and the diastolic volume.
Histology
Fibrosis imaging was performed in the diaphragm by analyzing trichrome (Masson)-stained, formalin-fixed sections (8-μm thickness) with a stain kit (HT-15-1KT; Sigma-Aldrich) and in the heart with picrosirius red stains per standard protocol (27). Color light microscope images of collagen were acquired from 5 randomly selected fields for each diaphragm section at ×200 with an Olympus BX61 microscope and F-View camera (Olympus America, Center Valley, PA, USA). Similar assessments were performed for basal heart sections from 5 treated and 5 untreated mdx. Custom software (written as a plugin for ImageJ) was used to quantify the area of each image associated with collagen-labeled pixels relative to the area of each image associated with tissue of any type.
For fluorescence histological imaging, 14-wk-old mdx mice were given i.v. injections (1 ml/kg) of dual-labeled fluorescent NPs and sacrificed 4 h postinjection following systemic saline perfusion to wash out any residual circulating NPs. NP and/or rapamycin tissue distribution was demonstrated in the biceps, diaphragm, gluteus, and myocardium of mice injected with fluorophore-loaded emulsions in excised and frozen [optimal cutting temperature (OCT) medium; Tissue-Tek, Sakura Finetek, Torrance, CA, USA] or paraffin-embedded tissues. Sectioning (8 μm) was performed on 10 consecutive slides from each tissue sample (a total of 60 slides were prepared), and random sections were chosen for fluorescence microscopy in a blinded procedure. Adjacent slides were saved for hematoxylin and eosin (H&E) staining. Multiple images were acquired from each section with an Olympus BX51 fluorescence microscope image system, using an Infinity3 camera (Lumenera Corp., Ottawa, ON, Canada) for optical image recording and digitization, and an Olympus F-View II black-and-white CCD camera for fluorescent images.
Pharmacokinetic studies
To obtain sufficient measurements of the concentration of circulating PFC NPs to enable robust biexponential fits to calculate distribution and clearance kinetics from single mice, we employed in vivo fluorine (19F) magnetic resonance spectroscopy (MRS) to register the perfluoro-15-crown-5 ether (crown ether) signal in the tail of the mouse at multiple time points after i.v. injection. Because the measured 19F signal magnitude emanating from the core of the NPs is directly proportional to the concentration of NPs in selected ROIs (voxels; refs. 28, 29), these data can be used as a surrogate for blood draw measurements when more finely sampled data are required. Furthermore, we also drew blood for coarser temporal sampling to compare against the fitted 19F signals as an additional check on accuracy.
PFC NPs were prepared as described in previous work (21, 24, 25, 28–30). Briefly, the emulsions contained 20% (v/v) perfluoro-15-crown-5 ether (Exfluor Research Corp., Round Rock, TX, USA), 2% (w/v) of a surfactant mixture, 1.7% (w/v) glycerin, and water for the balance. The surfactant, including 98 mol% egg yolk phosphotidylcholine (Avanti Polar Lipids) and 2 mol% phosphatidylethanolamine (Avanti Polar Lipids) in chloroform:methanol (3:1), was dried under vacuum to form a lipid film. The surfactant components were combined with the crown ether and distilled deionized water, and emulsified (Microfluidics, Newton, MA) at 20,000 psi for 4 min. Particle sizes were measured to be 200 ± 25 nm at 25°C using a laser light-scattering submicrometer particle analyzer (Brookhaven Instruments, Holtsville, NY, USA).
NPs were administered as a bolus through the jugular vein to 5 C57/BL6 mice at a dose of 1 ml/kg body weight after ketamine (87 mg/kg)/xylazine (13 mg/kg) anesthesia. 19F MR signal from the tail was measured on a Varian 4.7-T imaging system (Varian Medical Systems Inc., Palo Alto, CA, USA), while the mouse was maintained under anesthesia [ketamine (54 mg/kg/h)/xylazine (8.2 mg/kg/h)]. Spectra were obtained with a custom-built 4-turn solenoid coil via nonlocalized 19F spin-echo spectroscopy (256 signal averages, TR=1.05 s; TE=1 ms). Periodic blood draws were performed for ex vivo spectroscopic measurement of 19F concentration and plotted against the in vivo spectroscopic 19F measurements.
Tissue NP biodistribution
Nontargeted 19F PFC-core NPs were administered to 14 mdx mice (18 wk old) by tail vein injection; and tissue and organ harvesting was conducted at either 2, 12, or 24 h. Tissues harvested for measurement of NP concentrations were acquired after in situ perfusion fixation of the mouse that was initiated by cardiac puncture and flow-through of buffer solution until venous effluent was clear (∼10 min). This washout procedure ensured that blood contamination was not a factor in the estimation of tissue NP content. We also note that the tissue distributions and clearances for similar particles are not affected by drug loading and have been reported before for mice and rabbits (24, 25, 31). The specimens were blotted dry, weighed, fixed in formalin, and then subjected to fluorine spectroscopy with the use of an 11.7-T Varian MRI/MRS scanner and a custom-built single-turn solenoid coil. 19F spectra signal from each specimen and from a standard of perfluoro-15-crown-5 ether were obtained via nonlocalized 90-FID spectroscopy (2.5-s pulse TR, and 128, 256, or 512 signal averages NT). The unique and quantitative fluorine signature was expressed as total tissue fluorine content per gram. This methodology fortunately avoids the necessity for extraction of the target compound (PFC in this case) with its attendant inefficiencies, since the total amount of 19F signal in the entire unprocessed tissue can be registered nondestructively. Because there is a direct and linear relationship between the 19F signal and the NP concentration, the analytical method yields essentially the NP concentration per unit tissue mass (28).
Creatine kinase (CK)
Blood was obtained by left ventricular cardiocentesis, and serum was extracted by centrifugation. Serum CK levels were measured spectrophotometrically with a Vitros DT60 dry reagent chemistry analyzer (OrthoClinical Diagnostics, Raritan, NJ, USA).
Autophagy and autophagic flux measurements
Muscle tissues were extracted from the mdx mice and homogenized in 0.5 ml of RIPA buffer with 1 mM PMSF, Complete protease inhibitor cocktail (Roche, Indianapolis, IN, USA), and phosphatase inhibitor cocktail (Roche) using a glass grinding tube. The tissue homogenates were centrifuged at 10,000 g for 10 min at 4°C, and the supernatant was stored at −80°C.
Proteins in tissue homogenates were resolved on a NuPAGE Novex 4–12% Bis-Tris Gel (Invitrogen, Carlsbad, CA, USA), transferred to 0.2 μM nitrocellulose, blocked in 5.0% milk/TBS-T, and incubated with the following primary antibodies: rabbit anti-pS6 (1:1000; 4857; Cell Signaling, Boston, MA, USA), rabbit anti-S6 (1:1000; 2217; Cell Signaling), rabbit anti-LC3B (1:2000; Sigma-Aldrich), rabbit anti-Beclin1 (1:1000; Cell Signaling), goat anti-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Corresponding secondary antibodies were used at 1:5000. Protein bands were visualized using either ECL Western blotting substrate (32106; Pierce, Rockford, IL, USA) or Super Signal West Femto maximum sensitivity substrate (34095; Pierce). Western blot imaging was used to calculate relative protein expression using densitometry with ImageJ. Values were normalized to myosin loading control and are reported as fold change compared to the control, nontreated sample.
To illustrate how this approach might apply to actual primary human cells and be potentially generalizable, tumor cells [HIM3 line from the Washington University Human and Mouse Linked Evaluation of Tumors (HAMLET) Core] were derived from a biopsy specimen that was procured from a triple-negative primary breast tumor and were passaged through immunodeficient animals in a method described previously (32). Cells were maintained in McCoy's 5A medium (Invitrogen) supplemented with 10% FBS (Sigma-Aldrich) and penicillin/streptomycin (Invitrogen) in a 37°C humidified tissue culture incubator with 5% carbon dioxide. Lentivirus containing the LC3-GFP fusion was obtained from C. Weihl (Washington University). The cells were transfected using a standard spinfection method with protamine sulfate. After recovery, the cells were sorted on a BD Aria II high-speed cell sorter (BD Biosciences, San Jose, CA, USA) 3 times with 1-wk recovery periods between each sort. The stable cell lines were then treated for 24 h with DMSO (vehicle for rapamycin), rapamycin (100 nM), NPs (3.09×1010 particles/ml), or RNPs (3.09×1010 particles/ml=100 nM rapamycin). After treatment, the cells were washed, fixed with paraformadehyde, and imaged with a Zeiss LSM510 microscope (Carl Zeiss, Oberkochen, Germany). GFP-positive puncta were counted by a observer in a blinded procedure. Other treated cells were prepared for transmission electron microscopy (TEM) per standard protocols.
For in vivo autophagic flux assessment, we used a recently developed autophagic flux assay that measures turnover of LC3B-II in mouse skeletal muscle by comparing mice treated with and without a potent blocker of autophagic protein degradation (colchicine; ref. 33). This method avoids the use of other protein degradation blockers (bafilomycin, chloroquine) that can manifest confounding systemic toxicities. Colchicine was administered over 48 h in two doses (0.4 mg/kg) delivered intraperitoneally, followed by diaphragm or heart tissue collection. Tissue homogenates were probed as above.
A separate set of experiments using oral prednisolone as the active agent was conducted to delineate potential mechanistic similarities between RNP and steroids with respect to their effects on autophagy for an agent that is known to improve strength experimentally and in patients. Four mdx mice were treated with oral prednisolone and 4 control mdx were treated with vehicle for 42 wk (starting at age 4 wk), after which muscle tissues [tibialis anterior (TA)] were extracted for autophagy measurements as above.
Statistics
Statistics were generated as noted below for selected ANOVA, t test, and groupwise comparison procedures using SAS software (SAS Institute, Cary, NC, USA) and the open-source statistical package R (R Foundation for Statistical Computing, Vienna, Austria, http://www.r-project.org).
RESULTS
Rapid enhancement of global physical performance with RNPs
Wild-type and mdx mice (14 wk old) were treated with 8 doses of i.v. NPs over 4 wk, and standardized grip strength measurements were obtained on each animal before and after therapy. No animals demonstrated apparent adverse effects from the treatment at any time point, and all animals completed the study. Figure 1A shows that grip strength increased significantly when RNPs were employed as therapy (30% absolute increase in mean strength of group). Unexpectedly, when NPs alone were employed as therapy, grip strength exhibited a trend toward a modest strength increase of 8% (Fig. 1A).
Figure 1.

RNP treatment improves strength in mdx mice. A) Treatment with i.v. RNPs for 4 wk significantly increased mean weight-normalized grip strength in 14-wk-old mdx animals (n=16) vs. groups given plain NPs intravenously (n=18), equivalent oral doses of rapamycin (n=6), or oral placebo (n=6) (P=0.012, P=0.027, and P=0.016, respectively, using Fisher's protected least significant difference at 5% significance level). *P < 0.05. B) No significant difference in mean strength was observed for age-matched wild-type mice given either NPs (n=4) or RNPs intravenously (n=5) or oral rapamycin (n=6). C) Increase in weight-normalized strength in a subset of mdx mice (n=8) treated with i.v. RNPs occurred in both young (14–18 wk) and old (34–38 wk) animals (P=0.008 using a linear contrast model comparing pretreatment to posttreatment differences at a 5% significance level). Absolute values of the mean change in strength between time points are shown in columns below trend data. D) Wild-type mice (n=5) given similar treatment and drug holiday exhibit no significant difference between pretreatment and posttreatment strength (P=0.598). Bar graphs show mean ± sem values.
To determine whether the grip strength improvement was due to the drug alone, animals were treated with oral rapamycin administered at 10× the equivalent i.v. NP dose to account for an expected 10% oral absorption rate (34–37). As shown in Fig. 1A, the oral rapamycin did not improve grip strength. Age-matched wild-type mice exhibited no significant increase in grip strength when treated with RNPs or oral rapamycin (Fig. 1B).
To elucidate whether grip strength increase was repeatable, a subset of treated young animals were given a drug holiday from wk 18 to 34, and then the NP dosing was repeated. Figure 1C shows that over this washout interval, strength declined as anticipated. After the repeat dosing of i.v. RNP, grip strength once again improved in the older mdx animals (P=0.008, linear contrast model). No significant trend was observed in wild-type mice (Fig. 1D). Interestingly, RNP treatment of a cohort of 17-mo-old mdx (n=8) mice treated for 1 mo in similar fashion revealed a more muted effect on strength (P=0.201, 2-tailed paired t test; Fig. 2A).
Figure 2.
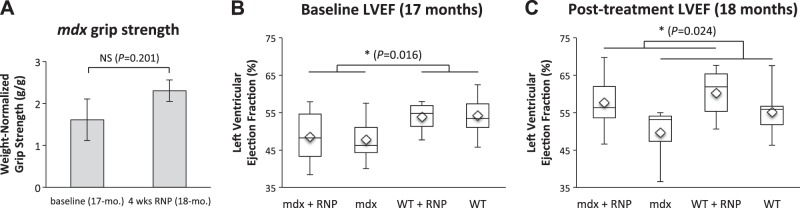
Aged mdx mice show muted responses in grip strength after treatment with RNP, but marked improvement in cardiac function commensurate with wild-type animals. A) Treatment of 17-mo-old mdx (n=8) for 4 wk yielded no significant increase in grip strength (P=0.201, 2-tailed paired t test). B) LVEF in wild-type mice is greater than mdx before treatment (P=0.016), as expected. C) Cardiac function significantly improved after RNP therapy, irrespective of animal type (16% difference for mdx and 9% difference for WT; P=0.024). NS, not significant. *P < 0.05.
Rapid enhancement of cardiac performance with RNPs in aged mice
In the same cohort of 17 mo-old mdx and wild-type mice, 1 mo of biweekly RNPs vs. no treatment yielded two compelling findings: LVEF in mdx was significantly lower than that of wild-type mice before treatment (P=0.016) as expected (Fig. 2B); and LVEF improved significantly after RNP therapy, irrespective of animal type (P=0.024).
NP pharmacokinetics and tissue distribution
Supplemental Fig. S1 shows an example of the PFC NP clearance curves for a single mouse. In 5 individual trials, the observed decay of in vivo 19F signal intensity was fitted to a biexponential curve. The obtained exponents were interpreted as half-lives in a 2-compartment model. Median distribution half-life of crown ether NPs was 8.23 min. Assuming a γ distribution, distribution half-life exhibited a 90% confidence interval of 0.08–50.67 min. Assuming a normal distribution, mean clearance half-life was 181.3 ± 40.7 min. Clearance half-life exhibited a 90% confidence interval of 142.5–220.0 min. These values for half-lives in the blood were comparable to those reported in previous work as delineated from blood sampling (29, 31, 38).
To ensure that the particles were being delivered to the muscles in the mdx animals, fluorine spectroscopy was performed as a means to quantify the 19F perfluorooctyl bromide (PFOB) core of the NPs, which directly reports the biodistribution and actual concentration of NPs in muscles and other tissues. Baseline levels of 19F in tissues are negligible, so it can be concluded that the spectroscopy signal is due to the presence of particles alone. Substantial uptake was observed in diverse tissue groups (Supplemental Fig. S2), and 2-tailed t-tests showed no differences in particle uptake when rapamycin was incorporated into the particles for all muscles tested (Fig. 3A). For example, on the basis of the fluorine data, ∼0.25% of the injected dose of NPs was taken up by the diaphragm, roughly equivalent to 0.1 μg rapamycin/g muscle.
Figure 3.

NPs and rapamycin are effectively delivered to skeletal and cardiac muscle in mdx animals. A) 19F spectroscopy was performed on excised tissues to determine the levels of PFOB in mdx muscle. Each of the muscles surveyed showed NP (PFOB) uptake 24 h after last systemic injection. There was no significant difference in uptake between NPs and RNPs in any of the muscles tested (2-tailed t test, 5% significance level). B) mdx mice were given i.v. injections (1 ml/kg) of dual-labeled fluorescent NPs and euthanized 4 h postinjection following saline perfusion. Biceps (top panels), diaphragm, (middle panels), and gluteus (bottom panels) were excised, and adjacent sections were prepared for staining with H&E (left panels) and fluorescence microscopy (middle and right panels). NP lipid labeled with rhodamine is evident from detected fluorescence within skeletal muscle (middle panels), as is rapamycin within same tissue (right panels). C) NP lipid (rhodamine, displayed as blue) and rapamycin (Cy7.5, labeled as red) are colocalized in mdx cardiac tissue.
In addition, fluorescence microscopy was performed with fluorophore-labeled rapamycin and, separately, fluorophore-labeled lipids contained in the outer NP shell (Fig. 3B). These data indicate that the particles and drug cargo traverse muscle boundaries and distribute widely within the skeletal muscle tissue. It is also clear that the particles penetrate heart tissue to deliver rapamycin locally (Fig. 3C). The labeled rapamycin appears to achieve an even greater penetration into tissues than do the NPs, arguing for some degree of local rapamycin release and more complete passive diffusion. We note that the use of the near-IR fluorophore avoids the problem of background autofluorescence, thereby permitting confident assessment of the more diffuse penetration of rapamycin into muscle tissues beyond the loci of NP deposition. The observation of diffuse rapamycin penetration into tissues beyond the NP carriers (Fig. 3B) was further supported by analyzing both blank slides and unstained muscle tissue specimens that were microscopically digitized at the same settings to rule out background signals of any type that might have overlapped with the rapamycin signal, and none were observed (data not shown).
Effects on skeletal and cardiac mTOR and autophagic processes
To further demonstrate that the drug reaches muscle tissue and then acts on a known target, lysates from the muscle were probed for phosphorylated S6. S6 is downstream of mTORC1, so that blocking mTORC1 with rapamycin should cause a decrease in pS6 levels. Indeed, in diaphragm, triceps, biceps, and gluteus, S6 levels were decreased in mdx animals treated with RNPs when compared to mice treated with NPs alone (Fig. 4A). Interestingly, the NPs themselves caused a modest increase in pS6 levels when compared with saline treated animals. Thus, the particle itself may induce signaling through mTORC1, but adding rapamycin to the particle then decreases the level of signaling, yet does not cause a return to baseline levels.
Figure 4.

mdx animals exhibit a defect in autophagy that is restored by NP treatment. A) RNPs cause a decrease in S6 levels in mdx animals when compared to NP treatment. However, compared to saline treatment, the NPs increased S6 phosphorylation in all muscle groups tested, a trend not observed in the muscles from wild-type animals. B) Proteins were extracted from mdx and age-matched control animal muscle. Western blot analysis for p62 (n=5/group) and BNIP3 (n=3/group) reveal a defect in autophagy as mdx animals exhibited higher levels of p62 and lower BNIP3 when compared to wild-type (Student's t test at 5% significance level). Myosin was used as the loading control. C) RNA was isolated from the TA muscle of age matched mdx (n=5) and control mice (n=3). Quantitative PCR was performed and normalized to GAPDH. Graph demonstrates fold change in the following transcripts vs. control: there was no change in p62, BNIP3, or beclin. β2-Microglobin is increased as expected, because it is part of MHCI, which is known to be up-regulated in mdx muscle. Actin levels (normal) serve as an internal control. D) Representative Western blots demonstrate that both NPs and RNPs increase the levels of LC3B-II in mdx animals, whereas saline-treated animals exhibit a low level of LC3B-II expression, even when blocked with colchicine. Scatterplots show results from individual animals.
Our data confirm that mdx mice harbor a defect in autophagy in muscle tissues, as there is an increase in p62 protein (Fig. 4B), with no difference in p62 transcription (Fig. 4C). This expression pattern suggests that p62 is not being consumed in the process of autophagy in the mdx animals, as it is in the wild-type animals. Further, BNIP3, a protein that is known to trigger autophagy, is found at lower levels in mdx animals when compared to wild type (Fig. 4B, C). Additional in vivo confirmation of the effects of RNPs on autophagy revealed that basal autophagic flux was diminished in mdx mouse muscle as demonstrated by comparing the change in LC3II protein levels with and without colchicine treatment (Fig. 4D). Confirmatory evidence of increased autophagy in primary human cells treated with RNPs was observed by TEM (Supplemental Fig. S3A) and by fluorescence imaging of punctate staining for LC3-GFP (Supplemental Fig. 3B), thereby illustrating the effect on actual human tissues and the potential for generalization to other tissue types.
Additional probing of the TA and heart muscle in the 17-mo-old mice treated for 1 mo with RNPs was performed to assess the autophagic responses. Figure 5A illustrates significant effects of RNPs in TA on p62 (decrease), BNIP3 (increase), and LC3II (increase). Figure 5B confirms that RNPs also enhance autophagy in the heart in the same manner. Figure 5C shows autophagic responses to prednisolone in the TA of mdx mice, where changes in p62, BNIP3, and LC3II are all similar in direction and magnitude to those reported above for RNPs, further suggesting that autophagy responses to either RNPs or steroids in the mdx represents a likely candidate mechanism for the observed augmentation of muscle strength and cardiac function. Taken together, these data point to a rapid and widespread induction of autophagy in diverse muscle tissues after RNP treatment.
Figure 5.

RNPs alter autophagic biomarkers in heart and skeletal muscle of mdx mice similar to that after steroid therapy. A) TA muscle in 17-mo-old mdx mice treated for 1 mo with RNPs exhibited significant effects of RNPs on p62 (decrease), BNIP3 (increase), and LC3II (increase). B) Cardiac data confirm that RNPs also enhance autophagy in the heart in the same manner. C) Young mdx mice given 42 wk of steroid therapy show similar autophagic responses in TA muscle. *P < 0.05.
Tissue structural responses
The amount of collagen within skeletal and cardiac muscles (Fig. 6A–D) was not different between the treatment groups (P=0.900 for diaphragm using mixed-model nested ANOVA, P=0.664 for heart using unpaired 2-tailed t test). Figure 6E demonstrates no significant difference between CK levels in saline-treated animals when compared to those treated with NPs or RNPs (P=0.682, 1-way ANOVA). From these data, it does not appear that the NPs are exerting their beneficial effect on strength by altering the turnover of myocytes or the replacement of myocytes with collagen in the short term, as might be expected after only 8 doses of therapy.
Figure 6.

Intravenously injected NPs do not affect short-term muscle cell destruction or fibrosis. A) Masson trichrome stains performed on diaphragm sections and visualized with light microscopy (×200) showed increased fibrosis (blue) for mdx animals relative to WT. B) No significant difference occurred over the short 4-wk test interval in mdx collagen levels after treatment with either RNPs, plain NPs, or saline (mixed model nested ANOVA, P=0.90). C, D) Picrosirius red stains of mdx mice (C) show diffuse heterogeneous fibrosis with no significant difference (P=0.664; D) when treated with RNPs. E) Serum CK levels for mdx mice exhibited no significant difference after treatment with either RNPs, NPs, or saline (ANOVA, P=0.71). Bar graphs show mean ± sd values.
DISCUSSION
The present findings indicate that rapid improvement in physical performance of either skeletal and cardiac muscle can be achieved after only 8 doses of RNPs in conjunction with augmented autophagy. In contrast, oral rapamycin in pharmacological doses exerted no significant effect on strength within the same interval. The observations that the improvement in grip strength is repeatable after a drug holiday in the same mice and that muscle inflammation per se is not the short-term target, together offer further support for the salutary role of RNPs on enhancing autophagy, global physical performance, and cardiac function in muscular dystrophy. The muted response to strength testing in aged mdx mice as compared to the clear enhancement in cardiac function reveals a not unexpected limitation on what might be achievable after initiation of therapy late in the disease process.
Recently published work suggests that autophagy induction may be beneficial to mdx animals, whether induced by rapamycin or by dietary protein reduction (13). Moreover, a decrease in autophagic flux in vivo has been previously reported for an animal model of Bethlem myopathy and Ullrich congenital muscular dystrophy due to mutations in Col6a−/−, suggesting that disturbed autophagy might occur in other muscular dystrophies (39), yet this mechanism has not been evaluated previously for aged DMD/mdx animals, as we show here. Furthermore, no prior studies have demonstrated improvements in global physical performance in vivo or cardiac function after rapamycin therapy in aged animals that might be attributable to enhanced autophagy.
The observation of a muted response to short-term RNPs in the older 17-mo-old mice for skeletal muscle performance in the face of a clear improvement in myocardial LVEF and clear induction of autophagy points out certain divergent pathophysiological features of the disease. It is recognized that the heart disease in DMD appears later than the skeletal muscle weakness, but emerges ultimately as a major source of morbidity and mortality. Why heart muscle is relatively more resistant to disease progression than is skeletal muscle despite continuous contractile activity remains unknown. Interestingly, the disease in the heart evolves regionally (e.g., base>apex) despite the ubiquitous presence of the genetic defect (40). Furthermore, even while LVEF appears normal at earlier ages in patients despite severe skeletal muscle weakness, subtle defects in cardiac strain parameters are revealed by cardiac MRI tagging procedures that indicate silent onset of the disease process, where early intervention might alter the natural history of overt manifestations such as diastolic dysfunction or reduced LVEF (41).
Although recent retrospective analyses of steroids in DMD are highly suggestive of a beneficial long-term effect on cardiac function (42), the present data are the first to illustrate such rapid improvement with systemic rapamycin in a prospective trial invoking a unique mechanism of action. Parenthetically, our observation of improved LVEF even in wild type subjects (Fig. 2) accords with a recent report by Flynn et al. (43) noting better LVEF in 24-mo-old female nondystrophic animals treated continuously for 3 mo with rapamycin in an oral high-delivery formulation. The attenuated response of skeletal muscle strength to RNPs in older mdx mice in our study suggests either that longer-term or more aggressive therapy might be required at this stage to enhance strength, or that certain limitations may exist for this (or even any) form of therapy, neither of which is especially surprising. Interestingly, Flynn et al. (43) also reported that oral rapamycin increased endurance but not intensity of activity in 24 mo-old female mice, suggesting a muted effect on strength improvement in older subjects, in general, which accords with our observation on grip strength for the 17-mo-old dystrophic mice. Regardless, the ability of RNPs to rapidly improve cardiac performance suggests that a single agent might be useful for both clinical sequelae.
The cumulative dose of rapamycin administered to the animals in the RNP treatment group was within the limits of recommended oral doses for patients receiving immunosuppressive therapy when accounting for ∼20% oral absorption in humans (44). The rationale for comparing i.v. RNP therapy with oral rapamycin delivery stems from the conventional practice of oral delivery as standard of care in patients requiring immune suppression, which is known to be effective and well tolerated at the recommended dose levels. We accounted for the well-known and previously reported bioavailability for oral dosing [rough estimates: ∼10% in mice (refs. 34–37)] by augmenting the doses that were delivered to mice by oral gavage to ensure adequate delivery. Typical clinical maintenance dosing for oral delivery of sirolimus in organ-transplant scenarios for body mass > 40 kg is 2 mg/d (Rapamune; Pfizer, Philadelphia, PA, USA). Assuming 20% bioavailability, this equates to ∼0.4 mg of rapamycin in circulation per day, or ∼0.006 mg/kg/d for a 70-kg patient. The maximum permissible daily dose of rapamycin is 40 mg, which equates to 0.11 mg/kg/d. Mice treated with i.v. RNPs in the current study were given 0.067 mg/kg rapamycin biweekly for a total of 8 times over 4 wk, or the equivalent of 0.002 mg/kg/d, within the clinical bounds for regular therapeutic use.
The i.v. dose used here was ∼7-fold less than the calculated absorbed oral dose administered to mdx mice by Eghtesad et al. (11) to improve muscle histopathology. Moreover, Eghtesad et al. (11) also reported that oral or injected rapamycin exerted no effect on the TA muscle, in contrast to the marked up-regulation of autophagy reported here in the TA after RNP therapy (see Fig. 6). Taken together, these data indicate that at clinically relevant doses of oral rapamycin for mdx animals, which scale up to 4-fold greater than the recommended oral dose for patients, no improvement in skeletal muscle strength might be expected. In contrast, a clinically approved cumulative dose of rapamycin administered in NP formulations rapidly elicited marked improvement in both global physical performance and cardiac function after only 8 i.v. treatments, which could represent an advantage for periodic, short-term dosing.
The enhancement of S6 phosphorylation seen in the mdx animals after NP treatment suggests that the particles themselves may drive the mTORC1 cell signaling pathway. Previously, it has been shown that protein synthesis is shut off in the high intracellular calcium environment found in muscles of patients with muscular dystrophy and in mouse models (45, 46). The enhancement of S6 phosphorylation is particularly surprising given that the diaphragm of mdx mice is reported to be unable to increase mTOR activity with age (47). The addition of rapamycin to the particle decreased the level of pS6 but did not reduce it to baseline, suggesting that some signal promotion was still occurring in these cells, which may be one factor contributing to an increase in physical performance.
Furthermore, NPs alone did not increase autophagic flux in wild-type mice, whereas RNPs did. While rapamycin is known to promote autophagy via an mTOR-dependent mechanism, the mechanism by which NPs influence autophagy is not known. It is intriguing that NPs were unable to enhance autophagy in wild-type mice with presumably normal sarcolemmal structure, whereas in mdx mice that have defective sarcolemmal integrity due to dystrophin mutations, NPs were effective.
Rapamycin also exhibits mTORC1-independent effects when it binds to FKBP12 on the ryanodine receptor. Previous studies suggest that binding of rapamycin disrupts the ryanodine receptor's functions and, therefore, would be expected to elicit dysfunctional calcium handling (48, 49). However, in mdx animals, the ryanodine receptor itself appears to be dysfunctional (50). It may be the case that the addition of rapamycin acts to modulate some of these receptors in a favorable manner and effect more normal calcium handling or else acts to decrease the expression of the receptors, as has been previously reported (51). Regardless, it is clear that both NPs and rapamycin NPs are affecting fundamental cellular signaling pathways in ways not heretofore reported, and which remain to be elucidated.
The effectiveness of NPs and RNPs for stimulating autophagy in mdx mouse muscle mirrors that of their efficacy in rapidly improving grip strength and cardiac function. Although these data may not establish absolute proof of cause and effect for the relationship between autophagy and strength, they suggest an intriguing candidate mechanism for the decline in strength that might be manageable clinically with an approved drug and a new delivery system. The observation that steroids also augment autophagy (Fig. 5C) supports this contention and suggests a complementary and synergistic approach to that of steroid monotherapy.
Although the exact mechanism responsible for rapamycin's entry into muscle tissue remains to be clarified, the effect on autophagy is clear and seems dependent on NP-linked depot delivery, because oral therapy is ineffective at the doses that were administered. The sustained presence of NPs and rapamycin in skeletal and cardiac tissues is clear from the 19F spectroscopy and the fluorescence imaging data. Prior pharmacokinetic studies in both rabbits and mice using PFC NPs have confirmed a clearance half-time (or, elimination half-life) of ∼3–5 h in normal mice and rabbits used in various cancer models (25, 31), irrespective of molecular targeting, suggesting that there should be ample opportunity to penetrate into the damaged tissues. We previously have demonstrated rapid permeation of these NPs in other situations where leaky or disrupted vasculature pertains, such as advanced atherosclerosis in cholesterol-fed rabbits, and even in atherosclerotic human carotid endarterectomy specimens (21, 52). It is instructive to note that in dystrophic subjects, recent and prior publications have pointed out the widespread vascular dysfunction and damage that permits egress of circulating components into muscle interstitium (53). Dystrophic subjects also harbor defects in vascular endothelium that render lining cells susceptible to apoptosis and impaired angiogenesis (54), creating a situation of barrier dysfunction that even applies to the blood-brain barrier (55). We further note that the tissue distributions of rapamycin (widespread) and NPs (more patchy) shown in Fig. 3B differ considerably, indicating the possibility of trapped NPs serving as a depot for subsequent local delivery of the rapamycin agent based on a sustained local diffusion gradient. Additional immunohistological sections that were stained for macrophages and vasculature also were not colocalized with the broader extent of rapamycin permeation, nor even with the fluorescent NP signatures (data not shown).
Regarding the translational potential of this approach, we note that these beneficial effects on both heart and skeletal muscle were achieved at a cumulative dose of rapamycin that accords with clinically recommended dosing. Although the side effects of rapamycin on glucose tolerance, weight gain, blood pressure, and immune responsiveness are recognized, the drug is generally well tolerated over many years by patients with organ transplantation. The fact that both prednisone and rapamycin appear to induce autophagy as a possible mechanism of action raises the possibility that the potential deleterious consequences of either agent might be mitigated if used in lower doses in combination, or at least if RNP could be employed for a steroid sparing action.
Supplementary Material
Acknowledgments
The authors acknowledge contributions of other colleagues to this study: Kefeng Wang (statistical analysis); John Allen, Cordelia Caradine, and Xiaoxia Yang (animal preparation); Huiying Zhang and Noriko Yanaba (histology); and Ralph W. Fuhrhop, Michael J. Scott, and Angana Senpan (NP formulation).
This work was supported by U.S. National Institutes of Health (NIH) grants R01 AR056223 and HL073646 (S.A.W.); NIH HL112518, HL113392, and NS073457ß (G.M.L.); and NIH K02 AG042095 (C.C.W.); the Muscular Dystrophy Association (C.C.W., A.M.C., and P.T.G.); and American Heart Association graduate fellowship award 12PRE9310044 (Y.-J.C.).
Footnotes
- CK
- creatine kinase
- DMD
- Duchenne muscular dystrophy
- H&E
- hematoxylin and eosin
- IR
- infrared
- LVEF
- left ventricular ejection fraction
- MRS
- magnetic resonance spectroscopy
- mTORC1
- mammalian target of rapamycin complex 1
- NP
- nanoparticle
- PFC
- perfluorocarbon
- PFOB
- perfluorooctyl bromide
- RF
- radio frequency
- RNP
- rapamycin-loaded nanoparticle
- ROI
- region of interest
- TA
- tibialis anterior
- TEM
- transmission electron microscopy
REFERENCES
- 1. Hoffman E. P., Brown R. H., Jr., Kunkel L. M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 [DOI] [PubMed] [Google Scholar]
- 2. Moxley R. T., 3rd, Ashwal S., Pandya S., Connolly A., Florence J., Mathews K., Baumbach L., McDonald C., Sussman M., Wade C. (2005) Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 64, 13–20 [DOI] [PubMed] [Google Scholar]
- 3. Markham L. W., Spicer R. L., Khoury P. R., Wong B. L., Mathews K. D., Cripe L. H. (2005) Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr. Cardiol. 26, 768–771 [DOI] [PubMed] [Google Scholar]
- 4. Biggar W. D., Harris V. A., Eliasoph L., Alman B. (2006) Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. 16, 249–255 [DOI] [PubMed] [Google Scholar]
- 5. Balaban B., Matthews D. J., Clayton G. H., Carry T. (2005) Corticosteroid treatment and functional improvement in Duchenne muscular dystrophy: long-term effect. Am. J. Phys. Med. Rehab. 84, 843–850 [DOI] [PubMed] [Google Scholar]
- 6. Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M. E., Bourke J., Wells D. J., Dickson G., Wood M. J., Wilton S. D., Straub V., Kole R., Shrewsbury S. B., Sewry C., Morgan J. E., Bushby K., Muntoni F. (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goemans N. M., Tulinius M., van den Akker J. T., Burm B. E., Ekhart P. F., Heuvelmans N., Holling T., Janson A. A., Platenburg G. J., Sipkens J. A., Sitsen J. M., Aartsma-Rus A., van Ommen G. J., Buyse G., Darin N., Verschuuren J. J., Campion G. V., de Kimpe S. J., van Deutekom J. C. (2011) Systemic administration of PRO051 in Duchenne's muscular dystrophy. N. Engl. J. Med. 364, 1513–1522 [DOI] [PubMed] [Google Scholar]
- 8. Kinali M., Arechavala-Gomeza V., Feng L., Cirak S., Hunt D., Adkin C., Guglieri M., Ashton E., Abbs S., Nihoyannopoulos P., Garralda M. E., Rutherford M., McCulley C., Popplewell L., Graham I. R., Dickson G., Wood M. J., Wells D. J., Wilton S. D., Kole R., Straub V., Bushby K., Sewry C., Morgan J. E., Muntoni F. (2009) Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Deutekom J. C., Janson A. A., Ginjaar I. B., Frankhuizen W. S., Aartsma-Rus A., Bremmer-Bout M., den Dunnen J. T., Koop K., van der Kooi A. J., Goemans N. M., de Kimpe S. J., Ekhart P. F., Venneker E. H., Platenburg G. J., Verschuuren J. J., van Ommen G. J. (2007) Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686 [DOI] [PubMed] [Google Scholar]
- 10. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., Al-Younes H. M., Al-Zeer M. A., Albert M. L., Albin R. L., Alegre-Abarrategui J., Aleo M. F., Alirezaei M., Almasan A., Almonte-Becerril M., Amano A., Amaravadi R., Amarnath S., Amer A. O., Andrieu-Abadie N., Anantharam V., Ann D. K., Anoopkumar-Duki S., Aoki H., Apostolova N., Arancia G., Aris J. P., Asanuma K., Asare N. Y., Ashida H., Askanas V., Askew D. S., Auberger P., Baba M., Backues S. K., Baehrecke E. H., Bahr B. A., Bai X. Y., Bailly Y., Baiocchi R., Baldini G., Balduini W., Ballabio A., Bamber B. A., Bampton E. T., Bánhegyi G., Bartholomew C. R., Bassham D. C., Bast R. C, Jr., Batoko H., Bay B. H., Beau I., Béchet D. M., Begley T. J., Behl C., Behrends C., Bekri S., Bellaire B., Bendall L. J., Benetti L., Berliocchi L., Bernardi H., Bernassola F., Besteiro S., Bhatia-Kissova I., Bi X., Biard-Piechaczyk M., Blum J. S., Boise L. H., Bonaldo P., Boone D. L., Bornhauser B. C., Bortoluci K. R., Bossis I., Bost F., Bourquin J. P., Boya P., Boyer-Guittaut M., Bozhkov P. V., Brady N. R., Brancolini C., Brech A., Brenman J. E., Brennand A., Bresnick E. H., Brest P., Bridges D., Bristol M. L., Brookes P. S., Brown E. J., Brumell J. H., Brunetti-Pierri N., Brunk U. T., Bulman D. E., Bultman S. J., Bultynck G., Burbulla L. F., Bursch W., Butchar J. P., Buzgariu W., Bydlowski S. P, Cadwell K., Cahová M., Cai D., Cai J., Cai Q., Calabretta B., Calvo-Garrido J., Camougrand N., Campanella M., Campos-Salinas J., Candi E., Cao L., Caplan A. B., Carding S. R., Cardoso S. M., Carew J. S, Carlin C. R., Carmignac V., Carneiro L. A., Carra S., Caruso R. A., Casari G., Casas C., Castino R., Cebollero E., Cecconi. F., Celli J., Chaachouay H., Chae H. J., Chai C. Y., Chan D. C., Chan EY, Chang RC, Che C. M., Chen C. C., Chen G. C., Chen G. Q., Chen M., Chen Q., Chen S. S., Chen W., Chen X., Chen X., Chen X., Chen Y. G., Chen Y., Chen Y., Chen Y. J., Chen Z., Cheng A., Cheng C. H., Cheng Y., Cheong H., Cheong J. H., Cherry S., Chess-Williams R., Cheung Z. H., Chevet E., Chiang H. L., Chiarelli R., Chiba T., Chin L. S., Chiou S. H., Chisari F. V., Cho C. H., Cho D. H., Choi A. M., Choi D., Choi K. S., Choi M. E., Chouaib S., Choubey D., Choubey V., Chu C. T., Chuang T. H, Chueh S. H, Chun T., Chwae Y. J., Chye M. L., Ciarcia R., Ciriolo M. R., Clague M. J., Clark R. S., Clarke P. G., Clarke R., Codogno P., Coller H. A., Colombo M. I., Comincini S., Condello M., Condorelli F., Cookson M. R., Coombs G. H., Coppens I., Corbalan R., Cossart P., Costelli P., Costes S., Coto-Montes A., Couve E., Coxon F. P., Cregg J. M., Crespo J. L., Cronjé M. J., Cuervo A. M., Cullen J. J., Czaja M. J., D'Amelio M., Darfeuille-Michaud A., Davids L. M., Davies F. E., De Felici M., de Groot J. F., de Haan C. A., De Martino L., De Milito A., De Tata V., Debnath J., Degterev A., Dehay B., Delbridge L. M., Demarchi F., Deng Y. Z., Dengjel J., Dent P., Denton D., Deretic V., Desai S. D., Devenish R. J., Di Gioacchino M., Di Paolo G., Di Pietro C., Díaz-Araya G., Díaz-Laviada I., Diaz-Meco M. T., Diaz-Nido J., Dikic I., Dinesh-Kumar S. P., Ding W. X., Distelhorst C. W., Diwan A., Djavaheri-Mergny M., Dokudovskaya S., Dong Z., Dorsey F. C., Dosenko V., Dowling J. J., Doxsey S., Dreux M, Drew M. E., Duan Q., Duchosal M. A., Duff K., Dugail I., Durbeej M., Duszenko M., Edelstein C. L., Edinger A. L., Egea G., Eichinger L., Eissa N. T., Ekmekcioglu S., El-Deiry W. S., Elazar Z., Elgendy M., Ellerby L. M., Eng K. E., Engelbrecht A. M., Engelender S., Erenpreisa J., Escalante R., Esclatine A., Eskelinen E. L., Espert L., Espina V., Fan H., Fan J., Fan Q. W., Fan Z., Fang S., Fang Y., Fanto M., Fanzani A., Farkas T., Farré J. C., Faure M., Fechheimer M., Feng C. G., Feng J., Feng Q., Feng Y., Fésüs L., Feuer R., Figueiredo-Pereira M. E., Fimia G. M., Fingar D. C., Finkbeiner S., Finkel T., Finley K. D., Fiorito F., Fisher E. A., Fisher P. B., Flajolet M., Florez-McClure M. L., Florio S., Fon E. A., Fornai F., Fortunato F., Fotedar R., Fowler D. H., Fox H. S., Franco R., Frankel L. B., Fransen M., Fuentes J. M., Fueyo J., Fujii J., Fujisaki K., Fujita E., Fukuda M., Furukawa R. H., Gaestel M., Gailly P., Gajewska M., Galliot B., Galy V., Ganesh S., Ganetzky B., Ganley I. G., Gao F. B., Gao G. F., Gao J., Garcia L., Garcia-Manero G., Garcia-Marcos M., Garmyn M., Gartel A. L., Gatti E., Gautel M., Gawriluk T. R., Gegg M. E., Geng J., Germain M., Gestwicki J. E., Gewirtz D. A., Ghavami S., Ghosh P., Giammarioli A. M., Giatromanolaki A. N., Gibson S. B., Gilkerson R. W., Ginger M. L., Ginsberg H. N., Golab J., Goligorsky M. S., Golstein P., Gomez-Manzano C., Goncu E., Gongora C., Gonzalez C. D., Gonzalez R., González-Estévez C., González-Polo R. A., Gonzalez-Rey E., Gorbunov N. V., Gorski S., Goruppi S., Gottlieb R. A., Gozuacik D., Granato G. E., Grant G. D., Green K. N., Gregorc A., Gros F., Grose C., Grunt T. W., Gual P., Guan J. L., Guan K. L., Guichard S. M., Gukovskaya A. S., Gukovsky I., Gunst J., Gustafsson A. B., Halayko A. J., Hale A. N., Halonen S. K., Hamasaki M, Han F., Han T., Hancock M. K., Hansen M., Harada H., Harada M., Hardt S. E., Harper J. W., Harris A. L., Harris J., Harris S. D., Hashimoto M., Haspel J. A., Hayashi S., Hazelhurst L. A., He C., He Y. W., Hébert M. J., Heidenreich K. A., Helfrich M. H., Helgason G. V., Henske E. P., Herman B., Herman P. K., Hetz C., Hilfiker S., Hill J. A., Hocking L. J., Hofman P., Hofmann T. G., Höhfeld J., Holyoake T. L., Hong M. H., Hood D. A., Hotamisligil G. S., Houwerzijl E. J., Høyer-Hansen M., Hu B., Hu C. A., Hu H. M., Hua Y., Huang C., Huang J., Huang S., Huang W. P., Huber T. B., Huh W. K., Hung T. H., Hupp T. R., Hur G. M., Hurley J. B., Hussain S. N., Hussey P. J., Hwang J. J., Hwang S., Ichihara A., Ilkhanizadeh S., Inoki K., Into T., Iovane V., Iovanna J. L., Ip N. Y., Isaka Y., Ishida H., Isidoro C., Isobe K., Iwasaki A., Izquierdo M., Izumi Y., Jaakkola P. M., Jäättelä M., Jackson G. R., Jackson W. T, Janji B., Jendrach M., Jeon J. H., Jeung E. B., Jiang H., Jiang H., Jiang J. X., Jiang M., Jiang Q., Jiang X., Jiang X., Jiménez A., Jin M., Jin S., Joe C. O., Johansen T., Johnson D. E., Johnson G. V., Jones N. L., Joseph B., Joseph S. K., Joubert A. M., Juhász G., Juillerat-Jeanneret L., Jung C. H., Jung Y. K., Kaarniranta K., Kaasik A., Kabuta T., Kadowaki M., Kagedal K., Kamada Y., Kaminskyy V. O., Kampinga H. H., Kanamori H., Kang C., Kang K. B., Kang K. I., Kang R., Kang Y. A., Kanki T., Kanneganti T. D., Kanno H., Kanthasamy A. G., Kanthasamy A., Karantza V., Kaushal G. P., Kaushik S., Kawazoe Y., Ke P. Y., Kehrl J. H., Kelekar A., Kerkhoff C., Kessel D. H., Khalil H., Kiel J. A., Kiger A. A., Kihara A., Kim D. R., Kim D. H., Kim D. H., Kim E. K., Kim H. R., Kim J. S., Kim J. H., Kim J. C., Kim J. K., Kim P. K., Kim S. W., Kim Y. S., Kim Y., Kimchi A., Kimmelman A. C., King J. S., Kinsella T. J., Kirkin V., Kirshenbaum L. A., Kitamoto K., Kitazato K., Klein L., Klimecki W. T., Klucken J., Knecht E., Ko B. C., Koch J. C., Koga H., Koh J. Y., Koh Y. H., Koike M., Komatsu M, Kominami E., Kong H. J., Kong W. J., Korolchuk V. I., Kotake Y., Koukourakis M. I., Kouri Flores J. B., Kovács A. L., Kraft C., Krainc D., Krämer H., Kretz-Remy C., Krichevsky A. M., Kroemer G., Krüger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, László L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee A. J., Lee B. W., Lee G. M., Lee J., Lee J. H., Lee M., Lee M. S., Lee S. H., Leeuwenburgh C., Legembre P., Legouis R., Lehmann M., Lei H. Y., Lei Q. Y., Leib D. A., Leiro J., Lemasters J. J., Lemoine A., Lesniak M. S., Lev D., Levenson V. V., Levine B., Levy E., Li F., Li J. L., Li L., Li S., Li W., Li X. J., Li Y. B., Li Y. P., Liang C., Liang Q., Liao Y. F., Liberski P. P., Lieberman A., Lim H. J., Lim K. L., Lim K., Lin C. F., Lin F. C., Lin J, Lin J. D., Lin K., Lin W. W., Lin W. C., Lin Y. L., Linden R., Lingor P., Lippincott-Schwartz J., Lisanti M. P., Liton P. B., Liu B., Liu C. F., Liu K., Liu L., Liu Q. A., Liu W., Liu Y. C., Liu Y., Lockshin R. A., Lok C. N., Lonial S., Loos B., Lopez-Berestein G., López-Otín C., Lossi L., Lotze M. T., Lőw P., Lu B., Lu B., Lu B., Lu Z., Luciano F., Lukacs N. W., Lund A. H., Lynch-Day M. A., Ma Y., Macian F., MacKeigan J. P., Macleod K. F., Madeo F., Maiuri L., Maiuri M. C., Malagoli D., Malicdan M. C., Malorni W., Man N., Mandelkow E. M., Manon S., Manov I., Mao K., Mao X., Mao Z., Marambaud P., Marazziti D., Marcel Y. L., Marchbank K., Marchetti P., Marciniak S. J., Marcondes M., Mardi M., Marfe G., Mariño G., Markaki M., Marten M. R., Martin S. J., Martinand-Mari C., Martinet W., Martinez-Vicente M., Masini M., Matarrese P., Matsuo S., Matteoni R., Mayer A., Mazure N. M., McConkey D. J., McConnell M. J., McDermott C., McDonald C., McInerney G. M., McKenna S. L., McLaughlin B., McLean P. J., McMaster C. R., McQuibban G. A., Meijer A. J., Meisler M. H., Meléndez A., Melia T. J., Melino G., Mena M. A., Menendez J. A., Menna-Barreto R. F., Menon M. B., Menzies F. M., Mercer C. A., Merighi A., Merry D. E., Meschini S., Meyer C. G., Meyer T. F., Miao C. Y., Miao J. Y., Michels P. A., Michiels C., Mijaljica D., Milojkovic A., Minucci S., Miracco C., Miranti C. K., Mitroulis I., Miyazawa K., Mizushima N., Mograbi B., Mohseni S., Molero X., Mollereau B., Mollinedo F., Momoi T., Monastyrska I., Monick M. M., Monteiro M. J., Moore M. N., Mora R., Moreau K., Moreira P. I., Moriyasu Y., Moscat J., Mostowy S., Mottram J. C., Motyl T., Moussa C. E., Müller S., Muller S., Münger K., Münz C., Murphy L. O., Murphy M. E., Musarò A., Mysorekar I., Nagata E., Nagata K., Nahimana A., Nair U., Nakagawa T., Nakahira K., Nakano H., Nakatogawa H., Nanjundan M., Naqvi N. I., Narendra D. P., Narita M., Navarro M., Nawrocki S. T., Nazarko T. Y., Nemchenko A., Netea M. G., Neufeld T. P., Ney P. A., Nezis I. P., Nguyen H. P., Nie D., Nishino I., Nislow C., Nixon R. A., Noda T., Noegel A. A., Nogalska A., Noguchi S., Notterpek L., Novak. I., Nozaki T., Nukina N., Nürnberger T., Nyfeler B., Obara K., Oberley T. D., Oddo S., Ogawa M., Ohashi T., Okamoto K., Oleinick N. L., Oliver F. J., Olsen L. J., Olsson S., Opota O., Osborne T. F., Ostrander G. K., Otsu K., Ou J. H., Ouimet M., Overholtzer M., Ozpolat B., Paganetti P., Pagnini U., Pallet N., Palmer G. E., Palumbo C., Pan T., Panaretakis T., Pandey U. B., Papackova Z., Papassideri I., Paris I., Park J., Park O. K., Parys J. B., Parzych K. R., Patschan S., Patterson C., Pattingre S., Pawelek J. M., Peng J., Perlmutter D. H., Perrotta I., Perry G., Pervaiz S., Peter M., Peters G. J., Petersen M., Petrovski G., Phang J. M., Piacentini M., Pierre P., Pierrefite-Carle V., Pierron G., Pinkas-Kramarski R., Piras A., Piri N., Platanias L. C., Pöggeler S., Poirot M., Poletti A., Poüs C., Pozuelo-Rubio M., Prætorius-Ibba M., Prasad A., Prescott M., Priault M., Produit-Zengaffinen N., Progulske-Fox A., Proikas-Cezanne T., Przedborski S., Przyklenk K., Puertollano R., Puyal J., Qian S. B., Qin L., Qin Z. H., Quaggin S. E., Raben N., Rabinowich H., Rabkin S. W., Rahman I., Rami A., Ramm G., Randall G., Randow F., Rao V. A., Rathmell J. C., Ravikumar B., Ray S. K., Reed B. H., Reed J. C., Reggiori F., Régnier-Vigouroux A., Reichert A. S., Reiners J. J, Jr., Reiter R. J., Ren J., Revuelta J. L., Rhodes C. J., Ritis K., Rizzo E., Robbins J., Roberge M., Roca H., Roccheri M. C., Rocchi S., Rodemann H. P., Rodríguez deCórdoba S., Rohrer B., Roninson I. B., Rosen K., Rost-Roszkowska M. M., Rouis M., Rouschop K. M., Rovetta F., Rubin B. P., Rubinsztein D. C., Ruckdeschel K., Rucker E. B., 3rd, Rudich A., Rudolf E., Ruiz-Opazo N., Russo R., Rusten T. E., Ryan K. M., Ryter S. W., Sabatini D. M., Sadoshima J., Saha T., Saitoh T., Sakagami H., Sakai Y., Salekdeh G. H., Salomoni P., Salvaterra P. M., Salvesen G., Salvioli R., Sanchez A. M., Sánchez-Alcázar J. A., Sánchez-Prieto R., Sandri M., Sankar U., Sansanwal P., Santambrogio L., Saran S., Sarkar S., Sarwal M., Sasakawa C., Sasnauskiene A., Sass M., Sato K., Sato M., Schapira A. H., Scharl M., Schätzl H. M., Scheper W., Schiaffino S., Schneider C., Schneider M. E., Schneider-Stock R., Schoenlein P. V., Schorderet D. F., Schüller C., Schwartz G. K., Scorrano L., Sealy L., Seglen P. O., Segura-Aguilar J., Seiliez I., Seleverstov O., Sell C., Seo J. B., Separovic D., Setaluri V., Setoguchi T., Settembre C., Shacka J. J., Shanmugam M., Shapiro I. M., Shaulian E., Shaw R. J., Shelhamer J. H., Shen H. M., Shen W. C., Sheng Z. H., Shi Y., Shibuya K., Shidoji Y., Shieh J. J., Shih C. M., Shimada Y., Shimizu S., Shintani T., Shirihai O. S., Shore G. C., Sibirny A. A., Sidhu S. B, Sikorska B., Silva-Zacarin E. C., Simmons A., Simon A. K., Simon H. U., Simone C., Simonsen A., Sinclair D. A., Singh R., Sinha D., Sinicrope F. A., Sirko A., Siu P. M., Sivridis E., Skop V., Skulachev V. P., Slack R. S., Smaili S. S., Smith D. R., Soengas M. S., Soldati T., Song X., Sood A. K., Soong T. W., Sotgia F., Spector S. A., Spies C. D., Springer W., Srinivasula S. M., Stefanis L., Steffan J. S., Stendel R., Stenmark H., Stephanou A., Stern S. T., Sternberg C., Stork B., Strålfors P., Subauste C. S., Sui X., Sulzer D., Sun J., Sun S. Y., Sun Z. J., Sung J. J., Suzuki K., Suzuki T., Swanson M. S., Swanton C., Sweeney S. T., Sy L. K., Szabadkai G., Tabas I., Taegtmeyer H., Tafani M., Takács-Vellai K., Takano Y., Takegawa K., Takemura G., Takeshita F., Talbot N. J., Tan K. S., Tanaka K., Tanaka K., Tang D., Tang D., Tanida I., Tannous B. A., Tavernarakis N., Taylor G. S., Taylor G. A., Taylor J. P., Terada L. S., Terman A., Tettamanti G., Thevissen K., Thompson C. B., Thorburn A., Thumm M., Tian F., Tian Y., Tocchini-Valentini G., Tolkovsky A. M., Tomino Y., Tönges L., Tooze S. A., Tournier C., Tower J., Towns R., Trajkovic V., Travassos L. H., Tsai T. F., Tschan M. P., Tsubata T., Tsung A., Turk B., Turner L. S., Tyagi S. C., Uchiyama Y., Ueno T., Umekawa M., Umemiya-Shirafuji R., Unni V. K., Vaccaro M. I., Valente E. M., Van den Berghe G., van der Klei I. J., van Doorn W., van Dyk L. F., van Egmond M., van Grunsven L. A., Vandenabeele P., Vandenberghe W. P., Vanhorebeek I., Vaquero E. C., Velasco G., Vellai T., Vicencio J. M., Vierstra R. D., Vila M., Vindis C., Viola G., Viscomi M. T., Voitsekhovskaja O. V., von Haefen C., Votruba M., Wada K., Wade-Martins R., Walker C. L., Walsh C. M., Walter J., Wan X. B., Wang A., Wang C., Wang D., Wang F., Wang F., Wang G., Wang H., Wang H. G., Wang H. D., Wang J., Wang K., Wang M., Wang R. C., Wang X., Wang X., Wang Y. J., Wang Y., Wang Z., Wang Z. C., Wang Z., Wansink D. G., Ward D. M., Watada H., Waters S. L., Webster P., Wei L., Weihl C. C., Weiss W. A., Welford S. M., Wen L. P., Whitehouse C. A., Whitton J. L., Whitworth A. J., Wileman T., Wiley J. W., Wilkinson S., Willbold D., Williams R. L., Williamson P. R., Wouters B. G., Wu C., Wu D. C., Wu W. K., Wyttenbach A., Xavier R. J., Xi Z., Xia P., Xiao G., Xie Z., Xie Z., Xu D. Z., Xu J., Xu L., Xu X., Yamamoto A., Yamamoto A., Yamashina S., Yamashita M., Yan X., Yanagida M., Yang D. S., Yang E., Yang J. M., Yang S. Y., Yang W., Yang W. Y., Yang Z., Yao M. C., Yao T. P., Yeganeh B, Yen W. L., Yin J. J., Yin X. M., Yoo O. J., Yoon G., Yoon S. Y., Yorimitsu T., Yoshikawa Y., Yoshimori T., Yoshimoto K., You H. J., Youle R. J., Younes A., Yu L., Yu L., Yu S. W., Yu W. H., Yuan Z. M., Yue Z., Yun C. H., Yuzaki M., Zabirnyk O., Silva-Zacarin E., Zacks D, Zacksenhaus E., Zaffaroni N., Zakeri Z., Zeh H. J., 3rd, Zeitlin S. O., Zhang H., Zhang H. L., Zhang J., Zhang J. P., Zhang L., Zhang L., Zhang M. Y., Zhang X. D., Zhao M., Zhao Y. F., Zhao Y., Zhao Z. J., Zheng X., Zhivotovsky B., Zhong Q., Zhou C. Z., Zhu C., Zhu W. G., Zhu X. F., Zhu X., Zhu Y., Zoladek T., Zong W. X., Zorzano A., Zschocke J., Zuckerbraun B. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eghtesad S., Jhunjhunwala S., Little S. R., Clemens P. R. (2011) Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol. Med. 17, 917–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Palma C., Morisi F., Cheli S., Pambianco S., Cappello V., Vezzoli M., Rovere-Querini P., Moggio M., Ripolone M., Francolini M., Sandri M., Clementi E. (2012) Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Disease 3, e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pauly M., Daussin F., Burelle Y., Li T., Godin R., Fauconnier J., Koechlin-Ramonatxo C., Hugon G., Lacampagne A., Coisy-Quivy M., Liang F., Hussain S., Matecki S., Petrof B. J. (2012) AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 181, 583–592 [DOI] [PubMed] [Google Scholar]
- 14. Thomson A. W., Turnquist H. R., Raimondi G. (2009) Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vilquin J. T., Asselin I., Guerette B., Kinoshita I., Roy R., Tremblay J. P. (1995) Successful myoblast allotransplantation in mdx mice using rapamycin. Transplantation 59, 422–426 [PubMed] [Google Scholar]
- 16. Adelman S. J. (2010) Sirolimus and its analogs and its effects on vascular diseases. Curr. Pharm. Des. 16, 4002–4011 [DOI] [PubMed] [Google Scholar]
- 17. Shigemitsu K., Tsujishita Y., Hara K., Nanahoshi M., Avruch J., Yonezawa K. (1999) Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J. Biol. Chem. 274, 1058–1065 [DOI] [PubMed] [Google Scholar]
- 18. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 19. Etheridge M. L., Campbell S. A., Erdman A. G., Haynes C. L., Wolf S. M., McCullough J. (2013) The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine 9, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keeling R. M., Golumbek P. T., Streif E. M., Connolly A. M. (2007) Weekly oral prednisolone improves survival and strength in male mdx mice. Muscle Nerve 35, 43–48 [DOI] [PubMed] [Google Scholar]
- 21. Cyrus T., Zhang H., Allen J. S., Williams T. A., Hu G., Caruthers S. D., Wickline S. A., Lanza G. M. (2008) Intramural delivery of rapamycin with αvβ3-targeted paramagnetic nanoparticles inhibits stenosis after balloon injury. Arterioscler. Thromb. Vasc. Biol. 28, 820–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wickline S., Neubauer A., Winter P., Caruthers S., Lanza G. (2006) Applications of nanotechnology to atherosclerosis, thrombosis, and vascular biology. Arterioscler. Thromb. Vasc. Biol. 26, 435–441 [DOI] [PubMed] [Google Scholar]
- 23. Wickline S. A., Mason R. P., Caruthers S. D., Chen J., Winter P. M., Hughes M. S., Lanza G. M. (2010) Fluorocarbon agents for multimodal molecular imaging and targeted therapeutics. In: Molecular Imaging: Principles and Practice, (Weissleder R., Ross B. D., Rehemtulla A., Gambhir S. S., eds.) pp. 542–573, Peoples Medical Publishing House, Shelton, CT, USA [Google Scholar]
- 24. Soman N. R., Lanza G. M., Heuser J. M., Schlesinger P. H., Wickline S. A. (2008) Synthesis and characterization of stable fluorocarbon nanostructures as drug delivery vehicles for cytolytic peptides. Nanoletters 8, 1131–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soman N. R., Baldwin S. L., Hu G., Marsh J. N., Lanza G. M., Heuser J. E., Arbeit J. M., Wickline S. A., Schlesinger P. H. (2009) Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J. Clin. Invest. 119, 2830–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flaim S. (1994) Pharmacokinetics and side effects of perfluorocarbon-based blood substitutes. Artif. Cells Blood Substit. Immobil. Biotechnol. 22, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 27. Dolber P. C., Spach M. S. (1987) Picrosirius red staining of cardiac muscle following phosphomolybdic acid treatment. Stain Technol. 62, 23–26 [DOI] [PubMed] [Google Scholar]
- 28. Morawski A. M., Winter P. M., Yu X., Fuhrhop R., Scott M., Hockett F., Robertson J. D., Gaffney P. J., Lanza G. M., Wickline S. A. (2004) Quantitative “magnetic resonance immunohistochemistry” with ligand-targeted 19F nanoparticles. Magn. Reson. Med. 52, 1255–1262 [DOI] [PubMed] [Google Scholar]
- 29. Neubauer A. M., Sim H., Winter P. M., Caruthers S. D., Williams T. A., Robertson J. D., Sept D., Lanza G. M., Wickline S. A. (2008) Nanoparticle pharmacokinetic profiling in vivo using magnetic resonance imaging. Magn. Reson. Med. 60, 1353–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan H., Marsh J. N., Christenson E. T., Soman N. R., Ivashyna O., Lanza G. M., Schlesinger P. H., Wickline S. A. (2012) Postformulation peptide drug loading of nanostructures. Methods Enzymol. 508, 17–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu G., Lijowski M., Zhang H., Partlow K. C., Caruthers S. D., Kiefer G., Gulyas G., Athey P., Scott M. J., Wickline S. A., Lanza G. M. (2007) Imaging of Vx-2 rabbit tumors with ανβ3-integrin-targeted 111In nanoparticles. Intl. J. Cancer 120, 1951–1957 [DOI] [PubMed] [Google Scholar]
- 32. Ding L., Ellis M. J., Li S. J., Larson D. E., Chen K., Wallis J. W., Harris C. C., McLellan M. D., Fulton R. S., Fulton L. L., Abbott R. M., Hoog J., Dooling D. J., Koboldt D. C., Schmidt H., Kalicki J., Zhang Q., Chen L., Lin L., Wendl M. C., McMichael J. F., Magrini V. J., Cook L., McGrath S. D., Vickery T. L., Appelbaum E., Deschryver K., Davies S., Guintoli T., Lin L., Crowder R., Tao Y., Snider J. E., Smith S. M., Dukes A. F., Sanderson G. E., Pohl C. S., Delehaunty K. D., Fronick C. C., Pape K. A., Reed J. S., Robinson J. S., Hodges J. S., Schierding W., Dees N. D., Shen D., Locke D. P., Wiechert M. E., Eldred J. M., Peck J. B., Oberkfell B. J., Lolofie J. T., Du F., Hawkins A. E., O'Laughlin M. D., Bernard K. E., Cunningham M., Elliott G., Mason M. D., Thompson D. M, Jr., Ivanovich J. L., Goodfellow P. J., Perou C. M., Weinstock G. M., Aft R., Watson M., Ley T. J., Wilson R. K., Mardis E. R. (2010) Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ju J. S., Miller S. E., Jackson E., Cadwell K., Piwnica-Worms D., Weihl C. C. (2009) Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy 5, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Napoli K. L., Wang M. E., Stepkowski S. M., Kahan B. D. (1997) Distribution of sirolimus in rat tissue. Clin. Biochem. 30, 135–142 [DOI] [PubMed] [Google Scholar]
- 35. Stepkowski S. M. (2003) Preclinical results of sirolimus treatment in transplant models. Transplant. Proc. 35, 219S–226S [DOI] [PubMed] [Google Scholar]
- 36. O'Reilly T., McSheehy P. M., Kawai R., Kretz O., McMahon L., Brueggen J., Bruelisauer A., Gschwind H. P., Allegrini P. R., Lane H. A. (2010) Comparative pharmacokinetics of RAD001 (everolimus) in normal and tumor-bearing rodents. Cancer Chemother. Pharmacol. 65, 625–639 [DOI] [PubMed] [Google Scholar]
- 37. Crowe A., Bruelisauer A., Duerr L., Guntz P., Lemaire M. (1999) Absorption and intestinal metabolism of SDZ-RAD and rapamycin in rats. Drug Metab. Dispos. 27, 627–632 [PubMed] [Google Scholar]
- 38. Winter P. M., Neubauer A. M., Caruthers S. D., Harris T. D., Robertson J. D., Williams T. A., Schmieder A. H., Hu G., Allen J. S., Lacy E. K., Zhang H., Wickline S. A., Lanza G. M. (2006) Endothelial α(v)β3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 26, 2103–2109 [DOI] [PubMed] [Google Scholar]
- 39. Grumati P., Coletto L., Sabatelli P., Cescon M., Angelin A., Bertaggia E., Blaauw B., Urciuolo A., Tiepolo T., Merlini L., Maraldi N. M., Bernardi P., Sandri M., Bonaldo P. (2010) Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 16, 1313–1320 [DOI] [PubMed] [Google Scholar]
- 40. Cheng Y. J., Lang D., Caruthers S. D., Efimov I. R., Chen J., Wickline S. A. (2012) Focal but reversible diastolic sheet dysfunction reflects regional calcium mishandling in dystrophic mdx mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 303, H559–H568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashford M. W., Jr., Liu W., Lin S. J., Abraszewski P., Caruthers S. D., Connolly A. M., Yu X., Wickline S. A. (2005) Occult cardiac contractile dysfunction in dystrophin-deficient children revealed by cardiac magnetic resonance strain imaging. Circulation 112, 2462–2467 [DOI] [PubMed] [Google Scholar]
- 42. Schram G., Fournier A., Leduc H., Dahdah N., Therien J., Vanasse M., Khairy P. (2013) All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 61, 948–954 [DOI] [PubMed] [Google Scholar]
- 43. Flynn J. M., O'Leary M. N., Zambataro C. A., Academia E. C., Presley M. P., Garrett B. J., Zykovich A., Mooney S. D., Strong R., Rosen C. J., Kapahi P., Nelson M. D., Kennedy B. K., Melov S. (2013) Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 12, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferron G. M., Conway W. D., Jusko W. J. (1997) Lipophilic benzamide and anilide derivatives as high-performance liquid chromatography internal standards: application to sirolimus (rapamycin) determination. J. Chromatogr. B. Biomed. Sci. Appl. 703, 243–251 [DOI] [PubMed] [Google Scholar]
- 45. Williams D. A., Head S. I., Bakker A. J., Stephenson D. G. (1990) Resting calcium concentrations in isolated skeletal muscle fibres of dystrophic mice. J. Physiol. 428, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeman R. J., Bernstein P. L., Ludemann R., Etlinger J. D. (1986) Regulation of Ca2+-dependent protein turnover in skeletal muscle by thyroxine. Biochem. J. 240, 269–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mouisel E., Vignaud A., Hourde C., Butler-Browne G., Ferry A. (2010) Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signaling in skeletal muscles of venerable (18–24-month-old) dystrophic mdx mice. Muscle Nerve 41, 809–818 [DOI] [PubMed] [Google Scholar]
- 48. Avila G., Lee E. H., Perez C. F., Allen P. D., Dirksen R. T. (2003) FKBP12 binding to RyR1 modulates excitation-contraction coupling in mouse skeletal myotubes. J. Biol. Chem. 278, 22600–22608 [DOI] [PubMed] [Google Scholar]
- 49. Ahern G. P., Junankar P. R., Dulhunty A. F. (1997) Ryanodine receptors from rabbit skeletal muscle are reversibly activated by rapamycin. Neurosci. Lett. 225, 81–84 [DOI] [PubMed] [Google Scholar]
- 50. Bellinger A. M., Reiken S., Carlson C., Mongillo M., Liu X., Rothman L., Matecki S., Lacampagne A., Marks A. R. (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Avila G., Dirksen R. T. (2005) Rapamycin and FK506 reduce skeletal muscle voltage sensor expression and function. Cell Calcium 38, 35–44 [DOI] [PubMed] [Google Scholar]
- 52. Zhang H., Zhang L., Myerson J., Bibee K., Scott M., Allen J., Sicard G., Lanza G., Wickline S. (2011) Quantifying the evolution of vascular barrier disruption in advanced atherosclerosis with semipermeant nanoparticle contrast agents. PLoS One 6, e26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vidal B., Ardite E., Suelves M., Ruiz-Bonilla V., Janue A., Flick M. J., Degen J. L., Serrano A. L., Munoz-Canoves P. (2012) Amelioration of Duchenne muscular dystrophy in mdx mice by elimination of matrix-associated fibrin-driven inflammation coupled to the αMβ2 leukocyte integrin receptor. Hum. Mol. Genet. 21, 1989–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palladino M., Gatto I., Neri V., Straino S., Smith R. C., Silver M., Gaetani E., Marcantoni M., Giarretta I., Stigliano E., Capogrossi M., Hlatky L., Landolfi R., Pola R. (2013) Angiogenic impairment of the vascular endothelium: a novel mechanism and potential therapeutic target in muscular dystrophy. Arterioscler. Thromb. Vasc. Biol. 33, 2867–2876 [DOI] [PubMed] [Google Scholar]
- 55. Lien C. F., Mohanta S. K., Frontczak-Baniewicz M., Swinny J. D., Zablocka B., Gorecki D. C. (2012) Absence of glial α-dystrobrevin causes abnormalities of the blood-brain barrier and progressive brain edema. J. Biol. Chem. 287, 41374–41385 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.