

Abstract
Background
Prophages, integral components of many bacterial genomes, play significant roles in cognate host bacteria, such as virulence, toxin biosynthesis and secretion, fitness cost, genomic variations, and evolution. Many prophages and prophage-like elements present in sequenced bacterial genomes, such as Bifidobacteria, Lactococcus and Streptococcus, have been described. However, information for the prophage of Mycobacterium remains poorly defined.
Results
In this study, based on the search of the complete genome database from GenBank, the Whole Genome Shotgun (WGS) databases, and some published literatures, thirty-three prophages were described in detail. Eleven of them were full-length prophages, and others were prophage-like elements. Eleven prophages were firstly revealed. They were phiMAV_1, phiMAV_2, phiMmcs_1, phiMmcs_2, phiMkms_1, phiMkms_2, phiBN42_1, phiBN44_1, phiMCAN_1, phiMycsm_1, and phiW7S_1. Their genomes and gene contents were firstly analyzed. Furthermore, comparative genomics analyses among mycobacterioprophages showed that full-length prophage phi172_2 belonged to mycobacteriophage Cluster A and the phiMmcs_1, phiMkms_1, phiBN44_1, and phiMCAN_1 shared high homology and could be classified into one group.
Conclusions
To our knowledge, this is the first systematic characterization of mycobacterioprophages, their genomic organization and phylogeny. This information will afford more understanding of the biology of Mycobacterium.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-243) contains supplementary material, which is available to authorized users.
Keywords: Prophage, Mycobacterioprophage, Phylogeny, Comparative genomics
Background
Phages can be divided into virulent or temperate based on their relationship with the host. Temperate phage inserts and integrates into its host genome upon infection, and can reside as quiescent prophage. Prophage does not infect its host and maintains the dormant state [1]. Whole-genome sequencing reveals that prophage DNAs are widespread among bacterial genomes, even up to 20% of the host genome content [2]. Prophages are important genetic components transferred horizontally that can impart bacterial genome variability, evolution, and virulence [1, 3]. Some prophage genes contribute to the adaptation of bacteria to their specific ecological niches [3]. This has been demonstrated in many bacteria [1, 4, 5], but a little is known for Mycobacterium prophages.
There is huge gap between the number of mycobacteriophages isolated and cognate prophages found within mycobacteria. To date, there are 3427 mycobacteriophages isolated and 448 of them with genome sequenced. They can be assembled into 20 clusters (A-T) and seven of them are singletons [6, 7]. In contrast with large number of sequenced mycobacteriophages, their cognate prophages are poorly defined. Only the following mycobacterioprophage sequences have been described. Two prophage-like elements, phiRv1 and phiRv2, have been detected in Mycobacterium tuberculosis H37Rv genome [8]; two prophage-like elements, PhiMU01 and PhiMU02, are found within M. ulcerans Agy99 genome [9]; 10 putative prophages, named phiMmar01–10, are found in M. marinum M and two of them, phiMmar02 and phiMmar08, are full-length prophages [10]; the M. abscessus ATCC 19977 chromosome contains a full-length prophage and three prophage-like elements [11]; prophage Araucaria is found in M. abscessus subsp. bolletii BD genome [6]; two prophages are found in pathogen M. abscessus Strain 47J26 [12]; a potential prophage in M. abscessus M93 is described [13]; M. massiliense Strain M172 contains putative mycobacteriophage [14]; a 55-kb region encodes a putative prophage in M. canettii STB-I [15]; a 40-kb prophage is predicted in addition to two prophage-like elements also are seen in M. simiae strain DSM 44165 [16]. Many Mycobacterium prophages remain to be characterized. Knowledge regarding their genomic composition, distribution can facilitate the elucidation of the biology of Mycobacterium.
In this study, we screened all available Mycobacterium complete genomes sequences from GenBank, shotgun assembly sequences from Whole Genome Shotgun (WGS) databases, and searched for mycobacterioprophages in published literatures. Together, 33 prophages were described in detail, and 11 of them were previously undocumented prophages among Mycobacterium genomes. The genomes, gene contents, comparative genomics studies and the relationships among them were characterized.
Results and discussion
Prophages in Mycobacteriumgenomes
Though the identification of prophages from sequenced bacterial genomes is difficult [1], prophage sequences can be found by several approaches. Integrases are well-recognized diagnostic markers for prophages within bacterial genomes [17–23]. Web servers and programs for prophages identification are available [24–28]. In this study, we used an integrated protocol to streamline the identification. Firstly, PHAST (PHAge Search Tool) was used to search Mycobacterium genomes. Secondly, the presence or absence of the integrase genes was tested to exclude negative results. Finally, mycobacterioprophage sequences were identified based on the homology between prophage ORFs (open reading frames) and known phage genes. Thirty mycobacterial complete genomes (see Additional file 1) were retrieved. Eleven new prophages were identified. The genomic features of these newly identified mycobacterioprophages are described in Table 1.
Table 1.
Genomic features of prophages in Mycobacterium genomes
| Prophages | Host | Coordinate | Insertion sites | Size | Putative att B regions of prophage-like elements | References |
|---|---|---|---|---|---|---|
| phiMAV_1 | M.avium 104 | 746,437-794,445 | - | 48.0 kb | GGACCTGCGGATTAAAAGTC | This study |
| phiMAV_2 | 1,446,840-1,463,298 | - | 16.5 kb | GTGGTCAGCTT | This study | |
| phiMmcs_1 | M.sp.MCS | 3,067,419-3,080,311 | tRNA-Pro | 12.9 kb | CCGTTGCCGT | This study |
| phiMmcs_2 | 4,054,082-4,063,334 | - | 9.3 kb | GGTGAGGGCGT | This study | |
| phiMkms_1 | M.sp.KMS | 3,085,307-3,098,199 | - | 12.9 kb | CCGTTGCCGT | This study |
| phiMkms_2 | 4,088,564-4,097,816 | - | 9.3 kb | GGTGAGGGCGT | This study | |
| phiBN42_1 | M.canettii CIPT 140070010 | 1,514,379-1,522,784 | tRNA-Arg | 8.4 kb | GTGCCCCCGGCAGGATTCG | This study |
| phiBN44_1 | M.canettii CIPT 140060008 | 3,433,693-3,444,793 | - | 11.1 kb | CCGGAGAAGAAGTCATGGTTCT | This study |
| phiMCAN_1 | M.canettii CIPT 140010059 | 1,180,578-1,191,783 | - | 11.2 kb | GTTCGAGTCCGACTGGGGGCAC | This study |
| phiMycsm_1 | M.smegmatis JS623 | 4,221,770-4,232,715 | - | 10.9 kb | GCCGACGACG | This study |
| phiW7S_1 | M.sp. MOTT36Y | 968,527-980,490 | tRNA-Ala | 12.0 kb | GTTCGCATCGAGTAGGTCAGGGGTTCGATTCCC | This study |
| phiRv1a | M.tuberculosis H37Rv | 1,779,267-1,788,525 | - | 9.3 kb | GGTTGGCCGTGG | [8] |
| phiRv2a | 2,970,063-2,980,853 | tRNA-Val | 10.8 kb | CCGCGCAATAAACGCGCAATA | [8] | |
| phiMU01a | M.ulcerans Agy99 | NM | NM | 18 kb | NM | [9] |
| phiMU02a | NM | NM | 24 kb | NM | [9] | |
| phiMmar01a | M.marinum M | 27,047- 33,095 | tRNA-Leu | 6.0 kb | NM | [10] |
| phiMmar02a | 4,812,334- 4,869,620 | tRNA- Lys | 57.8 kb | NM | [10] | |
| phiMmar03a | 5,460,072– 5,470,770 | tRNA- Leu | 10.7 kb | NM | [10] | |
| phiMmar04a | 5,628,721– 5,636,467 | tRNA- Arg | 7.7 kb | NM | [10] | |
| phiMmar05a | 5,884,651– 5,904,290 | tRNA-Phe | 19.6 kb | NM | [10] | |
| phiMmar06a | 2,567,503– 2,589,174 | - | 21.6 kb | NM | [10] | |
| phiMmar07a | 3,082,858– 3,100,046 | tRNA- Pro | 17.2 kb | NM | [10] | |
| phiMmar08a | 3,808,513-3,851,917 | tRNA-Leu | 43.0 kb | NM | [10] | |
| phiMmar09a | 688,611-695,966 | tRNA-Gly | 7.4 kb | NM | [10] | |
| phiMmar10a | 4,405,758- 4,430,810 | tRNA-Met | 25.1 kb | NM | [10] | |
| Prophage Araucariaa | M.abscessus subsp. bolletii BD Contig17 | NM (1,000-65,113) | - | 64.1 kb | NM | [6] |
| phiMAB_1# | M.abscessus ATCC 19977 | 1,754,551-1,835,095 | tRNA-Met | 80.5 kb | NM | [11] |
| phiMAB_2# | 233,621-247,981 | tRNA-Arg | 14.4 kb | NM | [11] | |
| phiMAB_3# | 770,916-778,753 | tRNA-Lys | 7.8 kb | NM | [11] | |
| phiMAB_4# | 4,909,957-4,959,626 | - | 49.7 kb | NM | [11] | |
| phiMAB47J26_1# | M.abscessus Strain 47 J26 Contig02 | NM (55,744-116,295) | NM (tRNA-Leu) | 60.5 kb | NM (GCGGACTTAAAATCCGCCAAGTGTCGGTTCGAGTCCGACTGGGGGCAC) | [12] |
| phiMAB47j26_2# | M.abscessus Strain 47 J26 Contig03 | NM (58,656-105,841) | - | 47.2 kb | NM | [12] |
| phiOUW_1# | M.abscessus M93 Contig09 | NM (267,663-342,276) | NM (tRNA-Arg) | 74.6 kb | NM (GTGCGCCCGAAGGGATTCGAACCCCTAACCTTCTG) | [13] |
| phiM172_1# | M.massiliense Strain M172 Contig06 | NM (358,576-422,610) | NM (tRNA-Val) | 64.0 kb | NM (TTGGTGGGCGCGGAGGGTTTCGAACCC) | [14] |
| phiM172_2# | NM (456,450-513,710) | - | 57.3 kb | NM (CAACCAGTCGGCCTGA) | [14] | |
| phiSTB-I_1# | M.canettii STB-1 (CIPT 140070007) | NM | tRNA-Lys | 55 kb | NM | [15] |
| phiDSM_1# | M.simiae DSM 44165 | NM | NM | 40 kb | NM | [16] |
| phiDSM_2# | NM | NM | 7 kb | NM | [16] | |
| phiDSM_3# | NM | NM | 18 kb | NM | [16] |
NM means that these data do not be mentioned; parentheses means that these data is shown in this study; - means these prophages are not integrating into tRNA genes; athose prophages has been described and named; #those prophages has been described, but did not be named.
In the WGS databases, some mycobacteria containing prophages are also reported [12–16]. Since the whole genome sequences of these mycobacteria and the specific information of these prophages are not available, we searched for prophages in five mycobacterial shotgun assembly sequences contigs (see Additional file 1) using the method mentioned above. The results showed that prophages were found in some sequences contigs of M. abscessus Strain 47J26, M.abscessus M93, and M.massiliense M172 (Table 1). Prophages previously reported in the genomes of M.canettii CIPT 140070007 and M.simiae DSM 44165 cannot be detected in our study. With annotated whole genomic sequence, this puzzle might be solved.
Some mycobacteria harboring prophages have been detailed in previous studies [6, 8, 10, 11], which are included in Table 1. Four of them contained in M.abscessus ATCC 19977 chromosome are not designated. We named them phiMAB_1, phiMAB_2, phiMAB_3, and phiMAB_4, respectively. We noted that two prophage, PhiMU01 and PhiMU02, mentioned in M.ulcerans Agy99 genome, lack specific information and cannot be detected.
Overall, thirty-three prophages were described, and six prophages had been mentioned, but without specific information. Eleven prophages were found from the complete genome database; five prophages were retrieved from the WGS databases; seventeen of them were reported prophages with specific sequence information. Their size range was from 6 kb to 80.5 kb. Based on the length of prophage genome (the length of mycobacteriophage genomes is 41,441 bp – 164,602 bp, http://phagesdb.org/), 11 prophages can be considered as full-length prophage. The remaining 22 prophages were prophage-like elements. The result showed that small prophage-like elements were more prevalent than putative full-length prophages. The small prophage-like elements might be more stable due to mutational decay and loss of some genes somehow involved in genome excision. Small prophage-like elements were more stable and can be more easily detected than the full-length prophages. Through the tRNA search tool, 19 prophages were integrated into tRNA genes (Table 1). The frequency of tRNA integration was tRNA-Leu (4/19), tRNA-Arg (4/19), tRNA-Val (2/19), tRNA-Lys (2/19), tRNA-Pro (2/19), tRNA-Met (2/19), tRNA-Phe (1/19), tRNA-Gly (1/19), tRNA-Ala (1/19). The genome of M.sp.KMS, M.sp.MCS, M.avium 104, M.tuberculosis H37Rv, M.marinum M, M.abscessus ATCC 19977, M.abscessus Strain 47J26, and M.massiliense Strain M172 was polylysogenic.
New prophages of Mycobacteriumgenomes
Full-length prophage phiMAV_1 in the genome of M. avium 104
Prophage phiMAV_1, spanning from MAV_0779 (integrase gene) to MAV_0841 (excisionase DNA binding protein), contains sixty-three ORFs (see Additional file 2), and is flanked by two 20-bp repeats (Table 1) reminiscent of attL and attR sites. There is no predicted tRNA within the prophage. PhiMAV_1 cannot be categorized into any known phage clusters and might represent new singleton type [29].
Based on Blast-p, 41 phiMAV_1 ORFs show more or less amino acid sequence similarity to other known phage genes, and 17 can be assigned functionalities based on homology (see Additional file 2). PhiMAV_1 genome consists of different functional modules (Figure 1).
Figure 1.
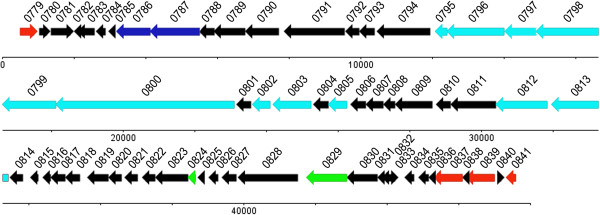
The genomic organization of M.avium 104 full-length prophage phiMAV_1. The red arrows represent lysogeny module; the blue arrows represent lysis module; the cyan arrows represent DNA packaging and structural modules; the green arrows represent DNA metabolism module. Numbers means the numbering of gene.
The lysis module consists of MAV_0786 and MAV_0787, which encode cutinase and glycosyl hydrolase respectively that can lyze bacterium and enable the release of progeny phages. The DNA packaging and structural modules extend from MAV_0795 to MAV_0813. MAV_0795, MAV_0797, and MAV_0803 all encode putative tail protein. MAV_0798 and MAV_0799 all encode putative structural protein. MAV_0800, MAV_0802, and MAV_0805 encode phage tail tape measure protein, tail assembly chaperone, and phage capsid and scaffold protein. MAV_0812 and MAV_0813 encode putative portal protein and phage terminase engaged in the phage head morphogenesis. The DNA metabolism module includes MAV_0824 and MAV_0829. MAV_0824 encodes exonuclease and MAV_0829 encodes recombination and repair protein RecT. The lysogeny module consists of MAV_0837, MAV_0839, MAV_0841 and MAV_0779. MAV_0779 and MAV_0841 encode phage integrase and excisionase DNA binding protein. Both MAV_0837 and MAV_0839 encode phage antirepressor protein.
In addition to ORFs similar to other phage genes, two ORFs show unexpected similarity to bacterial key proteins. MAV_0835 encodes type VI secretion protein IcmF (Intracellular Multiplication F), a core component of type VI secretion system in Pseudomonas aeruginosa, Vibrio cholerae or other pathogenic bacteria [30–32]. Based on Blast-p, type VI secretion system was not documented in mycobacteria except for M.avium 104 and M.parascrofulaceum. IcmF is involved in bacterial motility, adherence to epithelial cells, and conjugation frequency [31], and has been reported in an avian pathogenic Escherichia coli (APEC) strain [32]. In addition, MAV_0790 encodes PPE family protein, a widespread Mycobacterium unique protein. This implies that MAV_0835 and MAV_0790 play a role in the physiology and pathogenicity of M.avium 104.
Prophage-like elements phiMAV_2
Prophage phiMAV_2 (Figure 2), integrated into a hypothetical gene (MAV_1505) in M.avium 104, extends from MAV_1484 (integrase gene) to MAV_1504 (Phage terminase) and contains 21 ORFs (see Additional file 3) flanked by an 11-bp repeat (Table 1), indicative of attL and attR sites. No tRNA is found in the genome of phiMAV_2. Based on Blast-p, only nine ORFs have sequence similarity to other phage genes at the amino acid sequence level. Six ORFs of the phiMAV_2 prophage genome can be assigned function based on database search, namely the integrase gene (MAV_1484), response regulator receiver protein (MAV_1485), DNA primase/polymerase (MAV_1486), Y4cG protein (MAV_1493), transposase (MAV_1498) and phage terminase (MAV_1504). Other phiMAV_2 prophage ORFs similar to known bacterial functional proteins are also identified (see Additional file 3).
Figure 2.

Genomic organization of some defective prophage-like elements among mycobacteria. Numbers means the numbering of gene. The red arrows represent lysogeny module; the blue arrows represent lysis module; the cyan arrows represent DNA packaging and structural modules; the green arrows represent DNA metabolism module.
Prophage-like elements phiMmcs_1, phiMmcs_2, phiMkms_1, and phiMkms_2
There are two prophage-like elements in M.sp.MCS, phiMmcs_1 and phiMmcs_2. Prophage phiMmcs_1 (Figure 2), which is integrated into a tRNA-pro (Mmcs_R0021) in M.sp.MCS, extends from Mmcs_2923 (integrase gene) to Mmcs_2908 (transglycosylase-like protein) and contains sixteen ORFs (see Additional file 4) flanked by a 10-bp repeat (Table 1), indicative of attL and attR sites. No tRNA is found in the genome of phiMmcs_1. Only nine ORFs can be assigned function based on amino acid sequence homology. The prophage phiMmcs_1 genome contains 4 modules. The lysis module appeared to be limited to Mmcs_2908, whose protein product has 50% sequence identity to lysin of Rhodococcus phage REQ1. The structural module consists of Mmcs_2910 and Mmcs_2914. Mmcs_2910, Mmcs_2911, Mmcs_2913, and Mmcs_2914 encode phage major capsid protein, scaffolding protein, phage portal protein, and phage terminase, respectively. The DNA metabolism module has two genes (Mmcs_2915 and Mmcs_2918), whose predicted protein products are HNH endonuclease and DNA repair protein RadA, respectively. The lysogeny module consists of Mmcs_2921 (putative phage excisionase) and Mmcs_2923 (phage integrase).
The phiMmcs_2 prophage remnant inserts between Mmcs_3803 and Mmcs_3817. The prophage sequence contains 15 ORFs (see Additional file 5) and is flanked by two 11-bp repeats, indicating the existence of putative attL and attR sites. Based on Blast-p, only 8 ORFs have sequence similarity to other phage genes at the amino acid sequence level and 4 can be assigned function, namely Mmcs_3802 (HNH endonuclease), Mmcs_3805 (phage major capsid protein), Mmcs_3814 (HNH endonuclease domain-containing protein), and Mmcs_3816 (phiRv1 integrase).
PhiMkms_1 and phiMkms_2 (see Additional files 6 and 7) are prophage-like elements in M.sp.KMS. PhiMmcs_1 is identical to phiMkms_1 and represents same prophage. They also insert into the same location in host genome. PhiMmcs_2 and phiMkms_2 is just the same scenario as phiMkms_1 and phiMkms_2.
Prophage-like elements phiBN42_1, phiBN44_1, and phiMCAN_1
PhiBN42_1, phiBN44_1, and phiMCAN_1 are found in M. canettii CIPT 140070010, M.canettii CIPT 140060008, and M.canettii CIPT 140010059 respectively. Prophage phiBN42_1 (Figure 2), which is integrated into a tRNA-arg (BN42_tRNA41) in M.canettii CIPT 140070010, extends from BN42_21176 (integrase gene) to BN42_21185 (hypothetical protein) and contains only eight ORFs (see Additional file 8) flanked by a 19-bp repeat (Table 1), indicative of attL and attR sites. No tRNA is found in the genome of phiBN42_1. Only seven genes have sequence similarity to other phage genes, five of which can be assigned function. There are BN42_21176 (integrase), BN42_21178 (excisionase), BN42_21179 (DNA primase), BN42_21182 (phage prohead protease), and BN42_21183 (phage major capsid protein).
The phiBN44_1 prophage remnant is located between BN44_60546 and BN44_60559 in M.canettii CIPT 140060008, flanked by a 22-bp repeat (Table 1), representing candidates for the attL and attR sites. There are 11 ORFs in phiBN44_1 prophage genome (see Additional file 9). Eight are similar to other phage genes and can be assign function. There are BN44_60547 (phage major capsid protein), BN44_60548 (scaffolding protein), BN44_60550 (Phage portal protein), BN44_60551 (Phage Terminase), BN44_60552 (HNH endonuclease), BN44_60554 (DNA primase), BN44_60557 (XRE family transcriptional regulator), and BN44_60558 (phage integrase). Additionally, BN44_60555 encodes protein similar to Human adenovirus DNA polymerase and BN44_60556 encodes protein similar to K+ transporter of many bacteria.
Prophage phiMCAN_1 (Figure 2), which is integrated into between MCAN_10501 and MCAN_10621 in M.canettii CIPT 140010059, contains only 11 ORFs flanked (see Additional file 10) by a 22-bp repeat (Table 1), indicative of attL and attR sites. No tRNA is found in the genome of phiMCAN_1. Only 8 ORFs similar to other phage genes at the amino acid sequence level and seven genes have been assigned function. There are MCAN_10511 (phage integrase), MCAN_10521 (DNA-binding protein), MCAN_10541 (DNA primase), MCAN_10551 (HNH endonuclease), MCAN_10561 (phage terminase), MCAN_10571 (phage portal protein), and MCAN_10601 (phage major capsid protein).
Prophage-like elements phiMycsm_1 and phiW7S_1
Prophage phiMycsm_1 (Figure 2), inserted between Mycsm_04290 and Mycsm_04304 in M.smegmatis JS623, contains 13 ORFs (see Additional file 11) flanked by a 10-bp repeat (Table 1), indicative of attL and attR sites. No tRNA is found in the genome of phiMycsm_1. Nine ORFs show the protein sequence similarity to other phage genes, in which six ORFs have the descriptive function: Mycsm_04291 (phage integrase), Mycsm_04296 (DNA-binding protein), Mycsm_04298 (DNA primase), Mycsm_04299 (HNH endonuclease), Mycsm_04302 (phage terminase), and Mycsm_04303 (phage portal protein). Additionally, Mycsm_04293, whose protein product is similar to glycerate kinase, is also present in phiBN44_1.
Prophage phiW7S_1 (Figure 2) integrated into a tRNA-ala (W7S_t25871) in M.sp. MOTT36Y, extends from W7S_04825 (integrase gene) to W7S_04880 (hypothetical protein) and contains 12 ORFs (see Additional file 12) flanked by a 33-bp repeat (Table 1), indicative of attL and attR sites. No tRNA is found in the genome of phiW7S_1. Only six genes have sequence similarity to other phage genes and three of them have annotated function, which are W7S_04825 (integrase), W7S_04845 (pantothenate kinase), and W7S_04855 (transposase).
Grouping of full-length prophages
We searched all the literatures published so far about full-length mycobacterioprophages. Only one prophage Araucaria is assigned to a Dori-like prophage [6]. BlastN (http://phagesdb.org/blast/) and dot plot matrix of the genomes of full-length mycobacterioprophages and mycobacteriophage clusters (A-T and singletons) revealed that phi172_2 shared sequence similarity to cluster A (see Additional file 13); phiMAB_1 shared an even weaker sequence similarity to subcluster F1 (see Additional file 14); phiMAB47J26_1 shared an even weak sequence similarity to subcluster F1 and cluster N (see Additional file 15); phiMAB47J26_2 shared an even weak sequence similarity to cluster P, subcluster F1, and cluster N (see Additional file 16); phi172_1 shared an even weaker sequence similarity to subcluster F1 and cluster N (see Additional file 17). The remaining full-length prophages had no close relatives to any cluster. We proposed that phi172_2 was grouped into cluster A, and other full-length mycobacterioprophages did not belong to any mycobacteriophage clusters and were ‘singletons’.
Comparative genomics of prophage-like elements
Dot plot matrix was generated for the complete genomes of 22 mycobacterioprophage-like elements in this study (Figure 3). The figure displays that phiMmcs_1, phiMkms_1, phiBN44_1, and phiMCAN_1 are more closely related to each other than to other mycobacterioprophage-like elements, and can be classified as one group. In a simple NCBI ‘Align two sequences’comparison, the comparison between phiMmcs_1 (or phiMkms_1) and phiBN44_1 shows that one of the major segments less than 2801 bp has greater than 71% identity, and four segments less than 200 bp are reported to have 68% identity (Figure 4). The comparison between reverse complementary sequence of phiMCAN_1 and phiBN44_1 shows that one of the major segments 8952 bp has greater than 85% identity (Figure 4). Further analysis indicated a lack of homology between the prophage of M.tuberculosis H37Rv and other prophage-like elements.
Figure 3.
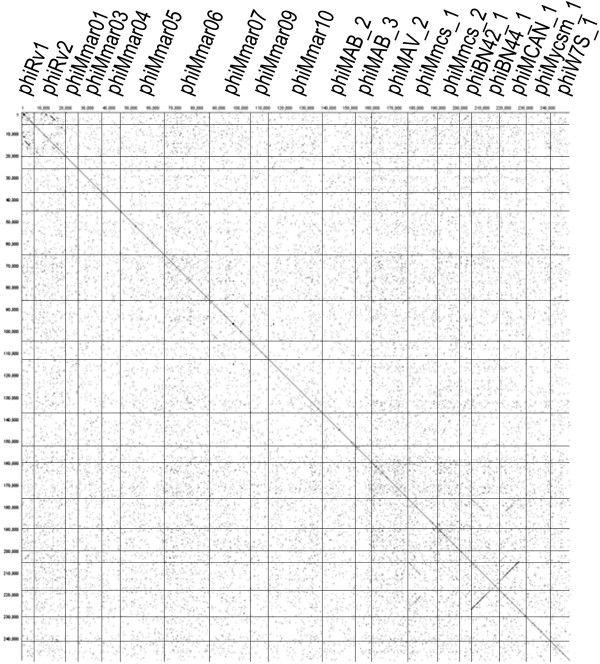
Comparative genomic analyses of prophage-like sequences. Dot plot matrix calculated for the complete genomes of all prophage-like sequences in Mycobacterium. The top x axis and the left y axis provide a scale in kilobases; and the top x axis identifies the prophage genomes that are compared in the corresponding square. The x and y axes are the identical sequences. The slash means that two DNA fragments are homologous to each other. The backslash means that one DNA fragment is homologous with the reverse sequence of other DNA fragment. The word length used is 12 bp.
Figure 4.

Global comparison of phiMmcs_1 (or phiMkms_1), phiBN44_1, and phiMCAN_1. Highly related sequences are shown by the red shadings. The blue shadings means that the DNA fragments are highly homologous to complementary sequence of other fragments.
Phylogeny of prophage integrases
Integrase can be found in virtually each prophage genome found in this study. And it can serve as good marker for the phylogeny of prophage phiRv1 element encodes a serine site-specific recombinase and phiRv2 encodes a tyrosine recombinase [33]. All integrases fall into the two categories (Figure 5). The serine recombinase division includes phiMycsm_1, phiMmcs_2 (phiMkms_2) and phiRv1. The tyrosine recombinase division includes the remaining prophages and phiRv2. PhiMmcs_1 (phiMkms_1), phiBN44_1, and phiMCAN_1 belong to the same clade, consistent with the comparative genomic result. The distance between prophages had little relevance to the phylogeny between their hosts, suggestive of independent evolutionary trajectory.
Figure 5.
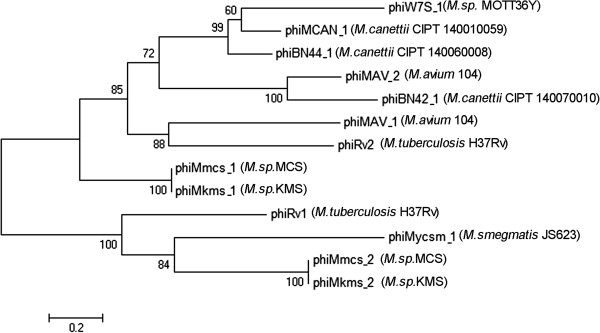
Phylogeny of prophage integrases. Unrooted phylogenetic relationships are represented using NJTree. Bootstrap values from 1,000 reiterations are shown.
Conclusions
In brief, we present here thirty-three mycobacterioprophages mined from sequenced mycobacterial genomes, the WGS databases, and some published literatures. Eleven prophages were newly identified prophages from complete genome database; five prophages were from the WGS databases; seventeen prophages were reported with specific sequence information. The genome sequences, gene contents of eleven newly identified prophages were analyzed. Comparative genomic analysis revealed that one full-length mycobacterioprophage phi172_2 belonged to cluster A and one group having recognizable sequence similarity was verified and contained four small prophage-like elements, including the phiMmcs_1, phiMkms_1, phiBN44_1, and phiMCAN_1. To our knowledge, this represents the first systematic analysis of mycobacterioprophages. With more forthcoming Mycobacterium genome sequences and thorough mycobacterioprophages screening, we can generate a more comprehensive picture of the role of prophages in mycobacterial evolution, adaptations and physiology.
Methods
Data collection and mycobacterioprophage identification
DNA sequences of bacteria for analysis were downloaded from multiple databases, such as NCBI (the National Center for Biotechnology Information). PHAST (http://phast.wishartlab.com/index.html) were firstly used for analyzing bacterial genome to find candidate prophages [24]. An integrase gene was screened from candidate prophage genome for in these results to drop false negative results [17–20]. Finally, prophages were identified on the basis of the presence of significant homology between ORFs (open reading frames) and known phage genes [17].
Analysis of mycobacterioprophage genome sequence
Prophage sequence was annotated using a variety of programs including Glimmer [34]. tRNA and tmRNA genes were identified using tRNA-Scan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/) [35] and ARAGORN (http://mbio-serv2.mbioekol.lu.se/ARAGORN/) [36]. BLAST analyses were performed remotely at the NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and the phagesdb.org site (http://phagesdb.org/blast/). Some data about mycobacteriophage genomes was downloaded from the phagesdb.org site (http://phagesdb.org/). DNAman was used to searching the flank of prophage to find attL and attR sites. Sequences were submitted entries to the GenBank sequence database by Sequin (http://www.ncbi.nlm.nih.gov/projects/Sequin/index.html). Comparative genomic analyses of prophage could be carried out by Blast-N for the global comparison of phiMmcs_1 (or phiMkms_1), phiBN44_1, and phiMCAN_1 and Geneious software for the dotplot of all the mycobacterioprophage-like sequences [37]. Multiple sequence alignment and the construct of phylogenetic trees were performed using ClustalW (http://embnet.vital-it.ch/software/ClustalW.html) or MEGA4 [38].
Electronic supplementary material
Additional file 1: Table S1: Mycobacterial genomes retrieved in this study. (DOC 56 KB)
Additional file 2: Table S2: Database matches for phiMAV_1. (DOC 85 KB)
Additional file 3: Table S3: Database matches for phiMAV_2. (DOC 44 KB)
Additional file 4: Table S4: Database matches for phiMmcs_1. (DOC 40 KB)
Additional file 5: Table S5: Database matches for phiMmcs_2. (DOC 38 KB)
Additional file 6: Table S6: Database matches for phiMkms_1. (DOC 40 KB)
Additional file 7: Table S7: Database matches for phiMkms_2. (DOC 39 KB)
Additional file 8: Table S8: Database matches for phiBN42_1. (DOC 30 KB)
Additional file 9: Table S9: Database matches for phiBN44_1. (DOC 34 KB)
Additional file 10: Table S10: Database matches for phiMCAN_1. (DOC 33 KB)
Additional file 11: Table S11: Ddatabase matches for phiMycsm_1. (DOC 34 KB)
Additional file 12: Table S12: Database matches for phiW7S_1. (DOC 34 KB)
Additional file 13: Figure S1-S11: Comparative genomic analyses of phi172_2 and cluster A (subcluster A1-A11) mycobacteriophage. (DOC 5 MB)
Additional file 14: Figure S12: Comparative genomic analyses of phiMAB_1 and subcluster F1 mycobacteriophage. (DOC 549 KB)
Additional file 15: Figure S13-S14: Comparative genomic analyses of phiMAB47J26_1, subcluster F1 and cluster N mycobacteriophage. (DOC 1 MB)
Additional file 16: Figure S15-S17: Comparative genomic analyses of phiMAB47J26_2, cluster P, subcluster F1 and cluster N mycobacteriophage. (DOC 2 MB)
Additional file 17: Figure S18-S19: Comparative genomic analyses of phi172_1, subcluster F1 and cluster N mycobacteriophage. (DOC 1 MB)
Acknowledgements
This work was supported by National Natural Science Foundation [grant numbers 81371851, 81071316, 81271882 and 81301394], New Century Excellent Talents in Universities [grant number NCET-11-0703], National Megaprojects for Key Infectious Diseases [grant numbers 2008ZX10003-006], Excellent PhD thesis fellowship of southwest university [grant numbers kb2010017, ky2011003], the Fundamental Research Funds for the Central Universities [grant numbers XDJK2011D006, XDJK2012D011, XDJK2012D007, XDJK2013D003 and XDJK2014D040], The Chongqing municipal committee of Education for postgraduates excellence program [grant numbers YJG123104], The undergraduates teaching reform program [grant numbers 2011JY052].
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XF participated in the design of the study, analyzed data and wrote the paper. LX and WL helped to modify the manuscript. JX designed the research and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Xiangyu Fan, Email: fxysnd@126.com.
Longxiang Xie, Email: xielongxiang123@126.com.
Wu Li, Email: liwu0503@swu.edu.cn.
Jianping Xie, Email: georgex@swu.edu.cn.
References
- 1.Varani AM, Monteiro-Vitorello CB, Nakaya HI, Van Sluys MA. The role of prophage in plant-pathogenic bacteria. Annu Rev Phytopathol. 2013;51:429–451. doi: 10.1146/annurev-phyto-081211-173010. [DOI] [PubMed] [Google Scholar]
- 2.Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49(2):277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- 3.Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67(2):238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou QH, Li QH, Zhu HY, Feng Y, Li YG, Johnston RN, Liu GR, Liu SL. SPC-P1: a pathogenicity-associated prophage of Salmonella paratyphi C. BMC Genomics. 2010;11:729. doi: 10.1186/1471-2164-11-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fortier LC, Sekulovic O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence. 2013;4(5):354–365. doi: 10.4161/viru.24498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sassi M, Bebeacua C, Drancourt M, Cambillau C. The first structure of a mycobacteriophage, the Mycobacterium abscessus subsp. bolletii phage Araucaria. J Virol. 2013;87(14):8099–8109. doi: 10.1128/JVI.01209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatfull GF. Complete genome sequences of 138 mycobacteriophages. J Virol. 2012;86(4):2382–2384. doi: 10.1128/JVI.06870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 9.Stinear TP, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T, Meurice G, Simon D, Bouchier C, Ma L, Tichit M, Porter JL, Ryan J, Johnson PD, Davies JK, Jenkin GA, Small PL, Jones LM, Tekaia F, Laval F, Daffe M, Parkhill J, Cole ST. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. 2007;17(2):192–200. doi: 10.1101/gr.5942807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, Clarke K, Cronin A, Davis P, Goodhead I, Holroyd N, Jagels K, Lord A, Moule S, Mungall K, Norbertczak H, Quail MA, Rabbinowitsch E, Walker D, White B, Whitehead S, Small PL, Brosch R, Ramakrishnan L, Fischbach MA, Parkhill J, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008;18(5):729–741. doi: 10.1101/gr.075069.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Macheras E, Heym B, Herrmann JL, Daffe M, Brosch R, Risler JL, Gaillard JL. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One. 2009;4(6):e5660. doi: 10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan J, Halachev M, Yates E, Smith G, Pallen M. Whole-genome sequence of the emerging pathogen Mycobacterium abscessus strain 47J26. J Bacteriol. 2012;194(2):549. doi: 10.1128/JB.06440-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell. 2013;49(2):237–248. doi: 10.1016/j.molcel.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choo SW, Yusoff AM, Wong YL, Wee WY, Ong CS, Ng KP, Ngeow YF. Genome analysis of Mycobacterium massiliense strain M172, which contains a putative mycobacteriophage. J Bacteriol. 2012;194(18):5128. doi: 10.1128/JB.01096-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, Majlessi L, Criscuolo A, Tap J, Pawlik A, Fiette L, Orgeur M, Fabre M, Parmentier C, Frigui W, Simeone R, Boritsch EC, Debrie AS, Willery E, Walker D, Quail MA, Ma L, Bouchier C, Salvignol G, Sayes F, Cascioferro A, Seemann T, Barbe V, Locht C, Gutierrez MC, et al. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. 2013;45(2):172–179. doi: 10.1038/ng.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sassi M, Robert C, Raoult D, Drancourt M. Non-contiguous genome sequence of Mycobacterium simiae strain DSM 44165(T.) Stand Genomic Sci. 2013;8(2):306–317. doi: 10.4056/sigs.3707349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ventura M, Zomer A, Canchaya C, O'Connell-Motherway M, Kuipers O, Turroni F, Ribbera A, Foroni E, Buist G, Wegmann U, Shearman C, Gasson MJ, Fitzgerald GF, Kok J, van Sinderen D. Comparative analyses of prophage-like elements present in two Lactococcus lactis strains. Appl Environ Microbiol. 2007;73(23):7771–7780. doi: 10.1128/AEM.01273-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ventura M, Turroni F, Lima-Mendez G, Foroni E, Zomer A, Duranti S, Giubellini V, Bottacini F, Horvath P, Barrangou R, Sela DA, Mills DA, van Sinderen D. Comparative analyses of prophage-like elements present in bifidobacterial genomes. Appl Environ Microbiol. 2009;75(21):6929–6936. doi: 10.1128/AEM.01112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ventura M, Canchaya C, Pridmore D, Berger B, Brüssow H. Integration and distribution of Lactobacillus johnsonii prophages. J Bacteriol. 2003;185(15):4603–4608. doi: 10.1128/JB.185.15.4603-4608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventura M, Canchaya C, Kleerebezem M, de Vos WM, Siezen RJ, Brüssow H. The prophage sequences of Lactobacillus plantarum strain WCFS1. Virology. 2003;316(2):245–255. doi: 10.1016/j.virol.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 21.Ventura M, Turroni F, Foroni E, Duranti S, Giubellini V, Bottacini F, van Sinderen D. Analyses of bifidobacterial prophage-like sequences. Antonie Van Leeuwenhoek. 2010;98(1):39–50. doi: 10.1007/s10482-010-9426-4. [DOI] [PubMed] [Google Scholar]
- 22.Ventura M, Lee JH, Canchaya C, Zink R, Leahy S, Moreno-Munoz JA, O'Connell-Motherway M, Higgins D, Fitzgerald GF, O'Sullivan DJ, van Sinderen D. Prophage-like elements in bifidobacteria: insights from genomics, transcription, integration, distribution, and phylogenetic analysis. Appl Environ Microbiol. 2005;71(12):8692–8705. doi: 10.1128/AEM.71.12.8692-8705.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Wang K, Ackermann HW, Halden RU, Jiao N, Chen F. Searching for a “hidden” prophage in a marine bacterium. Appl Environ Microbiol. 2010;76(2):589–595. doi: 10.1128/AEM.01450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fouts DE. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006;34(20):5839–5851. doi: 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics. 2008;24(6):863–865. doi: 10.1093/bioinformatics/btn043. [DOI] [PubMed] [Google Scholar]
- 27.Bose M, Barber RD. Prophage Finder: a prophage loci prediction tool for prokaryotic genome sequences. In Silico Biol. 2006;6(3):223–227. [PubMed] [Google Scholar]
- 28.Akhter S, Aziz RK, Edwards RA. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012;40(16):e126. doi: 10.1093/nar/gks406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J Mol Biol. 2010;397(1):119–143. doi: 10.1016/j.jmb.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol. 2012;66:453–472. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das S, Chakrabortty A, Banerjee R, Chaudhuri K. Involvement of in vivo induced icmF gene of Vibrio cholerae in motility, adherence to epithelial cells, and conjugation frequency. Biochem Biophys Res Commun. 2002;295(4):922–928. doi: 10.1016/S0006-291X(02)00782-9. [DOI] [PubMed] [Google Scholar]
- 32.de Pace F, Boldrin de Paiva J, Nakazato G, Lancellotti M, Sircili MP, Guedes Stehling E, Dias da Silveira W, Sperandio V. Characterization of IcmF of the type VI secretion system in an avian pathogenic Escherichia coli (APEC) strain. Microbiology. 2011;157(Pt 10):2954–2962. doi: 10.1099/mic.0.050005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bibb LA, Hatfull GF. Integration and excision of the Mycobacterium tuberculosis prophage-like element, phiRv1. Mol Microbiol. 2002;45(6):1515–1526. doi: 10.1046/j.1365-2958.2002.03130.x. [DOI] [PubMed] [Google Scholar]
- 34.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33(Web Server issue):W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32(1):11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Mycobacterial genomes retrieved in this study. (DOC 56 KB)
Additional file 2: Table S2: Database matches for phiMAV_1. (DOC 85 KB)
Additional file 3: Table S3: Database matches for phiMAV_2. (DOC 44 KB)
Additional file 4: Table S4: Database matches for phiMmcs_1. (DOC 40 KB)
Additional file 5: Table S5: Database matches for phiMmcs_2. (DOC 38 KB)
Additional file 6: Table S6: Database matches for phiMkms_1. (DOC 40 KB)
Additional file 7: Table S7: Database matches for phiMkms_2. (DOC 39 KB)
Additional file 8: Table S8: Database matches for phiBN42_1. (DOC 30 KB)
Additional file 9: Table S9: Database matches for phiBN44_1. (DOC 34 KB)
Additional file 10: Table S10: Database matches for phiMCAN_1. (DOC 33 KB)
Additional file 11: Table S11: Ddatabase matches for phiMycsm_1. (DOC 34 KB)
Additional file 12: Table S12: Database matches for phiW7S_1. (DOC 34 KB)
Additional file 13: Figure S1-S11: Comparative genomic analyses of phi172_2 and cluster A (subcluster A1-A11) mycobacteriophage. (DOC 5 MB)
Additional file 14: Figure S12: Comparative genomic analyses of phiMAB_1 and subcluster F1 mycobacteriophage. (DOC 549 KB)
Additional file 15: Figure S13-S14: Comparative genomic analyses of phiMAB47J26_1, subcluster F1 and cluster N mycobacteriophage. (DOC 1 MB)
Additional file 16: Figure S15-S17: Comparative genomic analyses of phiMAB47J26_2, cluster P, subcluster F1 and cluster N mycobacteriophage. (DOC 2 MB)
Additional file 17: Figure S18-S19: Comparative genomic analyses of phi172_1, subcluster F1 and cluster N mycobacteriophage. (DOC 1 MB)