

Summary
Background
Striate palmoplantar keratoderma (SPPK; OMIM #148700) is a rare autosomal dominant genodermatosis characterized by linear hyperkeratosis on the digits and hyperkeratosis on the palms and soles. SPPK is known to be caused by heterozygous mutations in either the desmoglein 1 (DSG1), desmoplakin (DSP), or keratin 1 (KRT1) genes.
Objective
To define the molecular basis of SPPK in five Pakistani families showing a clear autosomal dominant inheritance pattern of SPPK.
Methods
Based on previous reports of DSG1 mutations in SPPK, we performed direct sequencing of the DSG1 gene of all five families.
Results
Mutation analysis resulted in the identification of one recurrent mutation (p.R26X) and four novel mutations (c.Ivs4-2A>G, c.515C>T, c.Ivs9-3C>G, and c.1399delA) in the DSG1 gene. Each mutation is predicted to cause haploinsufficiency of DSG1 protein.
Conclusion
The results of our study further underscore the significance of the desmoglein gene family in diseases of epidermal integrity.
Keywords: Desmoglein 1, Desmosome, Striate palmoplantar keratoderma, Splice site mutation
1. Introduction
Palmoplantar keratoderma (PPK) is a heterogeneous group of skin diseases, which is characterized by thickening of epidermis on the palms of the hands and soles of the feet. PPK is further sub-classified based on the pattern of hyperkeratosis of the lesions. Of these, striate palmoplantar keratoderma (SPPK; OMIM #148700) is a rare autosomal dominant PPK, which typically shows linear hyperkeratosis on the digits, as well as focal hyperkeratosis on the palms and soles. The symptoms usually appear early in life. Areas of mechanical pressure on the soles are often severely affected. In addition, some environmental factors, such as manual labor, may exacerbate the phenotype on the hands [1].
The skin lesions observed in SPPK are thought to result from impaired function of desmosomes at sites exposed to high degree of mechanical stress, such as palms and soles, where these cell-cell junctions are critical for epidermal integrity [2]. Desmosomes are a type of anchoring junction found in many tissues including epidermis [3 4]. Major structural components of desmosome are encoded by the desmosomal cadherin gene family, which consists of the desmogleins (DSG1-4) and desmocollins (DSC1-3). Both DSGs and DSCs are transmembrane glycoproteins involved in Ca2+-mediated cell-cell adhesion, and connect with one other via their extracellular domains. The desmosome forms a dense plaque in the cytoplasm parallel to the cell membrane [3 5]. The desmosomal plaque is composed of three members, plakophilin (PKP), plakoglobin (JUP) and desmoplakin (DSP). These proteins function as linker molecules and anchor the keratin intermediate filaments at the cell membrane [6]. As such, the desmosomes connect cells to one another, and maintain tissue integrity.
SPPK has been shown to be caused by mutations in one of three genes: desmoglein 1 (DSG1) [7], desmoplakin (DSP) [8 9], or keratin 1 (KRT1) [10] gene. Importantly, all three genes encode a component of desmosomes. SPPK caused by mutations in either the DSG1 or KRT1 gene does not show any additional phenotypic manifestations. By contrast, it is noteworthy that SPPK caused by DSP mutations can be a symptom of Naxos disease which is a syndrome characterized by PPK, woolly hair and cardiomyopathy [11 12]. To date, the majority of mutations in non-syndromic SPPK have been identified in the DSG1 gene by our group and several others [7 2 13 14 15].
In this study, we have identified five distinct mutations in the DSG1 gene in five Pakistani families with SPPK.
2. Materials and Methods
2.1. Subjects
We analyzed five families of Pakistani origin (Families 1-5) affected with SPPK showing a clear autosomal dominant inheritance pattern (Fig. 1). All affected individuals have hyperkeratosis on the palm of the hands, with particular prominence on the creases of the palm, and linear hyperkeratosis along the flexor aspect of the fingers (Fig. 2). Focal hyperkeratosis was seen on the soles of the toes as well as the balls and heels of the feet (Fig. 2). The phenotype is more pronounced in areas that undergo frequent mechanical stress. There was no family history of either cardiomyopathy, early sudden death or cancers. In addition, none of the patients show signs of woolly hair. Skin biopsies were not available from any affected individuals.
Figure 1. Pedigrees of five Pakistani families with SPPK.
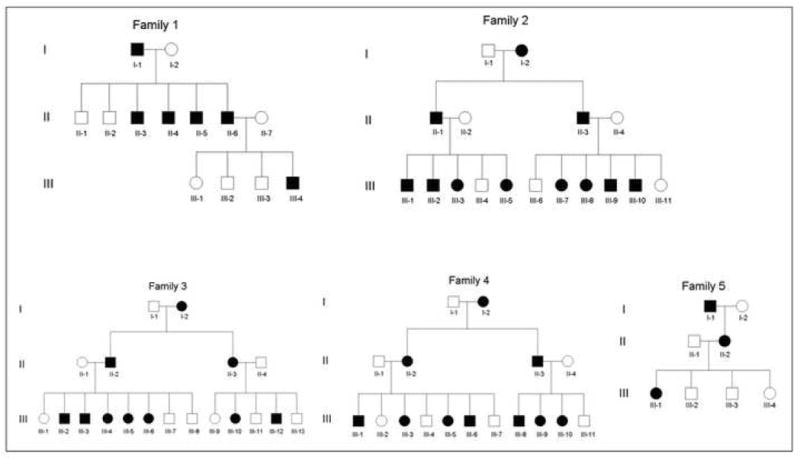
Note that all families show a clear autosomal dominant inheritance pattern.
Figure 2. Clinical appearance of SPPK.
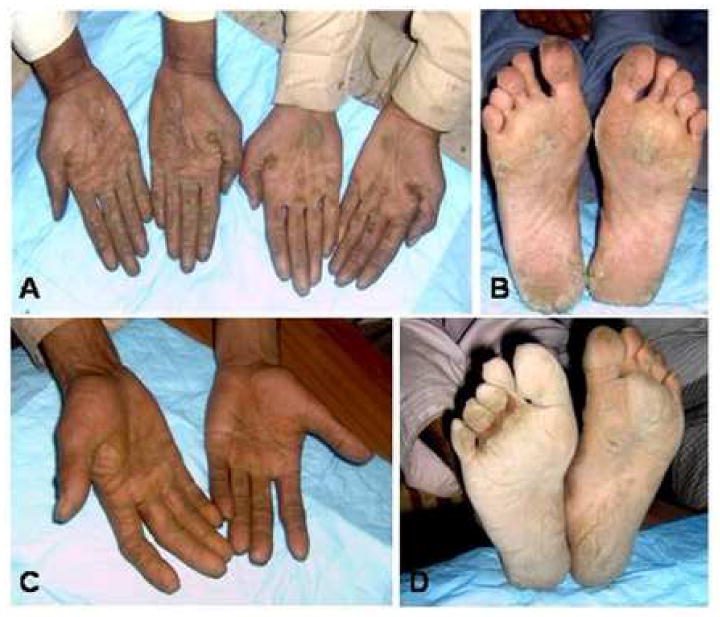
(A, B) Affected individuals in Family 1. (C, D) An affected individual in Family 2. Hyperkeratosis on the palm of the hands and linear hyperkeratosis on the digits (A,C) as well as focal hyperkeratosis on the soles (B,D) are observed in all affected individuals.
After obtaining informed consent, we collected peripheral blood samples from members of these families and 100 population-matched unrelated healthy control individuals in EDTA-containing tubes (under institutional approval and in adherence to the Declaration of Helsinki Principles). Genomic DNA was isolated from these samples according to standard techniques.
2.2. Mutation analysis of the DSG1 gene
Using the genomic DNA from members of Families 1-5, all exons with adjacent sequences of exon-intron borders of the DSG1 gene were amplified by PCR as previously described [16 2]. The amplified PCR products were sequenced directly in an ABI Prism 310 Automated Sequencer, using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems). The deleterious consequences of splice site mutations were predicted using a tool for automated splice site analyses provided by the University of Western Ontario (https://splice.uwo.ca).
2.3. Screening assays for the DSG1 mutation
The mutation c.Ivs4-2A>G generates an HpaII restriction enzyme site. Exon 5 and the adjacent intron sequences of the DSG1 gene were amplified by PCR. The PCR products were digested with the HpaII at 37°C overnight, and analyzed on 1.5% agarose gels. In order to analyze the mutation c.515C>T, the exon 5 – intron 5 border of the DSG1 gene was amplified by PCR using a mismatch forward primer 5’-CTACATTTGCAGGACAAATAGAAGAAAATTCGAATG-3’. Note that the T>G substitution was introduced into the primer to generate a BsmI restriction enzyme site only in the PCR product amplified from the wild type allele (shown in bold and underlined). The PCR products were digested by BsmI at 65°C overnight, and analyzed on 1.5% agarose gels. The mutation c.Ivs9-3C>G leads to generate a MnlI restriction enzyme site. Intron 9 – exon 10 border of the DSG1 gene was PCR-amplified, and subsequently digested by MnlI at 37°C overnight, and run on 1.5% agarose gels. The mutation c.1399delA generates a DdeI restriction enzyme site. Exon 10 and the adjacent intron sequences of the DSG1 gene were amplified by PCR. The PCR products were digested with the DdeI at 37°C overnight, and analyzed on 1.5% agarose gels.
3. Results
Identification of five distinct mutations in the DSG1 gene
We performed direct sequencing of the DSG1 gene in the five families and identified distinct mutations in the DSG1 in each family. First, affected individuals in Family 1 carry a heterozygous C-to-T transition at nucleotide 76 in exon 2 of DSG1, which results in a premature termination codon (PTC) at codon 26, designated p.R26X (Fig. 3A). This mutation was previously reported by others [2], thus represents a recurrent mutation. Second, all affected individuals in Family 2 carry a novel heterozygous A>G transition in the 3’ acceptor splice site of intron 4 of DSG1, designated c.Ivs4-2A>G (Fig. 3B). Restriction digestion assays with the enzyme HpaII showed that neither unaffected individuals in the family nor 100 control individuals carry this mutation (Fig. 3C). Third, we found a novel heterozygous C-to-T transition at nucleotide 515 in exon 5 of the DSG1 of Family 3, which lies 3 bp upstream of the exon 5 - intron 5 boundary, designated c.515C>T (Fig. 3D). Mismatch allele-specific PCR showed that all affected individuals in Family 3 are heterozygous for this nucleotide transition. None of the unaffected individuals in the family or the 100 control individuals carries it (Fig. 3E). Affected individuals in Family 4 have a novel heterozygous C-to-G transversion 3 bp upstream of intron 9 – exon 10 boundary, designated c.Ivs9-3C>G (Fig. 3F). Screening assays with the restriction enzyme MnlI showed that the 100 control individuals do not have the mutation (Fig. 3G). Finally, we have identified a novel heterozygous single base pair deletion mutation, c.1399delA, in exon 10 of DSG1 of Family 5 (Fig. 3H). Restriction enzyme analysis with DdeI excluded it from 100 control individuals (Fig. 3I).
Figure 3. Identification of mutations in the DSG1 gene.
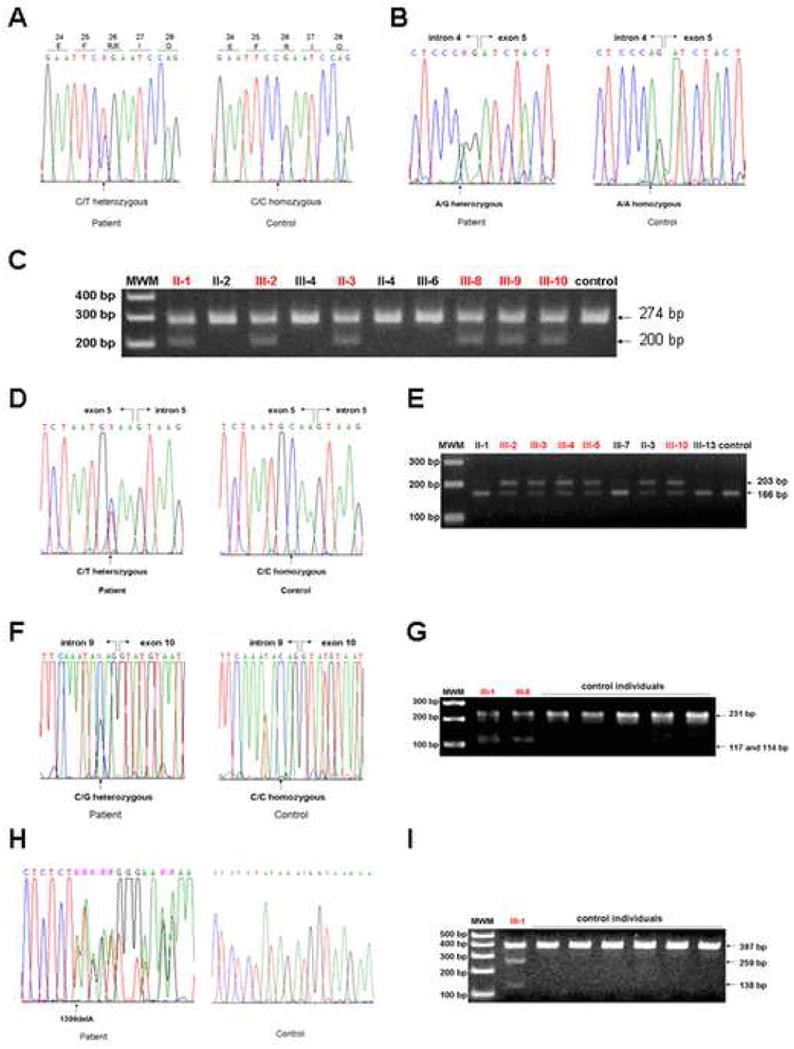
(A) Heterozygous mutation p.R26X in exon 2 of the DSG1 gene in Family 1. (B) Heterozygous mutation c.Ivs4-2A>G in intron 4 of the DSG1 gene in Family 2. (C) Screening assays for the mutation c.Ivs4-2A>G. PCR products from the mutant allele, 274 bp in size, were digested into 200 bp and 74 bp fragments by the restriction enzyme HpaII. The 74 bp fragments are not shown. (D) Heterozygous mutation c.515C>T in exon 5 of the DSG1 gene in Family 3. (E) Screening assays for the mutation c.515C>T. Mismatch PCR products from the wild type allele, 203 bp in size, were digested into 166 bp and 37 bp fragments by the restriction enzyme BsmI. The 37 bp fragments are not shown. (F) Heterozygous mutation c.Ivs9-3C>G in intron 9 of the DSG1 gene in Family 4. (G) Screening assays for the mutation c.Ivs9-3C>G. PCR products from the mutant allele, 231 bp in size, were digested into 117 bp and 114 bp fragments by the restriction enzyme MnlI. (H) Heterozygous mutation c.1399delA in exon 10 of the DSG1 gene in Family 5. (I) Screening assays for the mutation c.1399delA. PCR products from the mutant allele, 397 bp in size, were digested into 259 bp and 138 bp fragments by the restriction enzyme DdeI. In (C), (E), (G), and (I), affected individuals are colored in red. MWM, molecular weight markers.
4. Discussion
In this study, we analyzed five families of Pakistani origin affected with non-syndromic SPPK and identified heterozygous mutations in the DSG1 gene of all five families. The mutation p.R26X in Family 1 is a recurrent nonsense mutation which has been previously identified in a sporadic case of SPPK [2]. Since this nucleotide change exists in a CpG dinucleotide (Fig. 3A), it is most probably due to a spontaneous CpG deamination [17].
The novel mutation c.Ivs4-2A>G in Family 2 occurred in 3’-end of intron 4 of the DSG1 gene (Fig. 3B). Computational splice site analysis suggested that a novel potential splice acceptor site could be generated 1 bp upstream of the intron 4 - exon 5 junction (Fig. 4A). Thus, the mutation c.Ivs4-2A>G is predicted to cause abnormal splicing of exon 5, leading to a frameshift and a premature termination codon (PTC) in exon 5 (Fig. 4A).
Figure 4. Diagram illustrating the predicted consequences caused by three splice site mutations in the DSG1.
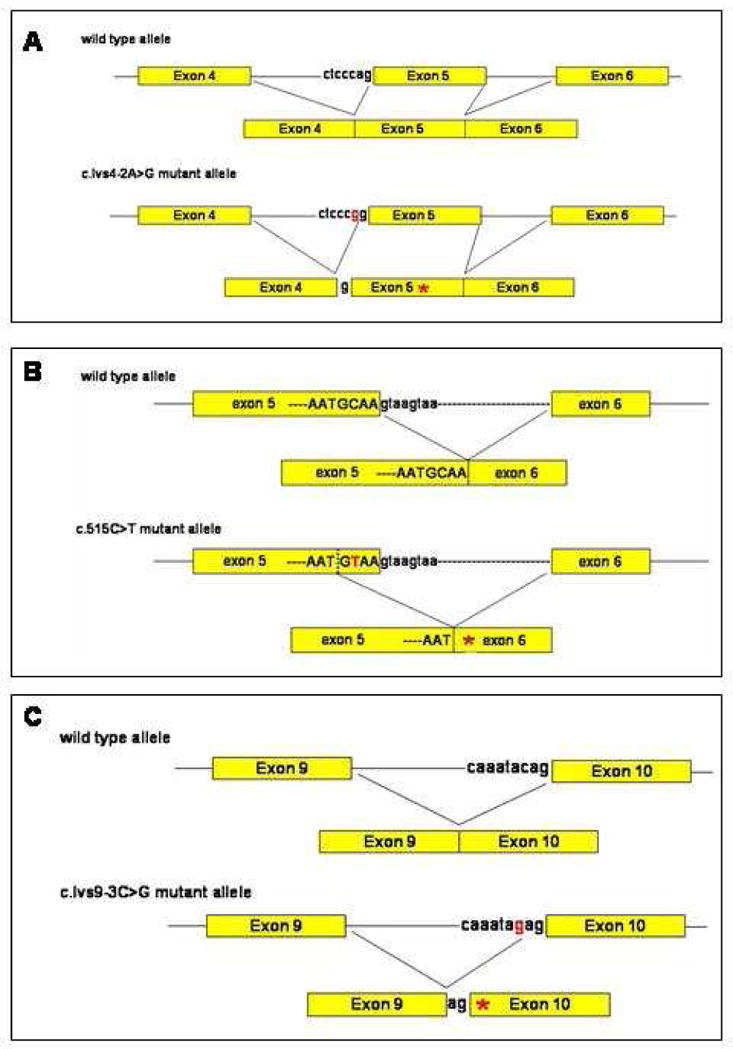
(A) c.Ivs4-2A>G. (B) c.515C>T. (C) c.Ivs9-3C>G. Mutations are colored in red. Asterisks indicate premature termination codons.
The novel mutation c.515C>T in Family 3 was detected in exon 5 of the DSG1 gene, which is located 3 bp upstream of the exon 5 - intron 5 junction (Fig. 3D). Although it could result in an amino acid change from alanine to valine at codon 172, computational analysis suggested that the c.515C>T mutation could generate a putative splice donor site at the mutation site, which is predicted to cause an aberrant splicing of exon 5, resulting in a frameshift and a PTC (Fig. 4B). It is well known that a point mutation near the ends of an exon can affect splicing [18]. Furthermore, most DSG1 mutations identified to date result in a PTC [2 13 14 15], which supports our interpretation that the c.515C>T is most likely to function as a splice site mutation, rather than a missense mutation.
We also identified a novel heterozygous mutation c.Ivs9-3C>G in the DSG1 gene in Family 4 (Fig. 3F). Computational splice site analysis indicated that the mutation would severely disrupt the splice acceptor sequence in intron 9, and instead, a putative splice acceptor site might be generated 2 bp upstream of the intron 9 – exon 10 junction, which is predicted to cause an abnormal splicing of exon 10 (Fig. 4C). Alternatively, the out-of-frame skipping of exon 10 (140 bp in size), might be another possible consequence caused by the mutation. Both possibilities would lead to a frameshift and a downstream PTC.
Finally, we found a novel heterozygous mutation c.1399delA in the DSG1 gene in Family 5. The mutation leads to a frameshift and generates a PTC at codon 467, three nucleotides downstream of the deletion (Fig. 3H).
Consistent with previous reports, all five mutations found in this study are also predicted to result in a PTC, albeit by different mechanisms (Fig. 5), which could theoretically generate truncated DSG1 proteins completely lacking the transmembrane and cytoplasmic domains. More likely, the aberrant DSG1 transcripts containing a PTC are predicted to be degraded due to nonsense-mediated messenger RNA decay [19 20]. Collectively, we conclude that the haploinsufficiency of DSG1 is most likely to be the predominant mechanism underlying SPPK in all five families. Our findings not only expand the allelic series of mutations in the DSG1 gene, but also enhance our understanding of the important role of desmosomes in maintaining epidermal integrity.
Figure 5. Schematic representation of DSG1 protein and the DSG1 mutations.
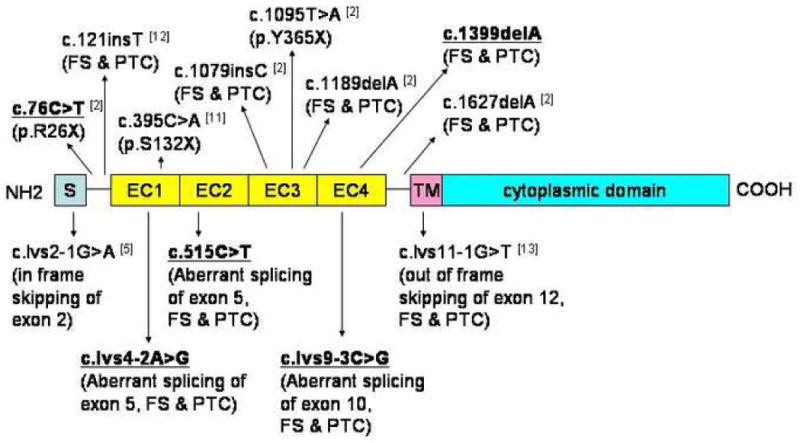
Mutations that were identified in this study are indicated in bold and underlined. Previously reported mutations are indicated for comparison. S, signal peptide; EC1-EC4, extracellular cadherin repeat domains; TM, transmembrane domain; FS, frameshift; PTC, premature termination codon.
Acknowledgments
We acknowledge the family members involved in this study. We also acknowledge thanks to Helen Lam for her excellent technical assistance. This study was supported by USPHS NIH grant from NIH/NIAMS RO1 AR44924 (to A.M.C.) and the support of the New York Times College Scholarship/New York-Presbyterian Hospital Summer Mentorship/Employee Program Grant (M.B.D-A).
Abbreviations
- SPPK
striate palmoplantar keratoderma
- PTC
premature termination codon
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.White KL. Striate palmoplantar keratoderma. Dermatol Online J. 2002;8:16. [PubMed] [Google Scholar]
- 2.Hunt DM, Rickman L, Whittock NV, Eady RA, Simrak D, Dopping-Hepenstal PJ, et al. Spectrum of dominant mutations in the desmosomal cadherin desmoglein 1, causing the skin disease striate palmoplantar keratoderma. Eur J Hum Genet. 2001;9:197–203. doi: 10.1038/sj.ejhg.5200605. [DOI] [PubMed] [Google Scholar]
- 3.McGrath JA. Inherited disorders of desmosomes. Australas J Dermatol. 2005;46:221–229. doi: 10.1111/j.1440-0960.2005.00188.x. [DOI] [PubMed] [Google Scholar]
- 4.Stokes DL. Desmosomes from a structural perspective. Curr Opin Cell Biol. 2007;5:565–571. doi: 10.1016/j.ceb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Amoudi A, Díez DC, Betts MJ, Frangakis AS. The molecular architecture of cadherins in native epidermal desmosomes. Nature. 2007;7171:832–837. doi: 10.1038/nature05994. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs E, Raghavan S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 2002;3:199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- 7.Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, et al. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet. 1999;8:971–976. doi: 10.1093/hmg/8.6.971. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong DKB, McKenna KE, Purkis PE, Green KJ, Eady RAJ, Leigh IM, et al. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8:143–148. doi: 10.1093/hmg/8.1.143. [DOI] [PubMed] [Google Scholar]
- 9.Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, et al. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol. 1999;113:940–946. doi: 10.1046/j.1523-1747.1999.00783.x. [DOI] [PubMed] [Google Scholar]
- 10.Whittock NV, Smith FJ, Wan H, Mallipeddi R, Griffiths WA, Dopping-Hepenstal P, et al. Frameshift mutation in the V2 domain of human Keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol. 2002;118:838–844. doi: 10.1046/j.1523-1747.2002.01750.x. [DOI] [PubMed] [Google Scholar]
- 11.Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JCR, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, wooly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 12.Norgett EE, Lucke TW, Bowers B, Munro CS, Leigh IM, Kelsell DP. Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol. 2006;126:1651–1654. doi: 10.1038/sj.jid.5700291. [DOI] [PubMed] [Google Scholar]
- 13.Kljuic A, Gilead L, Martinez-Mir A, Frank J, Christiano AM, Zlotogorski A. A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol. 2003;12:523–527. doi: 10.1034/j.1600-0625.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- 14.Milingou M, Wood P, Masouye I, McLean WH, Borradori L. Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatology. 2006;212:117–122. doi: 10.1159/000090651. [DOI] [PubMed] [Google Scholar]
- 15.Barber AG, Wajid M, Columbo M, Lubetkin J, Christiano AM. Striate palmoplantar keratoderma resulting from a frameshift mutation in the desmoglein 1 gene. J Dermatol Sci. 2007;45:161–166. doi: 10.1016/j.jdermsci.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank J, Cserhalmi-Friedman PB, Ahmad W, Panteleyev AA, Aita VM, Christiano AM. Characterization of the desmosomal cadherin gene family: genomic organization of two desmoglein genes on human chromosome 18q12. Exp Dermatol. 2001;10:90–94. doi: 10.1034/j.1600-0625.2001.010002090.x. [DOI] [PubMed] [Google Scholar]
- 17.Bestor TH, Coxon A. Cytosine methylation: the pros and cons of DNA methylation. Curr Biol. 1993;6:384–386. doi: 10.1016/0960-9822(93)90209-7. [DOI] [PubMed] [Google Scholar]
- 18.Pagenstecher C, Wehner M, Friedl W, Rahner N, Aretz S, Friedrichs N, et al. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet. 2006;119:9–22. doi: 10.1007/s00439-005-0107-8. [DOI] [PubMed] [Google Scholar]
- 19.Maquat LE. Defects in RNA splicing and the consequence of shortened translational reading frames. Am J Hum Genet. 1996;59:279–286. [PMC free article] [PubMed] [Google Scholar]
- 20.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]