

Summary
Traumatic events generate some of the most enduring forms of memories. Despite the elevated lifetime prevalence of anxiety disorders, effective strategies to attenuate long-term traumatic memories are scarce. The most efficacious treatments to diminish recent (i.e., day-old) traumata capitalize on memory updating mechanisms during reconsolidation that are initiated upon memory recall. Here, we show that, in mice, successful reconsolidation-updating paradigms for recent memories fail to attenuate remote (i.e., month-old) ones. We find that, whereas recent memory recall induces a limited period of hippocampal neuroplasticity mediated, in part, by S-nitrosylation of HDAC2 and histone acetylation, such plasticity is absent for remote memories. However, by using an HDAC2-targeting inhibitor (HDACi) during reconsolidation, even remote memories can be persistently attenuated. This intervention epigenetically primes the expression of neuroplasticity-related genes, which is accompanied by higher metabolic, synaptic, and structural plasticity. Thus, applying HDACis during memory reconsolidation might constitute a treatment option for remote traumata.
Introduction
Fear and other anxiety disorders develop after the experience of a traumatic event such as grave physical or psychological harm. Because of a strong emotional underpinning, traumatic memories are extraordinarily robust and difficult to treat, evidenced by an estimated lifetime prevalence of close to 29% (Kessler et al., 2005). Among the most efficacious treatments for anxiety disorders are exposure-based therapies (Cukor et al., 2010; Foa, 2000; Foa and Kozak, 1986), during which a patient is repeatedly confronted with the original fear-eliciting stimulus in a safe environment so that the once fearful stimulus can be newly interpreted as neutral or safe (Foa and Kozak, 1986). A fundamental element for successful exposure-based therapies is the reactivation of the traumatic memory (Foa and Kozak, 1986), which initiates a time-limited process called memory reconsolidation, during which a memory becomes susceptible to modification (Misanin et al., 1968; Nader et al., 2000).
In the context of exposure-based therapies, reconsolidation-updating approaches have proven effective to attenuate the response to fearful stimuli in humans and rodents alike (Monfils et al., 2009; Schiller et al., 2010), and similar paradigms have been successfully used to prevent drug craving and relapse (Xue et al., 2012). Based on the assumption of a period of updating or learning during memory reconsolidation (McKenzie and Eichenbaum, 2011; Nader and Hardt, 2009; Tronson and Taylor, 2007), several other studies have referred to pharmacological means in order to enhance this process (Kaplan and Moore, 2011). Among those, histone deacetylase inhibitors (HDACis) might be a particularly promising candidate to permanently modify fearful memories (Lattal and Wood, 2013) for two reasons. First, by modifying chromatin compaction, epigenetic mechanisms can have potentially stable and long-lasting effects on gene expression (Levenson and Sweatt, 2005), a required feature of long-term memories (Kandel, 2001); second, epigenetic mechanisms per se can target a vast variety of nuclear processes involved in neuronal plasticity (Gräff et al., 2011), such that their effect is not restricted to a particular signaling pathway.
Remarkably, almost all of these either purely behavioral or pharmacologically supported approaches to attenuate fearful responses have exclusively focused on recent, i.e., day-old, memories—leaving it unclear whether they would also be effective for remote, i.e., month-old, memories. As traumatic memories are oftentimes not readily amenable to immediate treatments (Kearns et al., 2012) and because remote memories are more stable than recent ones (Frankland et al., 2006; Inda et al., 2011; Milekic and Alberini, 2002; Suzuki et al., 2004), there is a clear need to investigate options to overcome remote fear memories.
Results
Despite Using Reconsolidation-Updating Paradigms, Remote Memories Cannot Be Persistently Attenuated
To test whether exposure-therapy-based approaches can be used to attenuate remote traumatic memories, we used Pavlovian fear conditioning in mice, a commonly employed method to study fear responses underlying traumatic memories such as posttraumatic stress disorder (PTSD) (Mahan and Ressler, 2012). In fear conditioning, an unconditioned stimulus (US)—an electrical footshock—is paired with a conditioned stimulus (CS)—either a specific tone or context for cued and contextual fear conditioning, respectively. When the animals are later tested for their fear memory by exposing them to the CS, the CS alone will elicit the conditioned response (CR), freezing. Specifically, we trained mice for either cued or contextual fear conditioning, 1 day and 30 days after which we attempted to attenuate their CR by different fear extinction paradigms (Figure 1A, Experimental Procedures, and Figure S1A available online). All of these paradigms take advantage of a transient period of memory lability, the reconsolidation window, that occurs between 1 and 6 hr following the memory recall (Monfils et al., 2009).
Figure 1. Remote Fear Memories Are Resistant to Attenuation despite Using Reconsolidation-Updating Mechanisms.

(A) Schematic of the experimental paradigm. For details, see text.
(B) Schematic of the massed extinction paradigm.
(C) Using the massed extinction paradigm for contextual fear memories, recent memories show no signs of spontaneous recovery (n = 10; repeated-measurements ANOVA; p ≤ 0.0001).
(D) Using the same massed extinction paradigm, remote memories spontaneously recover (n = 9; repeated-measurements ANOVA; p ≤ 0.0001).
(E) Using the same paradigm but without memory recall, remote memories spontaneously recover (n = 10; repeated-measurements ANOVA; p ≤ 0.0001).
(F) Schematic of the spaced extinction paradigm.
(G) Using the spaced extinction paradigm for contextual fear memories, recent memories show significant attenuation of fear and no signs of spontaneous recovery (n = 12; repeated-measurements ANOVA; p = 0.0038).
(H) Using the same spaced extinction paradigm, remote memories show no signs of attenuation (n = 16).
(I) Using the spaced extinction paradigm without memory recall, remote memories show no signs of attenuation (n = 12).
Error bars indicate ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 for Tukey's posthoc analysis. See also Figure S1 for results of remote cued fear memories.
The first protocol aimed at extinguishing the CR to a tone (the CS) by employing a massed extinction paradigm for cued fear conditioning (Figure S1B, expo.). We found that, for recent memories, this paradigm persistently attenuated the CR immediately and 1 day after extinction (Figures S1C and S1F). Importantly, there were no signs of spontaneous recovery (SR) or reinstatement (RI) of the fear (Figures S1C and S1F), the presence of which is indicative of incomplete fear extinction (Bouton, 1993, 2004; Bouton and Bolles, 1979; Pavlov, 1927; Rescorla, 2004; Rescorla and Heth, 1975). For remote memories, in contrast, we found that, although this paradigm was effective in decreasing the freezing response after the extinction session, there were significant RI (Figure S1D) and SR (Figure S1G), indicating persistence of the original memory. This finding is consistent with the occurrence of both RI and SR upon remote memory extinction without memory recall (Figures S1E and S1H).
The second protocol aimed at attenuating the CR to a context (the CS) by a massed extinction paradigm adopted for contextual fear conditioning (Figure 1B) (Sananbenesi et al., 2007). By using this paradigm, recent fear memories could be successfully and permanently attenuated (Figure 1C), but remote fear memories showed significant SR (Figure 1D). Similar results were obtained when no recall was presented (Figure 1E).
Furthermore, because increasing the intertrial interval during fear extinction training has been shown to facilitate fear extinction of recent memories (Urcelay et al., 2009), we reasoned that such spacing would also help in attenuating remote memories (Figure 1F). This paradigm significantly attenuated recent fear memories, and there were no signs of SR (Figure 1G). However, when applied to remote memories, the same paradigm failed to even temporarily reduce fear, and when tested for SR, freezing levels were comparable to those during memory recall (Figure 1H). Similar results were obtained in the absence of memory recall (Figure 1I).
Together, these results indicate that three different behavioral paradigms that successfully extinguish recent fear memories were not capable of doing so for remote fear memories, despite taking advantage of the transient period of memory lability induced by memory recall. The question arises as to why remote fear memories are more difficult to extinguish.
Recalling Remote Memories Is Not Salient Enough to Induce Histone Acetylation-Mediated Neuronal Plasticity in the Hippocampus
By definition, the reconsolidation window opened by memory recall allows previously acquired memories to be updated with new information and is thereby thought to initiate a new period of neuronal plasticity (Hartley and Phelps, 2010; McKenzie and Eichenbaum, 2011). We hypothesized that the same memory recall might not induce the same extent of neuronal plasticity for remote as for recent memories. For the subsequent analyses, we focused on contextual fear memories, the formation of which depends on the hippocampus, a brain area crucial for memory-related neuronal plasticity (Kandel et al., 2013) that is activated upon remote memory recall (Debiec et al., 2002). One fundamental mechanism that governs neuronal plasticity is the epigenetic modification of gene expression by acetylation of histone proteins (Levenson and Sweatt, 2005). Histone acetylation promotes a chromatin structure that is more permissive for gene transcription (Brownell and Allis, 1996) and thereby positively regulates transcription-dependent long-lasting forms of neuronal plasticity.
To test whether the same memory recall leads to differences in histone acetylation between remote and recent memories, we first used immunohistochemical analyses to assess the acetylation of histone 3 on lysine residues 9 and 14 (H3K9/14), a modification associated with facilitated neuronal plasticity (Gräff et al., 2011). We found that, 1 hr post memory recall, hippocampal H3K9/14 acetylation of remote memories was indistinguishable from behaviorally naive animals but was significantly smaller than that of recent memories (Figures 2A and 2B). Next, we carried out chromatin immunoprecipitation (ChIP) to determine the abundance of H3K9/14 acetylation at the promoter region of cFos, an immediate early gene regulated by neuronal activity and critical for neuronal plasticity (Tischmeyer and Grimm, 1999). Consistent with the overall histone acetylation pattern, we found the cFos promoter to be hypoacetylated after remote memory recall compared to after recent memory recall (Figure 2C). Accordingly, the expression of cFos was significantly reduced after remote memory recall (Figure 2D), an effect that was also observed at the protein level evidenced by the number cFos-positive cells (Figure 2E), as reported previously (Frankland et al., 2004).
Figure 2. The Recall of Remote Memories Is Not Salient Enough to Induce Neuroplasticity-Permitting Histone Acetylation Changes.
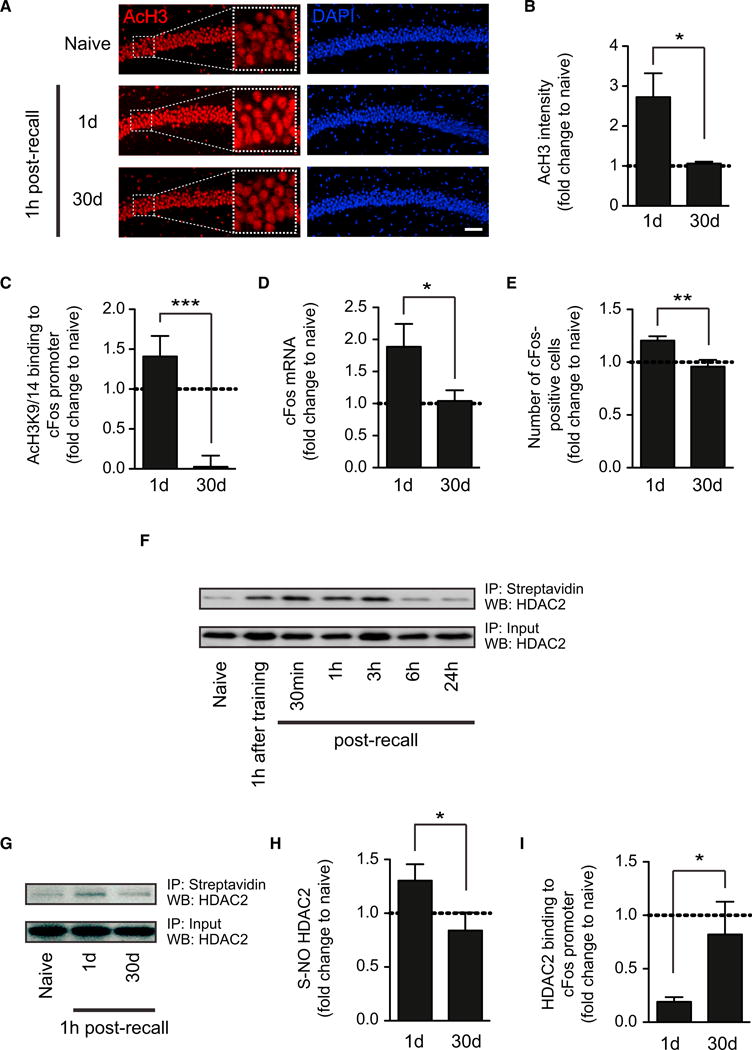
(A) Representative images of immunohistochemical labelings of acetylated H3K9/14 (“AcH3”) in hippocampal area CA1 1 hr after recent and remote memory recall as compared to behaviorally naive animals. Scale bar, 100 μm.
(B) Quantification thereof (n = 3–4 animals each).
(C) Quantitative PCR results of the abundance of acetylated H3K9/14 in the promoter region of cFos at the same time points (n = 8 animals each).
(D) Quantitative RT-PCR results of the expression of cFos in the hippocampus at the same time points (n = 4–5 animals each).
(E) Quantification of the number of cFos-positive cells in the hippocampus at the same time points (n = 3 animals each).
(F) Western blot analysis of S-nitrosylation of HDAC2 using the biotin-switch assay and streptavidin precipitation 1 hr after contextual fear conditioning and at different intervals after recent memory recall.
(G) Representative pictures of western blot analysis of S-nitrosylation of HDAC2 1 hr after recent and remote memory recall.
(H) Quantification thereof (n = 3–4 animals each).
(I) Quantitative PCR results of HDAC2 binding to the promoter region of cFos at the same time points (n = 8 animals each).
Error bars indicate ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Student's t test. See also Figure S2.
Increments in histone acetylation are dependent on neuronal activity (Levenson and Sweatt, 2005), and one pathway shown to mediate these changes in cultured neurons involves the dissociation of histone deacetylase 2 (HDAC2) from the chromatin following its nitrosylation on Cys262 and Cys274 (Nott et al., 2008). We therefore investigated whether the lack of increased histone acetylation following remote memory recall might be due to a lack of hippocampal HDAC2 nitrosylation. Using a biotin-switch assay, we found that, for recent memories, HDAC2 becomes nitrosylated between 30 and 60 min following memory recall, a state that lasted for no longer than 6 hr (Figure 2F). Thus, HDAC2 nitrosylation correlated with the limited period of memory lability during the reconsolidation window (Monfils et al., 2009). In contrast, we detected no increase in HDAC2 nitrosylation following remote memory recall (Figures 2G and 2H). Consistent with its nitrosylation status, the binding of HDAC2 to the promoter of cFos was reduced following recent memory recall, whereas HDAC2 was still bound to the chromatin after the recall of remote memories (Figure 2I). Importantly, memory recall was necessary to elicit all of the changes described above (Figure S2). Together, these results indicate that, in contrast to recent memories, the recall of remote memories fails to install HDAC2 nitrosylation and histone acetylation-mediated neuronal plasticity in the hippocampus.
S-Nitrosylation of HDAC2 in the Hippocampus Is Critical for Memory Updating during Reconsolidation
To further assess the role of nitrosylated HDAC2 in mediating such neuroplasticity, we treated fear-conditioned mice with Nω-Nitro-L-arginine methyl ester (L-NAME), an inhibitor of nitric oxide (NO) synthase, before the recall of recent memories and extinction training (Figure 3A). We reasoned that, if nitrosylation plays a role in facilitating the update of recent memories, its inhibition would prevent it. We found that, although L-NAME-treated animals showed significant attenuation of fear after extinction training, their fear spontaneously recovered (Figure 3B). In contrast, VEH-treated animals showed no SR (Figure 3B). L-NAME treatment did not affect the overall behavior of the animals (Figures S3A–S3D). Interestingly, L-NAME treatment also abolished the recall-induced nitrosylation of HDAC2 1 hr after memory recall (Figure 3C), suggesting that HDAC2 nitrosylation might be critical for recall-induced memory updating during reconsolidation.
Figure 3. Nitrosylation of HDAC2 Is Critical for Memory Updating during Reconsolidation.

(A) Schematic of the experimental paradigm. 1 hr before recent memory recall, L-NAME or its VEH was administered i.p.
(B) Using the massed extinction paradigm for contextual fear memories, L-NAME-treated animals show significant spontaneous recovery, whereas VEH-treated ones do not (n = 8 for each treatment; repeated-measurements ANOVA; p ≤ 0.0001 for VEH, p = 0.0034 for L-NAME).
(C) Representative pictures of western blot analysis of S-nitrosylation of HDAC2 1 hr after or without recall of recent memories in L-NAME and VEH-treated animals.
(D) Schematic of the experimental paradigm. For details, see text.
(E) Representative pictures of western blot analysis of S-nitrosylation of HDAC2 1 hr after remote memory recall in molsidomine- or VEH-treated HDAC2WT or HDAC2C262/274A-injected animals.
(F) Using the massed extinction paradigm for contextual fear memories, HDAC2WT-treated animals show no signs of spontaneous recovery, whereas HDAC2C262/274A-injected animals do (n = 7–10 for both groups; repeated-measurements ANOVA; p ≤ 0.0001 for HDAC2WT, p = 0.01 for HDAC2C262/274A).
Error bars indicate ± SEM. *p ≤ 0.05, **p≤0.01, ***p≤0.001 by Tukey's posthoc. See also Figure S3.
Then, to prove a causal implication of HDAC2 nitrosylation in memory updating, we used the following strategy. We treated mice before remote memory recall with the nitric oxide donor molsidomine (MOL), which should facilitate memory updating of even remote memories, and simultaneously overexpressed a non-nitrosylateable form of HDAC2, the double-alanine mutant HDAC2C262/274A(Nott et al., 2008), which should prevent remote memories from being updated in the presence of MOL. To this end, we injected short-term herpes simplex viral (HSV) vectors carrying either HDAC2WT or HDAC2C262/274A (Figure S3E) into hippocampal area CA1 (Figure S3F) 3 days before remote memory recall to assure maximal expression at the time of recall. Then, 1 hr prior to recall and subsequent extinction training, we administered MOL or its vehicle to both groups (Figure 3D). We found that, whereas MOL-administration increased HDAC2 nitrosylation in HSV-HDAC2WT-injected animals 1 hr after remote memory recall, HSV-HDAC2C262/274A-injected mice showed no HDAC2 nitrosylation despite MOL treatment (Figure 3E), indicating that, in HDAC2C262/274A-injected animals, HDAC2 is predominantly present in its non-nitrosylateable form. When tested for fear extinction, MOL-treated HSV-HDAC2WT-injected animals showed significant fear reduction after memory extinction and no signs of SR (Figure 3F), suggesting that the NO donor allowed for successful memory updating. In contrast, MOL-treated HDAC2C262/274A-injected animals showed significant SR despite successful memory extinction immediately and 1 day after extinction training (Figure 3F). Importantly, MOL treatment did not affect the animals' general behavior (Figures S3G– S3J). Together, these results indicate that nitrosylation of HDAC2 following memory recall is a critical event for remote memories to become amenable for effective updating during reconsolidation.
By Using HDAC2-Targeting HDAC Inhibitors, Remote Fear Memories Can Be Attenuated
Because MOL cannot be targeted to HDAC2 alone, we hypothesized that, by using an HDAC2-targeting HDACi, it might also be possible to overcome the lack of neuroplasticity-related histone hyperacetylation for remote memories and, consequently, to enable memory-updating mechanisms. For this, we used the benzamide-based HDACi CI-994 (Figure 4A), a selective inhibitor of class I HDACs, including HDAC2 (Figure 4B). Systemic, intraperitoneal administration of 30 mg per kg resulted in high (Cmax = 12.3 mM) and long-lasting (T1/2 = 3.7 hr) levels of CI-994 in the brain (Figure 4C) and did not affect overall behavior (Figures S4A–S4D). To confidently target the memory-updating mechanisms during the reconsolidation window, we administered CI-994 or its vehicle (VEH) to fear-conditioned animals 1 hr post memory recall and then performed the extinction paradigms used previously (Figure 4D).
Figure 4. Remote Fear Memories Become Amenable to Attenuation by CI-994.

(A) Chemical structure of CI-994, a benzamide-based HDAC inhibitor.
(B) Concentration-time curve of CI-994 in C57BL/6 mouse brain after a 30/10/1mg/kg i.p. dose.
(C) Pharmacokinetic parameters of CI-994 in C57BL/6 mouse brain (see Extended Experimental Procedures for additional details).
(D) Schematic of the experimental paradigm. 1 hr post remote memory recall, either CI-994 or its VEH was administered i.p., and 1 hr later, the different extinction procedures were applied.
(E) Using the massed extinction paradigm to the context, VEH-treated animals show significant spontaneous recovery of fear (n = 11; repeated-measurements ANOVA; p = 0.0001).
(F) Using the same paradigm, HDACi-treated animals show no spontaneous recovery (n = 16; repeated-measurements ANOVA; p ≤ 0.0053).
(G) Using the same paradigm but without memory recall, HDACi-treated animals show significant spontaneous recovery (n = 17; repeated-measurements ANOVA; p ≤ 0.0001).
(H) Using the spaced extinction paradigm to the context, VEH-treated animals do not show any attenuation in their fear response (n = 15).
(I) Using the same paradigm, HDACi-treated animals show significant reduction in their freezing response immediately and 1 day after extinction and show no signs of spontaneous recovery (n = 16; repeated-measurements ANOVA, p ≤ 0.0001).
(J) Using the same paradigm but without memory recall, HDACi-treated animals do not show any attenuation in their fear response (n = 12).
Error bars indicate ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Tukey's posthoc. See also Figure S4.
For the massed extinction paradigm, we found that, whereas VEH-treated animals showed significant SR (Figure 4E), CI-994-treated animals showed no signs thereof (Figure 4F). Furthermore, the fear-attenuating effect of the HDACi depended on memory recall, as we also observed SR when the HDACi was given without recall (Figure 4G). Next, when using the spaced extinction paradigm, which did not reduce remote fear memories (Figures 1H and 4H), we observed a significant reduction of the CR with CI-994, indicating memory extinction (Figure 4I). Importantly, we also observed a complete absence of SR, reflecting persistent memory extinction. This effect again depended on memory recall (Figure 4J). Of note, we observed the same beneficial effects with the HDACi for remote cued fear memories (data not shown).
Importantly, HDACi administration did not generalize fear extinction. When mice were both cued and contextual fear conditioned yet only the context as a CS underwent extinction (using the spaced extinction paradigm), fearful memories associated with the cue were still present after successful extinction of the context (Figures S4E–S4G). Together, these results suggest that, by using an HDACi in combination with extinction training during the reconsolidation window, even remote contextual fear memories become amenable to persistent and specific attenuation.
HDAC Inhibitors Epigenetically Prime the Hippocampal Transcriptome for Reinstated Neuroplasticity
Next, we investigated the mechanisms by which the HDACi operates, hypothesizing that extinction training in conjunction with CI-994 would allow for increased histone acetylation-mediated neuroplasticity. We focused on the spaced extinction paradigm (Figure 1F), as this paradigm was the only one in which VEH- and CI-994-treated animals showed different fear responses immediately following the extinction procedure (Figures 4H and 4I). We first measured hippocampal histone acetylation using immunohistochemistry 1 hr after the end of the extinction procedure. We found that, compared to the histone acetylation of VEH-treated animals and that of 1 hr after remote memory recall, CI-994-treated animals showed significantly higher acetylation levels (Figures 5A and 5B). This was consistent with reduced hippocampal HDAC2 activity upon HDACi-treatment (Figure 5C). In the prefrontal cortex, where remote memories are primarily stored (Bontempi et al., 1999; Frankland and Bontempi, 2005; Frankland et al., 2004), histone acetylation following extinction training in combination with CI-994 was also elevated relative to VEH-treated animals and was comparable to levels 1 hr after remote memory recall (Figure S5), which may indicate persistent engagement of cortical areas in HDACi-treated animals. For the following analyses, we nevertheless focused exclusively on the hippocampus, as this brain area is critical for the incorporation of new information into a given memory trace during reconsolidation of contextual fear memories (Debiec et al., 2002).
Figure 5. HDACis Prime a Transcriptional Program for Increased Neuroplasticity.
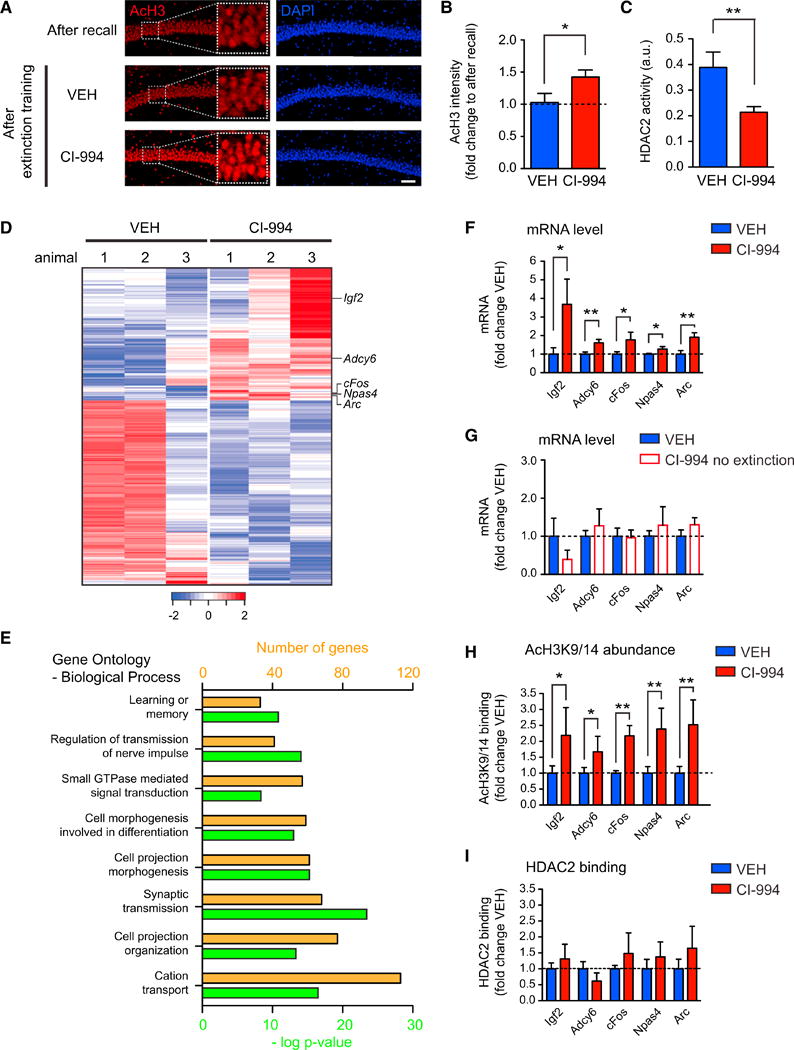
(A) Representative images of immunohistochemical labelings of acetylated H3K9/14 (“AcH3”) in hippocampal area CA1 1 hr after recall and 1 hr after completion of extinction in VEH- and HDACi-treated animals. Scale bar, 100 μm.
(B) Quantification thereof (n = 3–4 animals each).
(C) HDAC2 activity assay showing that CI-994 effectively inhibited hippocampal HDAC2 activity (n = 4 animals each).
(D) Heatmap depicting 475 differentially expressed genes (DEGs) determined by RNA-sequencing in the hippocampus between VEH-treated and HDACi-treated animals after remote memory attenuation. Each line represents a DEG, each row the gene expression per animal. Blue and red indicate low and high levels of expression, respectively.
(E) Histogram showing the biological processes to which the DEGs belong (orange, number of DEGs per biological process; green, significance of enrichment).
(F) Quantitative RT-PCR confirmation of the expression of several neuroplasticity-related genes detected under (D) (n = 5–9 animals each).
(H) Quantitative PCR results showing the abundance of AcH3K9/14 at the promoter region of the genes under (F) (n = 5–11 animals each).
(I) Quantitative PCR results showing the binding of HDAC2 to the promoter region of the genes under (F) (n = 5–6 animals each).
Error bars are ± SEM. *p ≤ 0.05, **p ≤ 0.01 by Student's t test. See also Figure S5 and Table S1.
Alongside histone acetylation, another critical aspect of neuroplasticity is the transcription of genes related to synaptic plasticity, learning, and memory (Kandel, 2001). To investigate those, we performed RNA sequencing of hippocampal extracts of both VEH and CI-994-treated animals 1 hr after completion of remote memory extinction. At an expression fold change cutoff of 1.4, we found 475 differentially expressed genes (DEGs) between CI-994 and VEH-treated animals, among which 199 genes showed higher expression in CI-994-treated animals (Figure 5D and Table S1). Upon generation of pathway and gene ontology analyses, we noticed these DEGs to be implicated in biological processes related to neuronal plasticity, such as learning or memory, regulation of transmission of nerve impulse, cell morphogenesis and projection, cation transport, and synaptic transmission (Figure 5E). These findings suggest that, by using CI-994 in combination with extinction training, neuroplasticity at the level of gene expression was enhanced.
Moreover, we detected several key regulators of neuronal plasticity to be significantly upregulated in CI-994-treated animals (Figure 5D). These genes include the immediate-early genes Arc and cFos, which are both critically involved in synaptic plasticity and memory-related processes (Korb and Finkbeiner, 2011; Shepherd and Bear, 2011; Tischmeyer and Grimm, 1999), Adcy6, an adenylate cyclase regulating neurite extension (Wu et al., 2011), Npas4, a transcription factor regulating contextual memory formation (Ramamoorthi et al., 2011; Lin et al., 2008), and Igf2 (insulin-like growth factor 2), which facilitates memory retention (Chen et al., 2011) and extinction of recent fear memories (Agis-Balboa et al., 2011). qRT-PCR analysis of independent CI-994 and VEH-treated samples confirmed the expression changes of these genes (Figure 5F). Importantly, no such expression changes were detected when the HDACi was administered without extinction training (Figure 5G), in line with the observation that the HDACi alone had no behavioral effect (Figure 4J).
The genes with higher expression also showed higher acetylation in their promoter region (Figure 5H). Intriguingly, such increased histone acetylation occurred despite HDAC2 being bound to their promoter (Figure 5I). These findings suggest that the HDACi treatment following remote memory recall can compensate for the absence of HDAC2 nitrosylation and its persistent binding to the chromatin, thereby increasing histone acetylation-mediated neuroplasticity at the level of gene expression.
HDAC Inhibitors Lead to Increased Neuroplasticity during Memory Extinction
Lastly, we conducted a series of experiments to assess whether such increased expression of neuroplasticity-related genes would also result in facilitated synaptic plasticity. We first measured hippocampal glucose utilization 1 hr after completion of remote memory extinction by means of radiolabeled [3H]2-deoxyglucose ([3H]2-DG) uptake, which reflects neuronal activity (Magistretti, 2006). We found that CI-994-treated animals showed higher hippocampal [3H]2-DG uptake compared to VEH-treated animals, depicting increased metabolic engagement of this brain area following memory extinction (Figures 6A and 6B). In contrast, [3H]2-DG measurements did not differ between VEH- and CI-994-treated animals in the piriform cortex, a brain region implicated in olfactory memories and thus unrelated to the formation of contextual memories (Figure S6A).
Figure 6. HDACis Increase Neuroplasticity after Remote Memory Attenuation.
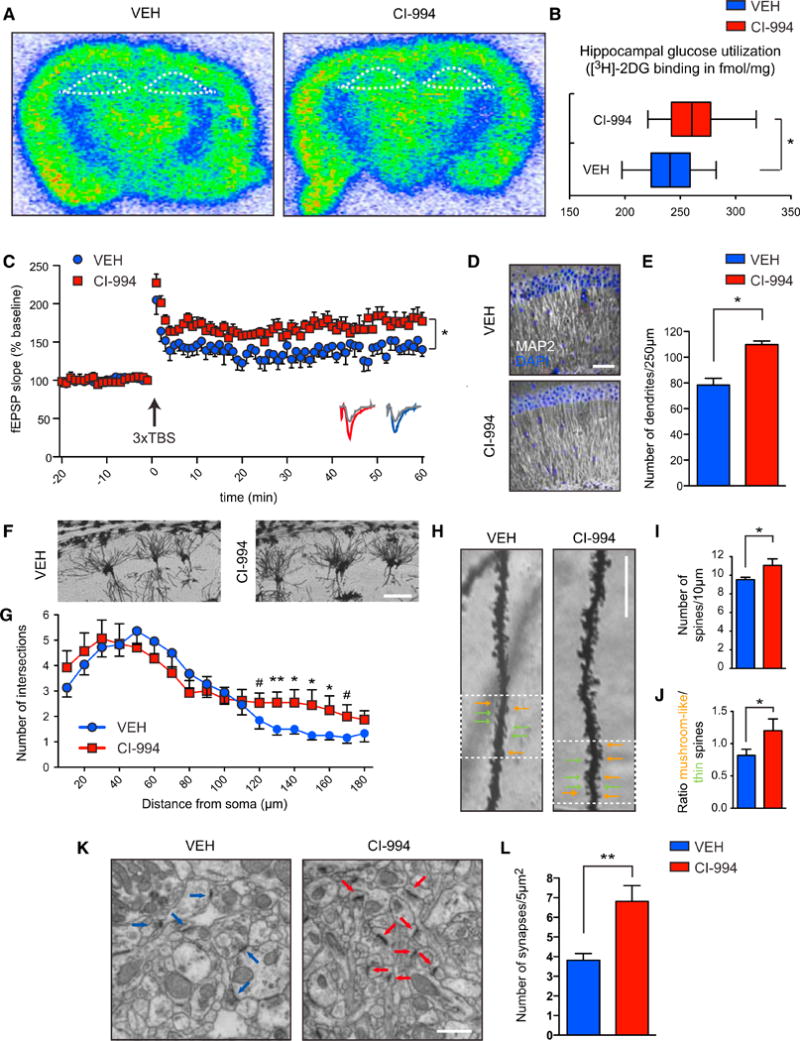
(A) Representative scans of [3H]-2DG uptake in coronal brain sections depicting higher metabolic activity in the hippocampus (outlined by white dotted lines) for CI-994-treated animals.
(B) Quantification thereof (n = 10 animals each).
(C) Field excitatory postsynaptic potential (fEPSP) slopes in hippocampal area CA1 of VEH- and CI-994-treated animals (n = 6 slices from four mice each); sample traces below the point chart represent fEPSPs at 1 min before (gray) and 1 hr after (colored) theta-burst stimulation (TBS).
(D) Representative images of MAP2-labeled hippocampal sections. Scale bar, 200 mm.
(E) Quantification thereof (n = 3 animals each).
(F) Representative images of Golgi-stained hippocampal pyramidal neurons used for Sholl analysis. Scale bar, 100 μm.
(G) Quantification of the number of dendritic branches per given distance from the soma (n = 8–12 neurons of 3 animals each).
(H) Representative images of Golgi-stained hippocampal pyramidal neurons. Scale bar, 10 μm.
(I) Quantification of (H) in terms of number of spines (n = 3 animals each).
(J) Quantification of (H) in terms of number of mature versus immature spines (n = 3 animals each).
(K) Representative images of hippocampal brain section (stratum radiatum) used for transmission electron microscopy. Arrows point to synapses. Scale bar, 1 μm.
(L) Quantification thereof (n = 3–4 animals each).
Error bars indicate ± SEM. #p ≤ 0.1, *p ≤ 0.05, **p ≤ 0.01 by Student's t test. See also Figure S6.
Next, to test whether such increased metabolic activity was indicative of increased neuronal plasticity, we assessed long-term potentiation (LTP) at Schaffer collaterals by electrophysiological recordings at the same time point. Using triple theta-burst stimulation (3 × TBS), we observed that CI-994-treated animals displayed facilitated LTP compared to VEH-treated ones (Figure 6C). In contrast, we found no difference in basal synaptic transmission, as the slope of field excitatory postsynaptic potential (fEPSP) elicited by a given presynaptic fiber volley did not differ between the two groups (Figure S6B). Likewise, we detected no significant difference in paired-pulse facilitation (Figure S6C). These results suggest that a combination of extinction training and CI-994 administration leads to increased synaptic plasticity.
Finally, we investigated whether such increased synaptic plasticity might also be accompanied by increased structural plasticity. For this, we measured the number of dendrites by microtubule-associated protein 2 (MAP2) immunohistochemistry, dendritic branching and the number of dendritic spines by Golgi staining, and the density of synapses by transmission electron microscopy and synaptophysin immunohistochemistry, a presynaptic marker, 1 hr after completion of memory extinction. These measurements reflect the degree of neuronal connectivity and thereby synaptic strength (Fiala et al., 2002). We found that CI-994-treated animals displayed an elevated number of MAP2-stained dendrites compared to VEH-treated ones in the stratum radiatum layer of the hippocampus (Figures 6D and 6E). Similarly, we detected a significant enhancement of dendritic branching between 120 and 170 μm from the soma as measured by Sholl analysis (Figures 6F and 6G), as well as a greater number of spines in CI-994-treated animals (Figures 6H and 6I). Of note, when analyzing the fine structure of these spines, we noticed a greater number of mushroom-like than thin spines upon HDACi-treatment (Figures 6H and 6J), an indicator of a more pronounced spine maturation process (Bourne and Harris, 2007). Lastly, the density of functional synapses was also markedly increased following CI-994 treatment (Figures 6K, 6L, and S6D, and S6E). Taken together, our findings demonstrate that, in addition to epigenetically enhancing neuroplasticity-related gene expression, the HDACi-treatment also resulted in increased synaptic and structural plasticity.
Discussion
To our knowledge, this is the first report successfully attempting to attenuate remote fear responses in an animal model of traumatic memories. Using different behavioral protocols, a previous study also found that, despite using reconsolidation-updating paradigms that successfully extinguish recent fear memories, remote memories are resistant to attenuation (Costanzi et al., 2011). One potential mechanism behind such resilience of remote memories to be attenuated by behavioral interventions alone might be the lack of histone acetylation-mediated hippocampal neuroplasticity for remote memories presented here. Whereas recent memory recall allows for histone hyperacetylation and the expression of neuroplasticity-related genes such as cFos by stimulating HDAC2 nitrosylation and its subsequent dissociation from the chromatin, remote memory recall fails to do so (Figures 7A and 7B). Interestingly, learning itself is known to be accompanied by increased NO signaling in the hippocampus (Harooni et al., 2009), whereas mice lacking the enzyme neuronal NO synthase (nNOS) display contextual fear memory deficits (Itzhak et al., 2012). Thus, as per yet to be determined mechanisms, reduced hippocampal NO signaling toward HDAC2 might prevent a phase of learning that is crucial for remote memory updating.
Figure 7. Working Model.
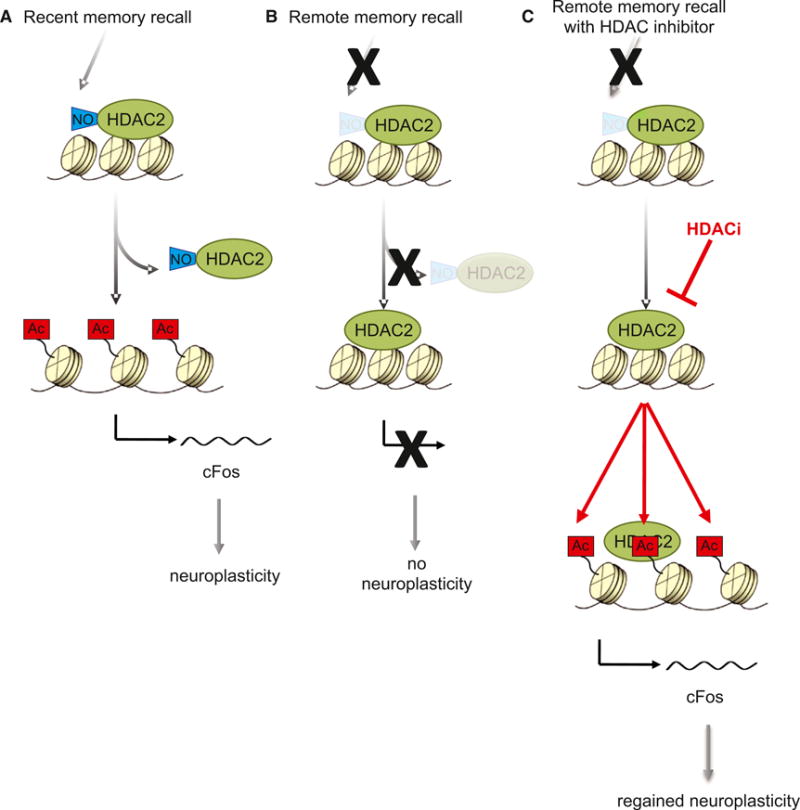
HDACis epigenetically prime the expression of neuroplasticity-related genes (e.g., cFos) to overcome the absence of hippocampal neuroplasticity upon remote memory recall and thereby the resilience of remote fear memories to successful extinction (for details, see text).
While our molecular analyses focused primarily on the hippocampus, we do not exclude a possible contribution of similar or different mechanisms in other brain regions such as the amygdala (see Clem and Huganir, 2010) or cortical areas (see Figure S5) to the attenuation of remote memories. This latter brain area in particular would warrant further studies, as it is widely acknowledged that, as a memory matures, it becomes increasingly represented in and dependent on cortical areas (Nadel and Moscovitch, 1997; Frankland and Bontempi, 2005). Our finding of a lack of hippocampal neuroplasticity upon remote memory recall indeed supports this concept of hippocampal disengagement for remote memories (Figure 7B). However, it has also been known that, upon memory recall, even supposedly cortex-dependent remote memories re-enter a phase of hippocampus dependency (Debiec et al., 2002), and artificially inhibiting hippocampal activity (in area CA1) during memory recall in effect abolishes the capacity to recall remote memories (Goshen et al., 2011). The present results extend these findings by showing that recalling remote fear in conjunction with extinction training alone does not lead to a level of neuroplasticity that is sufficient for fear memory updating. Rather, it is the combined use of HDAC2-targeting HDACis and extinction training during memory reconsolidation that reinstates hippocampal neuroplasticity and permits updating of even remote traumatic memories (Figure 7C). This finding is reminiscent of an fMRI study in humans showing that subjects with persisting traumatic memories display lower hippocampal activation upon memory recall than those for whom fear memory extinction training was successful (Milad et al., 2009).
We found the HDACi treatment to trigger the upregulation of a key set of neuroplasticity-related genes, which was accompanied by increased histone acetylation in their respective promoter region. This observation indicates that the HDACi may epigenetically prime the chromatin into a transcriptionally permissive state, thereby allowing for enhanced neuroplasticity. Of the genes that exhibited increased expression following remote memory extinction, cFos, Arc, and Igf2 were also found to be increased after the extinction of recent fear memories (Agis-Balboa et al., 2011), and conclusive evidence exists that—at least for recent fear memories—hippocampal Igf2 is necessary for successful contextual extinction (Agis-Balboa et al., 2011). However, the precise contribution of each of these genes to remote memory attenuation remains to be determined.
Previous studies have already demonstrated the usefulness of other HDACis in extinguishing recent memories (Bredy and Barad, 2008; Bredy et al., 2007; Fujita et al., 2012; Itzhak et al., 2012; Lattal et al., 2007; Stafford et al., 2012). Interestingly, in none of these studies were the extinction paradigms applied during the reconsolidation window, a procedure that by itself is sufficient to successfully attenuate fearful memories (Monfils et al., 2009). Together with our own observation that combining the HDACi treatment with successful reconsolidation-updating mechanisms for recent memories does not lead to further attenuation of the fear (data not shown), it seems that HDACis can facilitate memory updating in situations when extinction training alone is insufficient.
HDACi-supported extinction training led to increased neuroplasticity on a functional and structural level. For instance, we observed that hippocampal metabolic activity and LTP were enhanced. This is consistent with fMRI studies in humans, which show that successful extinction memory correlates with higher hippocampal activity (Milad et al., 2009). On a structural level, we found that memory extinction in combination with HDACi treatment was accompanied by increased synaptic density and dendritic branching and an elevated number of spines. These observations are consistent with a finding of increased spine numbers following recent memory extinction (Lai et al., 2012) and with earlier reports showing that HDACi treatment alone can lead to increased dendritic branching (Fischer et al., 2007). Therefore, the structural changes here are most likely the combined result of extinction training together with the HDACi application.
By extension, our finding of regained neuroplasticity strongly speaks in favor of a new period of learning during memory extinction (Bouton, 2004). Such learning has been postulated to either constitute a new memory trace of safety being laid over the original memory trace of fear or a re-learning of the original memory trace such that the association context-fear becomes re-associated toward one of context-safety (McKenzie and Eichenbaum, 2011; Nader and Hardt, 2009; Tronson and Taylor, 2007). It has been argued that, if the former is the case, the original memory of fear can still persist, which would become manifest in its SR, RI, or renewal, when the CS is presented outside of the extinction context (Bouton, 1993, 2004; Bouton and Bolles, 1979; Rescorla, 2004; Rescorla and Heth, 1975). Based on these purely behavioral results (Figures 4F and 4I) and the finding that the HDACi treatment in the absence of memory recall had no beneficial behavioral effect (Figures 4G and 4J), our results would therefore not only argue in favor of a permanent attenuation of the fear response, but also that it is the original memory trace that has been modified.
Experimental Procedures
All experiments were performed by a person unaware of treatment groups, whenever possible.
Animals
C57Bl/6 male mice were used for all experiments. Animals were 12–14 weeks old at the time of training. All animal work was performed in accordance with the guidelines of the Massachusetts Institute of Technology's Division of Comparative Medicine.
Behavior
Fear Conditioning
Freezing behavior was defined as the complete absence of movement except breathing.
Contextual Fear Conditioning and Extinction
Training consisted of a 3 min habituation of mice to the conditioning chamber (TSE systems) followed by three 2 s foot shocks (0.8 mA) with an intertrial interval (ITI) of 28 s. After the shocks, the animals remained in the chamber for an additional 15 s. 1 or 30 days later, the following extinction paradigms were used. For massed extinction, mice were re-exposed to the same chamber for 3 min without receiving the foot shock to recall the memory (expo.). One hour later (spent in the home cage), the animals were put back in the same chamber without shock for an additional 18 min (for a total of 6 × 3 min exposure), the last 3 min of which were employed to measure the animals' extinction memory (after extinction). For spaced extinction, mice were re-exposed to the same chamber for 3 min without receiving the foot shock to recall the memory and were returned to their home cage for 2 hr, after which they were once again exposed to the training chamber for 3 min. This procedure was repeated on three subsequent days, for a total of 4 days of spaced extinction. The second session of the fourth day was employed to measure freezing at the end of extinction (after extinction). For both paradigms, 24 hr after extinction, animals were exposed for 3 min to the context again to assess their 24 hr extinction memory (EM).
Spontaneous Recovery
Thirty days following massed or spaced extinction, mice were tested for spontaneous recovery of fear by exposing them to the training chamber for 3 min. See Extended Experimental Procedures for cued fear conditioning and extinction.
Open-Field Behavior
Open-field behavior was examined using the VersaMax system (Accuscan) during 20 min.
Experimental Manipulations
Drug Administration
L-NAME (10 mg/kg, Sigma), molsidomine (20 mg/kg, Sigma), and CI-994 (30 mg/kg, synthesized at the Broad Institute) was given intraperitoneally. CI-994 was synthesized at the Broad Institute with a purity of >95% by HPLC analysis.
Viral Administration
Mouse HDAC2WT or HDAC2C262/274A cDNAs were subcloned under the HSV IE 4/5 promoter into a short-term HSV p1005 vector that coexpresses GFP driven by a CMV promoter. 1 μl of high-titer (4 × 108 particles/ml) HSVs were stereotaxically introduced into both hemispheres of hippocampal area CA1 (see Extended Experimental Procedures).
Immunohistochemistry
Coronal brain slices (40 μm thickness) were incubated with AcH3K9/14 (Millipore), cFos (Santa Cruz), synaptophysin, MAP2 (Sigma-Aldrich), and GFP (Aves Labs) and were visualized (LSM 510, Zeiss) with fluorescently conjugated secondary antibodies (Molecular Probes). Images were quantified using ImageJ 1.42q (see Extended Experimental Procedures for additional details).
Chromatin Immunoprecipitation
Whole hippocampi (1 hemisphere) were crosslinked using formaldehyde, following which chromatin immunoprecipitation was performed using standard procedures with HDAC2 (Abcam) and AcH3K9/14 (Millipore) antibodies. The fluorescent signal of the amplified DNA (SYBR green, Biorad) was normalized to input with promoter-specific primers (Extended Experimental Procedures).
Detection of Protein S-Nitrosylation
To determine HDAC2 S-nitrosylation, a biotin-switch assay (Cayman Chemical Company), which visualizes nitrosylated cysteine residues, was performed essentially as per the manufacturer's instructions (Extended Experimental Procedures).
HDAC Activity Assay
HDAC2 activity was measured by a Fluor-de-Lys fluorometric enzymatic assay (Enzo Life Sciences; see Extended Experimental Procedures). HDAC2 activity was normalized to total HDAC2 protein levels as determined by western blotting with an HDAC2 (Abcam) antibody.
Gene Expression Analyses
For RNA sequencing, total mRNA was extracted (QIAGEN), quality controlled (Bioanalyzer, Agilent), prepared for sequencing (Illumina) as per the manufacturers' instructions, and sequenced at high-throughput (Illumina HiSeq 2000 platform). Three biological replicates per condition were used. Sequence reads were aligned and quality controlled (see Extended Experimental Procedures). A gene was considered differentially expressed with a fold change of ≥ 1.4 and a significance of p ≤ 0.05. For gene ontology analyses (http://www.geneontology.org), only biological pathways from hierarchical level 7 were compared with each other. Statistical tests were performed using Student's t test and the Benjamini-Hochberg false discovery rate to account for multiple comparisons. qRT-PCR was performed using standard procedures with exon-specific primers (Extended Experimental Procedures).
[3H]-2DG Autoradiography
Immediately after the last extinction session, animals were injected with 2 μCi/kg of radioactive 2-deoxyglucose ([3H]-2DG, PerkinElmer) and were returned to their home cage. One hour postinjection, animals were sacrificed using cervical dislocation and their brain dissected in 2-methylbutane (Sigma), stored at −80°C, and coronally cut at 30 μm thickness using a cryostat (Leica). Slides were air dried, made conductive by coating the free side with a copper foil tape, and placed into a gaseous chamber containing a mixture of argon and triethylamine (Sigma-Aldrich) as part of the Beta Imager 2000Z Digital Beta Imaging System (Biospace). Exposure for 20 hr yielded high-quality images. A [3H]-Microscale (American Radiolabeled Chemicals) was counted simultaneously as a reference for total radioactivity quantitative analysis. Quantitative analysis was performed with the program Beta-Vision Plus (BioSpace) for each anatomical region of interest. At least six brain sections of the same rostrocaudal position were used to obtain an average [3H]-2DG uptake per mouse.
Electrophysiology
To record field excitatory postsynaptic potentials, transverse hippocampal slices were prepared using standard procedures (Extended Experimental Procedures), and CA1 field potentials evoked by Schaffer collateral stimulation were measured. After recording of a stable baseline (at least 20 min), long-term potentiation was induced by three episodes of theta-burst stimulation (TBS) with 10 s intervals. TBS consisted of ten bursts (each with four pulses at 100 Hz) of stimuli delivered every 200 ms. Quantification was carried out with the average slopes of fEPSP during the last 10 min of recording.
Golgi Staining
Mice were perfused with 10% paraformaldehyde, and their entire brains were Golgi-Cox stained using the Rapid Golgistain Kit (FD NeuroTechnologies) as per the manufacturer's instructions (see also Extended Experimental Procedures). Mushroom spines were defined as having a clear round stubby head. An experimenter blind to treatment group counted the number and defined the types of apical and basal spines on hippocampal CA1 pyramidal neurons.
Sholl Analysis
z stack images from Golgi-Cox stained and isolated pyramidal neurons were acquired with 1 μm steps using a Zeiss LSM510 confocal microscope. Sholl analysis was performed by drawing concentric equidistant (10 μm) circles around the neuronal soma using Adobe Illustrator CS5 and by counting the number of intersecting branches per circle. Per condition, 8–12 neurons from 3 animals were analyzed.
Electron Microscopy
Electron microscopy on hippocampal brain sections was performed as described previously (Graff et al., 2013). Fifteen distinct apical regions (stratum radiatum) of CA1 ∼50-100 uM from the cell body layer of the hippocampus were imaged per animal. A synapse was defined as an electron dense postsynaptic density area juxtaposed to a presynaptic terminal filled with synaptic vesicles.
Statistics
Statistical analyses were performed using GraphPad Prism 5. Repeated-measurements ANOVAs followed by Tukey's posthoc analyses or by one-tailed Student's t tests were used. All data are represented as mean ± SEM. Statistical significance was set at p ≤ 0.05.
Supplementary Material
Acknowledgments
We thank MIT's BioMicroCenter for the RNA sequencing, Martin Kahn for help with the qRT-PCR and IHC experiments, and Theo Elfers for critical comments on the manuscript. This work was partially supported by NIH NINDS/NIA (NS078839) to L.-H.T. and NIH/NIDA (R01DA028301) to S.J.H., the Picower Neurological Disorder Fund (PNDRF), the Stanley Medical Foundation, HHMI, and a Bard Richmond fellowship to J.G.
Footnotes
Supplemental Information: Supplemental Information includes Extended Experimental Procedures, six figures, and one table and can be found with this article online at http://dx. doi.org/10.1016/j.cell.2013.12.020.
Accession Numbers: The RNA sequencing data has been deposited in the GEO Data Bank under ID code GSE53794.
References
- Agis-Balboa RC, Arcos-Diaz D, Wittnam J, Govindarajan N, Blom K, Burkhardt S, Haladyniak U, Agbemenyah HY, Zovoilis A, Salinas-Riester G, et al. A hippocampal insulin-growth factor 2 pathway regulates the extinction of fear memories. EMBO J. 2011;30:4071–4083. doi: 10.1038/emboj.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontempi B, Laurent-Demir C, Destrade C, Jaffard R. Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature. 1999;400:671–675. doi: 10.1038/23270. [DOI] [PubMed] [Google Scholar]
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull. 1993;114:80–99. doi: 10.1037/0033-2909.114.1.80. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- Bouton ME, Bolles RC. Role of conditioned contextual stimuli in reinstatement of extinguished fear. J Exp Psychol Anim Behav Process. 1979;5:368–378. doi: 10.1037//0097-7403.5.4.368. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell JE, Allis CD. Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Curr Opin Genet Dev. 1996;6:176–184. doi: 10.1016/s0959-437x(96)80048-7. [DOI] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi M, Cannas S, Saraulli D, Rossi-Arnaud C, Cestari V. Extinction after retrieval: effects on the associative and nonassociative components of remote contextual fear memory. Learn Mem. 2011;18:508–518. doi: 10.1101/lm.2175811. [DOI] [PubMed] [Google Scholar]
- Cukor J, Olden M, Lee F, Difede J. Evidence-based treatments for PTSD, new directions, and special challenges. Ann N Y Acad Sci. 2010;1208:82–89. doi: 10.1111/j.1749-6632.2010.05793.x. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Foa EB. Psychosocial treatment of posttraumatic stress disorder. J Clin Psychiatry. 2000;61(Suppl 5):43–48. discussion 49-51. [PubMed] [Google Scholar]
- Foa EB, Kozak MJ. Emotional processing of fear: exposure to corrective information. Psychol Bull. 1986;99:20–35. [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science. 2004;304:881–883. doi: 10.1126/science.1094804. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Ding HK, Takahashi E, Suzuki A, Kida S, Silva AJ. Stability of recent and remote contextual fear memory. Learn Mem. 2006;13:451–457. doi: 10.1101/lm.183406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Morinobu S, Takei S, Fuchikami M, Matsumoto T, Yamamoto S, Yamawaki S. Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J Psychiatr Res. 2012;46:635–643. doi: 10.1016/j.jpsychires.2012.01.026. [DOI] [PubMed] [Google Scholar]
- Goshen I, Brodsky M, Prakash R, Wallace J, Gradinaru V, Ramakrishnan C, Deisseroth K. Dynamics of retrieval strategies for remote memories. Cell. 2011;147:678–689. doi: 10.1016/j.cell.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Gräff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91:603–649. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- Gräff J, Kahn M, Samiei A, Gao J, Ota KT, Rei D, Tsai LH. A dietary regimen of caloric restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J Neurosci. 2013;33:8951–8960. doi: 10.1523/JNEUROSCI.5657-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harooni HE, Naghdi N, Sepehri H, Rohani AH. The role of hippocampal nitric oxide (NO) on learning and immediate, short- and long-term memory retrieval in inhibitory avoidance task in male adult rats. Behav Brain Res. 2009;201:166–172. doi: 10.1016/j.bbr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35:136–146. doi: 10.1038/npp.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Inda MC, Muravieva EV, Alberini CM. Memory retrieval and the passage of time: from reconsolidation and strengthening to extinction. J Neurosci. 2011;31:1635–1643. doi: 10.1523/JNEUROSCI.4736-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak Y, Anderson KL, Kelley JB, Petkov M. Histone acetylation rescues contextual fear conditioning in nNOS KO mice and accelerates extinction of cued fear conditioning in wild type mice. Neurobiol Learn Mem. 2012;97:409–417. doi: 10.1016/j.nlm.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ. Principles of Neural Science. New York: McGraw Hill; 2013. [Google Scholar]
- Kaplan GB, Moore KA. The use of cognitive enhancers in animal models of fear extinction. Pharmacol Biochem Behav. 2011;99:217–228. doi: 10.1016/j.pbb.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Kearns MC, Ressler KJ, Zatzick D, Rothbaum BO. Early interventions for PTSD: a review. Depress Anxiety. 2012;29:833–842. doi: 10.1002/da.21997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Korb E, Finkbeiner S. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci. 2011;34:591–598. doi: 10.1016/j.tins.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Wood MA. Epigenetics and persistent memory: implications for reconsolidation and silent extinction beyond the zero. Nat Neurosci. 2013;16:124–129. doi: 10.1038/nn.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008;455:1198–1204. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. 2012;35:24–35. doi: 10.1016/j.tins.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie S, Eichenbaum H. Consolidation and reconsolidation: two lives of memories? Neuron. 2011;71:224–233. doi: 10.1016/j.neuron.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milekic MH, Alberini CM. Temporally graded requirement for protein synthesis following memory reactivation. Neuron. 2002;36:521–525. doi: 10.1016/s0896-6273(02)00976-5. [DOI] [PubMed] [Google Scholar]
- Misanin JR, Miller RR, Lewis DJ. Retrograde amnesia produced by electroconvulsive shock after reactivation of a consolidated memory trace. Science. 1968;160:554–555. doi: 10.1126/science.160.3827.554. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel L, Moscovitch M. Memory consolidation, retrograde amnesia and the hippocampal complex. Curr Opin Neurobiol. 1997;7:217–227. doi: 10.1016/s0959-4388(97)80010-4. [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O. A single standard for memory: the case for reconsolidation. Nat Rev Neurosci. 2009;10:224–234. doi: 10.1038/nrn2590. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Neve RL, Lim F. Generation of high-titer defective HSV-1 vectors. Curr Protoc Neurosci. 2013;Chapter 4(Unit 13) doi: 10.1002/0471142301.ns0413s62. [DOI] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- Pavlov I. Conditioned reflexes. Oxford, UK: Oxford University Press; 1927. [Google Scholar]
- Ramamoorthi K, Fropf R, Belfort GM, Fitzmaurice HL, McKinney RM, Neve RL, Otto T, Lin Y. Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science. 2011;334:1669–1675. doi: 10.1126/science.1208049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA. Spontaneous recovery. Learn Mem. 2004;11:501–509. doi: 10.1101/lm.77504. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Wang X, Schrick C, Neve R, Radulovic J, Tsai LH. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat Neurosci. 2007;10:1012–1019. doi: 10.1038/nn1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D, Monfils MH, Raio CM, Johnson DC, Ledoux JE, Phelps EA. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature. 2010;463:49–53. doi: 10.1038/nature08637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry. 2012;72:25–33. doi: 10.1016/j.biopsych.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischmeyer W, Grimm R. Activation of immediate early genes and memory formation. Cell Mol Life Sci. 1999;55:564–574. doi: 10.1007/s000180050315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- Urcelay GP, Wheeler DS, Miller RR. Spacing extinction trials alleviates renewal and spontaneous recovery. Learn Behav. 2009;37:60–73. doi: 10.3758/LB.37.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Lin JT, Chien CL, Chang WC, Lai HL, Chang CP, Chern Y. Type VI adenylyl cyclase regulates neurite extension by binding to Snapin and Snap25. Mol Cell Biol. 2011;31:4874–4886. doi: 10.1128/MCB.05593-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, Zhu WL, Ding ZB, Bao YP, Shi J, et al. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science. 2012;336:241–245. doi: 10.1126/science.1215070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.