

Abstract
Background/Aim:
We aimed to assess the influence of Helicobacter pylori and its virulent factors, cytotoxin associated gene (cag) A and E, on portal hypertensive gastropathy (PHG) and the levels of interleukin (IL)-8, IL-10, and tumor necrosis factor-alpha (TNF-α).
Patients and Methods:
The patients with cirrhosis underwent screening endoscopy and the lesions related to PHG were graded. Biopsies were obtained for histology, and polymerase chain reaction (PCR) of H. pylori 16S rRNA, cagA, cagE, and tissue cytokine levels was carried out. Absent or mild PHG was compared with moderate to severe PHG.
Results:
One hundred and forty patients with cirrhosis were studied; males numbered 92 and the mean age of the patients was 50.3 ± 12.0 years, H. pylori positivity in 87 (62.1%) patients was associated with male gender (P = 0.032), younger age (P = 0.029), hepatitis D etiology (P = 0.005), higher serum albumin (0.000), lower Child Pugh score (P = 0.001), and lower portal vein diameter (P = 0.001). There was no significant difference in the levels of TNF-α and IL-8. However, a decrease in the anti-inflammatory cytokine IL-10 was noted with moderate to severe gastropathy. Four H. pylori strains were positive for both cagA and cagE, while four were positive for cagA only. All the four patients with both virulent factors had mild gastropathy only.
Conclusion:
The presence of H. pylori infection neither affected the severity of PHG nor augmented the IL-8 and TNF-α levels. There was a decline of virulent H. pylori strains and IL-10 levels in patients with advanced PHG.
Keywords: Cytokines, Helicobacter pylori, portal hypertension, virulence factors
Portal hypertensive gastropathy (PHG) results from increased resistance to portal blood flow in patients with liver disease (with and without cirrhosis) and the consequent elevation in portal pressure. Elevated portal pressure induces local hemodynamic changes, thus causing congestion in the stomach and gastric tissue damage.[1,2,3] However, the existence of any correlation of the severity of PHG with the degree of portal pressure, hyperkinetic state, and hepatocellular dysfunction is controversial.[4,5,6,7,8] There may be other factors involved in its physiopathology, including local factors connected with the gastric mucosa, alterations in local blood flow, nitric oxide, endothelins, or growth factors, as yet unidentified factors.[4]
In cirrhotic patients with portal gastropathy, there is activation of cytokines and growth factors, such as interleukin (IL)-6 and tumor necrosis factor-alpha (TNF-α).[9] TNF-α activates endothelial constitutive nitric oxide synthase and endothelin-1 in the portal hypertensive gastric mucosa.[3] Overexpressed nitric oxide synthase produces an excess of nitric oxide, which induces hyperdynamic circulation and peroxynitrite overproduction. Overexpression of peroxynitrite together with endothelin-1 affects gastric mucosal defenses, which are impaired in portal hypertension.
Helicobacter pylori is a major etiological factor of peptic ulcer disease (PUD). It is supposed to be a risk factor for the more frequently encountered PUD in patients with liver cirrhosis.[10] The prevalence of H. pylori in cirrhotics is similar to that in controls, and investigators have evaluated the effect of H. pylori on liver cirrhosis and PHG and have obtained controversial results.[11,12] Colonization of the gastric mucosa by H. pylori might have an indirect role in PHG as colonization is, at least theoretically, associated with inflammation. H. pylori virulence factors induce proinflammatory cytokines such as IL-1, IL-8, and TNF-α, which influence mucosal inflammation and/or gastric acid secretion.[13]
The aim of this study was to find if there is any correlation of H. pylori and its virulence factors, cytotoxin-associated gene (cag) A and E, with PHG severity as graded by Baveno classification and to assess the expression of the proinflammatory cytokines, IL-8 and TNF-α, and anti-inflammatory cytokine, IL-10, in patients with or without H. pylori infection.
PATIENTS AND METHODS
Patients
Consecutive patients with cirrhosis, who referred to the endoscopy unit of the Aga Khan University Hospital, were enrolled. The study was approved by the Ethics Review Committee of the hospital. Upper gastrointestinal (GI) endoscopy was performed as a part of the assessment of portal hypertension. Informed consent was obtained from the patients for participation in the study and for gastric biopsies from the antrum. Diagnosis of cirrhosis was based on clinical, biochemical, and radiological findings or on liver biopsy that was done previously. The severity of cirrhosis was assessed using the Child Pugh classification.[14] The etiology of the cirrhosis was defined as viral when hepatitis B surface antigen (HBsAg) or antibodies to hepatitis C virus (HCV) were present or alcoholic if there was a daily ethanol intake of >60 g/day for at least 5 years. Diagnosis of other less common diseases, such as non-alcoholic fatty liver disease, autoimmune liver disease, and primary biliary cirrhosis, was based on the latest diagnostic criteria.
Inclusion and exclusion criteria
The inclusion criteria for selection of the study population included patients undergoing screening endoscopy for varices, with or without history of previous upper GI bleeding. The exclusion criteria included the following: History of previous band ligation or sclerotherapy for esophageal varices; patients on beta blockers, nitrates, nonsteroidal anti-inflammatory drugs, aspirin, proton pump inhibitors, or histamine 2 receptor antagonists; pregnant and lactating females; patients with inflammatory bowel disease, celiac disease, or other systemic diseases affecting the GI tract; patients on antibiotics within last 2 weeks; patients with any ongoing infection; patients with ongoing ingestion of alcohol; patients with gastric neoplasia; patients who had undergone surgery for the correction of portal hypertension, gastrectomies, and/or vagotomies; patients with thrombosis of the portal or suprahepatic veins or inferior vena cava; patients not willing to participate; and patients with prothrombin time greater than 20 s.
Endoscopic examination
All patients had upper GI endoscopy (GIF 180, Olympus Tokyo, Japan) performed by a single experienced endoscopist to prevent interobserver variability. Sterilized biopsy forceps were used to obtain gastric biopsy specimens from the antrum. Two biopsy specimens were removed for histology and dispatched in a formalin-containing container. Gastric biopsy specimens obtained for RNA extraction were collected in an eppendorf containing TRIzol Reagent (Invitrogen Corporation, Carlsbad, California, USA) and stored in a liquid nitrogen container for transport to the laboratory and stored at −70°C for further use. The proforma section related to the endoscopic findings was filled by the assistant during the procedure.
Grading of portal hypertensive gastropathy
Baveno classification was used to classify portal gastropathy.[15,16] Mosaic pattern was defined as small polygonal areas demarcated by a distinct white to yellow border and with or without a slight central bulge, which had a mosaic, fish scale-like appearance upon endoscopy. The lesions were graded as were graded as mild (if the color of the mucosa was pink) and severe when diffuse erythema (redness) of the mucosa was present [Table 1]. Red markings defined as flat or slightly bulging red lesions were seen in the gastric mucosa. Such lesions included fine punctate hemorrhagic spots and discrete cherry red spots. The lesions were graded as (1) isolated discrete spots and cherry red spots and (2) when confluent areas of submucosal hemorrhage. Presence of flat or slightly raised red stripe-like lesions radiating from the pylorus to the antrum and the body of the stomach for a variable distance was described as gastric antral vascular ectasia (GAVE). Black and brown spots due to old submucosal hemorrhage were not scored. Presence of oozing of blood from gastropathy was also noted. Patients of absent to mild gastropathy (score 0-2) were compared with those of moderate to severe gastropathy (score 3-6).
Table 1.
Baveno scoring system for endoscopic appearance in portal hypertensive gastropathy

Documentation of varices
Esophageal varices were classified as small or large, where small esophageal varices were defined as varices that flattened with insufflations or minimally protruded into the esophageal lumen, while large esophageal varices were defined as varices that protruded into the esophageal lumen and touched each other (presence of confluence) or that filled at least 50% of the esophageal lumen. Gastric varices were documented according to the location, as described by Sarin et al.[17]
Assessment of portal hypertension and liver dysfunction
Noninvasive methods like portal vein diameter, splenic size on ultrasound, platelet count, and aspartate aminotransferase to platelet ratio index (APRI) and platelet spleen diameter ratio were used to assess portal hypertension. Severity of liver dysfunction was judged by Child Pugh score and Model for End-stage Liver Disease (MELD) score.
Diagnosis of H. pylori infection
Diagnosis of H. pylori infection was based on histopathology of gastric biopsy and polymerase chain reaction (PCR) for H. pylori 16S rDNA.[18]
Histology
Gastric biopsy specimens were fixed immediately in 10% formalin in sodium phosphate buffer (4 g/L monobasic and 6.5 g/L dibasic; Fisher, Morris Plains, NJ, USA) and sent to the Department of Pathology for processing. Biopsies were embedded in paraffin, and histological sections were stained with hematoxylin and eosin, and Giemsa for evaluation.
Extraction of DNA from tissue
DNA extraction from gastric biopsy tissue was carried out as described before.[18] Briefly, gastric tissue was homogenized to uniformity in 500 μl of sterile water and centrifuged at 12,000 × g for 3 min. Five hundred microliters of lysis buffer [100 mM NaCl, 10 mM Tris-HCl (pH 8.0), 25 mM ethylenediaminetetraacetic acid (EDTA), 0.5% sodium dodecyl sulfate] and 10 μl of proteinase K (10 mg/ml) were added. Incubation was carried out at 56°C for 20 hours; this was followed by phenol-chloroform extraction and ethanol precipitation. The resulting pellet was allowed to dissolve in 40 μl of TE buffer [10 mM Tris-HCl (pH 7.4) and 0.1 mM EDTA (pH 8.0)] for 20 hours at 37°C. Samples were stored at −20°C before PCR amplification was performed. DNA content and purity was determined by measuring the absorbance at 260 nm and 280 nm, respectively, using a spectrophotometer (Beckman DU-600, Brea, California, USA).
PCR for H. pylori 16S rDNA, cagA, and cagE
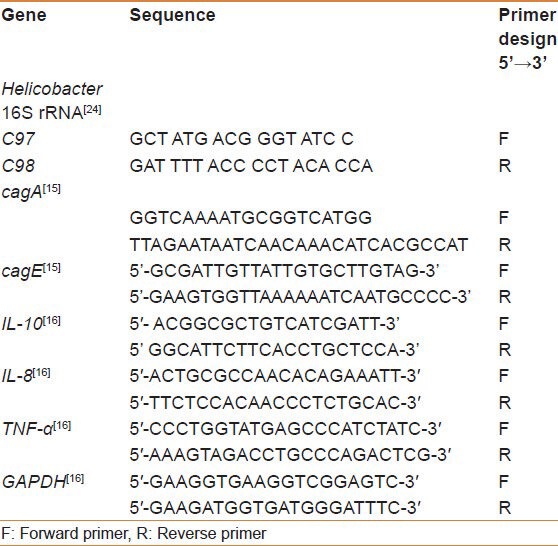
PCR was performed using extracted DNA as the template. Helicobacter genus specific 16S rRNA, cagA, and cagE primers[19] [Table 2] were used to amplify the respective gene products.[18,19] PCR amplification was carried out in a total volume of 50 μl containing 2 μl of 2 mM deoxyribonucleotide triphosphates (dNTPs), 1 μl containing 50 pmol of primer 1, 1 μl containing 50 pmol of primer 2 (synthesized by Applied Biosystems, Inc, Foster automatic synthesizer California, USA), 1 unit of Taq DNA polymerase (Promega Corporation, Madison, Wisconsin,USA), 5 μl of 10× PCR reaction buffer, 3 mM of MgCl2, and 2 μl of DNA template containing 0.5 ng of extracted DNA, and the total volume was rounded to 50 μl with double distilled water. The reaction was carried out in a Perkin Elmer 9700 thermal cycler. The amplification cycle consisted of an initial denaturation of target DNA at 94°C for 5 min and then denaturation at 94°C for 1 min, primer annealing at 56°C for 1 min, and extension at 72°C for 1 min. The final cycle included an extension step at 72°C for 5 min to ensure full extension of the product. Samples were amplified through 35 consecutive cycles. Negative reagent control reactions were performed with each batch of amplifications, consisting of tubes containing distilled water in place of the DNA samples. Five microliters of PCR product was electrophoresed on a 2% agarose gel to ensure homogeneity and yield. PCR amplification resulted in a homogeneous DNA fragment of the expected size of 400 bp for 16S rDNA of H. pylori.
Table 2.
Primer sequence
Tissue cytokines using real-time quantitative polymerase chain reaction with SBG
Total RNA was extracted from the endoscopic biopsy samples of gastric mucosa with TRIzol method described previously.[20] Reverse transcription of the extracted RNA was performed using RNase H–deficient reverse transcriptase (Superscript II; Life Technologies New York, USA) and oligo (dT) primers (Life Technologies). Aliquots (2 μl) of reverse transcription reaction mixture (20 μl) were used for quantification of IL-8, IL-10, TNF-α, and GAPDH Glyceraldehyde-3-Phosphate Dehydrogenase (GAPGH) gene expression by real-time PCR assays [Table 2]. The SYBR Green based quantitative real time polymerase chain reaction (SABG QRT- PCR) was used to quantify IL-8, IL-10, TNF-α, and GAPDH gene expression (PE Applied Biosystems, Foster City, CA, USA). The PCR reactions were performed using the SYBR Green (SBG) PCR kit (PE Applied Biosystems). After activation for 10 min at 95°C, 40 cycles of 15 s at 95°C and 1 min at 62°C were carried out in model icycler (Biorad Hercules, CA, USA). Real-time fluorescence measurements were recorded and the threshold cycle (Ct) value for each sample was calculated by the above sequence detector. For TNF-α and GAPDH, standard curves of Ct values were obtained from real-time PCR of pMFGhTNF and PCRII GAPDH (reference plasmids). Ct values for IL-8, IL-10, TNF-α, and GAPDH transcripts from clinical specimens were plotted on the standard curves, and the amounts (in picograms) of each transcript were calculated. The amounts of IL-8, IL-10, and TNF-α transcripts (pg) were expressed relative to that of GAPDH (pg). Each biopsy sample obtained from the same patient was tested in duplicate, and the average of two Ct values was used in this study.
Statistical analysis
The sample size was calculated by A-priori sample size calculator for studies using two-tailed Student's t values for comparison (http://www.danielsoper.com/statcalc). A sample of 128 analyzable subjects provides an 80% power at the 0.05 alpha level. So, the initial plan was to include 64 H. pylori positive and 64 negative patients. The Statistical Package for Social Science (SPSS, Release 19) was used for data analysis and Prism GraphPad (version 5) for graphics. The descriptive analysis was done for demographic and clinical features. Results were presented as a mean ± standard deviation for quantitative variables and number (percentage) for qualitative variables. Independent sample Student's t-test was used for continuous variables. Homogeneity of variances was assessed by Levene test and the corrected P value was used. Chi-squared test or Fisher's exact test was used for categorical variables. Factors found to be statistically significant in univariate analysis were included in a multivariate regression analysis. All P values were two sided and the significance level was defined as P < 0.05.
RESULTS
One hundred and forty patients with cirrhosis were studied, with males numbering 92, and the mean age of the patients was 50.3 ± 12.0 years. PHG was graded as negative in 16 (11.4%), mild in 48 (34.3%), moderate in 44 (31.4%), and severe in 32 (22.9%) patients. H. pylori was present in 87 (62.1%) of the patients. The presence of H. pylori was associated with male gender (P = 0.032), younger age (P = 0.029), hepatitis D etiology (P = 0.005), higher serum albumin (P = 0.000), lower Child Pugh score (P = 0.001), and lower portal vein diameter (P = 0.001). There were no significant differences in the levels of TNF-α, IL-8, and IL-10 in biopsies with or without H. pylori [Table 3 and Figure 1].
Table 3.
Comparison of Helicobacter pylori positive and negative patients with cirrhosis of liver
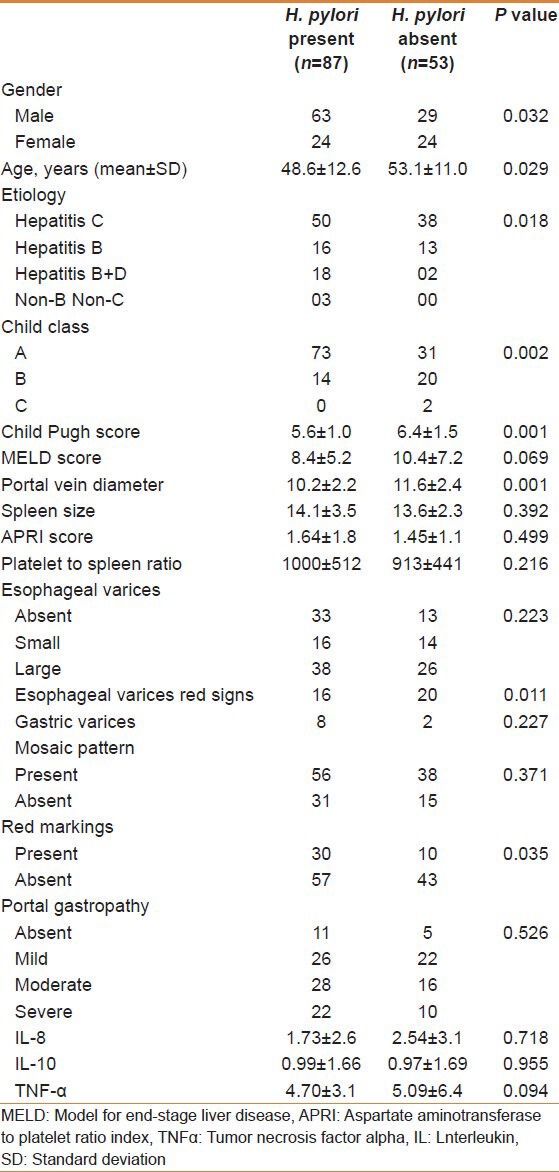
Figure 1.
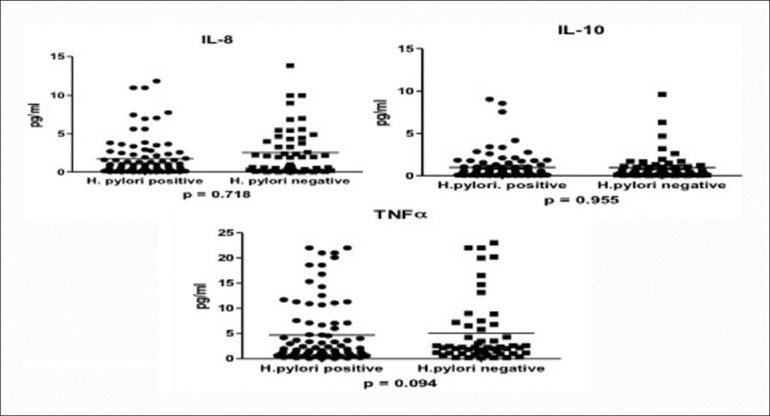
Interleukin 8, interleukin 10, and tumor necrosis factor alpha levels in Helicobacter pylori positive and negative patients
Moderate to severe gastropathy (n = 76) was associated more with male gender (P = 0.030), hepatitis B etiology (P = 0.020), larger spleen diameter (P = 0.000), greater APRI score (P = 0.021), lesser platelet to spleen ratio (P = 0.000), and presence of varices (P = 0.000) and gastric varices (P = 0.002). No effects of age, Child Pugh score, MELD score, and presence of H. pylori on the severity of PHG could be found. There was no significant difference in the levels of TNF-α and IL-8. However, a decrease in the anti-inflammatory cytokine IL-10 was noted with moderate to severe gastropathy [Table 4 and Figure 2]. Regression analysis showed that male gender and presence of esophageal and gastric varices were independently associated with moderate to severe PHG (P = 0.046, 0.05, and 0.042 respectively).
Table 4.
Comparison of patients with absent to mild portal gastropathy and patients with moderate to severe gastropathy
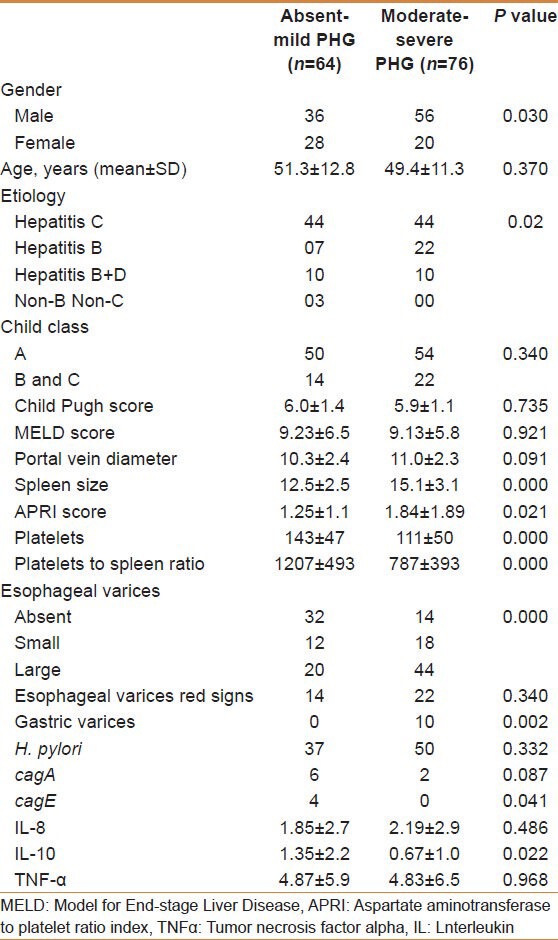
Figure 2.
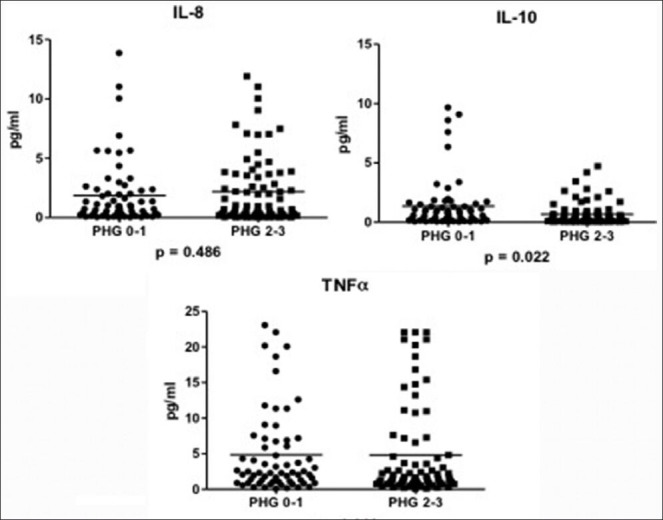
Interleukin 8, interleukin 10, and tumor necrosis factor alpha levels in absent to mild versus moderate to severe gastropathy
Four H. pylori strains were positive for both cagA and cagE, while four were positive for cagA only. All the four patients with both virulent factors had mild gastropathy only.
DISCUSSION
PHG can lead to chronic blood loss and transfusion-dependent anemia, as well as acute bleeding episodes. It is associated with dilation and tortuosity of the submucosal vessels, thinning of the vascular wall, and increased area of the gastric mucosa occupied by vessels.[21] It apparently results from elevated portal pressure which induces changes of local hemodynamics, thus causing congestion in the upper stomach and gastric tissue damage.
The relationship of the functional status of liver and the development of PHG is not straightforward and is controversial.[4,5,6,7,8] Altered vascular hemodynamics occurring in PHG may not be purely a local phenomenon of the stomach and may be a part of a generalized vascular abnormality of cirrhosis and portal hypertension.[6] The results of a study suggested that there were no statistically significant differences in the level of hepatic venous pressure gradient (HVPG) between patients with PHG and those without PHG and between patients with mild PHG and those with severe PHG.[4] Moreover, Child Pugh's classification, systemic vascular resistance index, and glucagon levels were also not different among the different grades of PHG.[4] Another study had a similar conclusion,[8] while a study conducted by Kim et al. concluded that PHG was associated with portal hypertension severity and prognosis in patients with cirrhosis.[5] In our study, the severity of PHG correlated with the variables known to reflect portal hypertension. However, we did not measure HVPG. We could not find any correlation of Child Pugh and MELD scores, which measure liver dysfunction, with the severity of PHG.
H. pylori is considered a risk factor for PUD in cirrhotic patients.[22] There is a significant increase in the inducible nitric oxide synthase (iNOS) production in patients with PHG. H. pylori also increases iNOS expression. However, there is no evidence of synergistic action between H. pylori and PHG on iNOS expression.[23] Patients with chronic liver disease showed higher risk of developing gastroduodenal lesions regardless of the presence of the H. pylori infection. Many studies failed to find a correlation between the severity of PHG and H. pylori infection.[24,25,26] During advanced gastropathy, the environment for the colonization of H. pylori becomes unfavorable, resulting in a decrease in the number of patients harboring this organism. In our study, the patients harboring H. pylori were of younger age, had higher serum albumin, lower Child Pugh score, and lower portal vein diameter. So, H. pylori associated changes in the mucosa are unlikely to contribute to the severity of PHG.
Previous studies have not addressed the issue of virulence factors and the role of cytokines in the presence or absence of H. pylori in the endoscopic severity of gastropathy. We found lower levels of IL-10 in the mucosa of patients with advanced PHG. IL-10 is an anti-inflammatory cytokine. We propose that IL-10 might have some protective role against the development of severe gastropathy, and as the levels decrease, gastropathy becomes severe. As discussed earlier, TNF-α may have a role in the pathogenesis of portal gastropathy. We did not compare its levels with those of non-cirrhotic controls. So, it is not possible to exactly find how much TNF-α level is higher in patients with PHG compared to non-cirrhotic controls. However, we could not find any significant difference in the levels of TNF-α and IL-8 in patients with none to mild versus moderate to severe gastropathy. Moreover, the H. pylori status did not influence the level of these cytokines. Another interesting finding was a decline in the H. pylori harboring virulent factors cagA and cagE in patients with moderate to severe gastropathy, which reflects a hostile microenvironment for H. pylori in patients with advanced PHG and no role of these virulence factors.
In summary, in our study, the severity of PHG did not correlate with the severity of liver disease as judged by Child Pugh and MELD scoring. The patients with moderate to severe PHG had larger spleen, larger esophageal varices, and gastric varices. The level of anti-inflammatory cytokine IL-10 was lower in advanced PHG, but the levels of proinflammatory cytokines TNF-α and IL-8 were not higher. The presence of H. pylori infection did not affect the severity of PHG and the levels of IL-8, IL-10, and TNF-α. H. pylori with virulent factors (e.g., cagA) was not associated with advanced PHG.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Ferraz JG, Wallace JL. Underlying mechanisms of portal hypertensive gastropathy. J Clin Gastroenterol. 1997;25:S73–8. doi: 10.1097/00004836-199700001-00012. [DOI] [PubMed] [Google Scholar]
- 2.Ohta M, Hashizume M, Higashi H, Ueno K, Tomikawa M, Kishihara F, et al. Portal and gastric mucosal hemodynamics in cirrhotic patients with portal hypertensive gastropathy. Hepatology. 1994;20:1432–6. doi: 10.1002/hep.1840200609. [DOI] [PubMed] [Google Scholar]
- 3.Ohta M, Yamaguchi S, Gotoh N, Tomikawa M. Pathogenesis of portal hypertensive gastropathy: A clinical and experimental review. Surgery. 2002;131:S165–70. doi: 10.1067/msy.2002.119499. [DOI] [PubMed] [Google Scholar]
- 4.Curvêlo LA, Brabosa W, Rhor R, Lanzoni V, Parise ER, Ferrari AP, et al. Underlying mechanism of portal hypertensive gastropathy in cirrhosis: A hemodynamic and morphological approach. J Gastroenterol Hepatol. 2009;24:1541–6. doi: 10.1111/j.1440-1746.2009.05871.x. [DOI] [PubMed] [Google Scholar]
- 5.Kim MY, Choi H, Baik SK, Yea CJ, Won CS, Byun JW, et al. Portal hypertensive gastropathy: Correlation with portal hypertension and prognosis in cirrhosis. Dig Dis Sci. 2010;55:3561–7. doi: 10.1007/s10620-010-1221-6. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Mishra SR, Sharma P, Sharma BC, Sarin SK. Clinical, laboratory, and hemodynamic parameters in portal hypertensive gastropathy: A study of 254 cirrhotics. J Clin Gastroenterol. 2010;44:294–300. doi: 10.1097/MCG.0b013e3181b37ea1. [DOI] [PubMed] [Google Scholar]
- 7.Merkel C, Schipilliti M, Bighin R, Bellini B, Angeli P, Bolognesi M, et al. Portal hypertension and portal hypertensive gastropathy in patients with liver cirrhosis: A haemodynamic study. Dig Liver Dis. 2003;35:269–74. doi: 10.1016/s1590-8658(03)00064-1. [DOI] [PubMed] [Google Scholar]
- 8.Bellis L, Nicodemo S, Galossi A, Guarisco R, Spilabotti L, Durola L, et al. Hepatic venous pressure gradient does not correlate with the presence and the severity of portal hypertensive gastropathy in patients with liver cirrhosis. J Gastrointestin Liver Dis. 2007;16:273–7. [PubMed] [Google Scholar]
- 9.Byl B, Roucloux I, Crusiaux A, Dupont E, Deviere J. Tumor necrosis factor alpha and interleukin 6 plasma levels in infected cirrhotic patients. Gastroenterology. 1993;104:1492–7. doi: 10.1016/0016-5085(93)90361-f. [DOI] [PubMed] [Google Scholar]
- 10.Dore MP, Mura D, Deledda S, Maragkoudakis E, Pironti A, Realdi G. Active peptic ulcer disease in patients with hepatitis C virus-related cirrhosis: The role of Helicobacter pylori infection and portal hypertensive gastropathy. Can J Gastroenterol. 2004;18:521–4. doi: 10.1155/2004/150674. [DOI] [PubMed] [Google Scholar]
- 11.Al Mofleh IA. Does Helicobacter pylori affect portal hypertensive gastropathy? Saudi J Gastroenterol. 2007;13:95–7. doi: 10.4103/1319-3767.32186. [DOI] [PubMed] [Google Scholar]
- 12.Urso G, Interlandi D, Puglisi M, Abate G, Bertino G, Raciti C, et al. Role of Helicobacter pylori in patients with portal hypertensive gastropathy by liver cirrhosis hepatitis C virus-related. Minerva Gastroenterol Dietol. 2006;52:303–8. [PubMed] [Google Scholar]
- 13.Patel MK, Trombly MI, Kurt-Jones EA. Innate immune responses to Helicobacter pylori infection: An overview. Methods Mol Biol. 2012;921:205–7. doi: 10.1007/978-1-62703-005-2_23. [DOI] [PubMed] [Google Scholar]
- 14.Child CG, Turcotte JG. Surgery and portal hypertension. In: Child CG, editor. The liver and portal hypertension. Philadelphia: Saunders; 1964. pp. 50–64. [Google Scholar]
- 15.De Franchis R. Evolving Consensus in Portal Hypertension Report of the Baveno IV Consensus Workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol. 2005;43:67–176. doi: 10.1016/j.jhep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Stewart CA, Sanyal AJ. Grading portal gastropathy: Validation of a gastropathy scoring system. Am J Gastroenterol. 2003;98:1758–65. doi: 10.1111/j.1572-0241.2003.07595.x. [DOI] [PubMed] [Google Scholar]
- 17.Sarin SK. Diagnostic issues: Portal hypertensive gastropathy and gastric varices. In: DeFranchis R, editor. Portal hypertension II. Proceedings of the second Baveno international consensus workshop on definitions, methodology and therapeutic strategies. Oxford: Blackwell Science; 1996. pp. 30–55. [Google Scholar]
- 18.Yakoob J, Abid S, Jafri W, Abbas Z, Islam M, Ahmad Z. Comparison of biopsy-based methods for the detection of Helicobacter pylori infection. Br J Biomed Sci. 2006;63:159–62. doi: 10.1080/09674845.2006.11732745. [DOI] [PubMed] [Google Scholar]
- 19.Yakoob J, Jafri W, Abbas Z, Abid S, Khan R, Jafri N, et al. Low prevalence of the intact cag pathogenicity island in clinical isolates of Helicobacter pylori in Karachi, Pakistan. Br J Biomed Sci. 2009;66:137–42. doi: 10.1080/09674845.2009.11730260. [DOI] [PubMed] [Google Scholar]
- 20.Tsukada Y, Nakamura T, Iimura M, Iizuka BE, Hayashi N. Cytokine profile in colonic mucosa of ulcerative colitis correlates with disease activity and response to granulocytapheresis. Am J Gastroenterol. 2002;97:2820–8. doi: 10.1111/j.1572-0241.2002.07029.x. [DOI] [PubMed] [Google Scholar]
- 21.McCormack TT, Sims J, Eyre-Brook I, Kennedy H, Goepel J, Johnson AG, et al. Gastric lesions in portal hypertension: Inflammatory gastritis or congestive gastropathy? Gut. 1985;26:1226–32. doi: 10.1136/gut.26.11.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vergara M, Calvet X, Roque M. Helicobacter pylori is a risk factor for peptic ulcer disease in cirrhotic patients. A meta-analysis. Eur J Gastroenterol Hepatol. 2002;14:717–22. doi: 10.1097/00042737-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Arafa UA, Fujiwara Y, Higuchi K, Shiba M, Uchida T, Watanabe T, et al. No additive effect between Helicobacter pylori infection and portal hypertensive gastropathy on inducible nitric oxide synthase expression in gastric mucosa of cirrhotic patients. Dig Dis Sci. 2003;48:162–8. doi: 10.1023/a:1021707103590. [DOI] [PubMed] [Google Scholar]
- 24.Pan WD, Xun RY, Chen YN. Correlation of portal hypertensive gastropathy of hepatitis B cirrhosis with other mfactors. Hepatobiliary Pancreat Dis Int. 2002;1:527–31. [PubMed] [Google Scholar]
- 25.Parikh SS, Desai SB, Prabhu SR, Trivedi MH, Shankaran K, Bhukhanwala FA, et al. Congestive gastropathy: Factors influencing development, endoscopic features, Helicobacter pylori infection and microvessel changes. Am J Gastroenterol. 1994;89:1036–42. [PubMed] [Google Scholar]
- 26.Bahnacy A, Kupcsulik P, Elés ZS, Jàray B, Flautner L. Helicobacter pylori and congestive gastropathy. Z Gastroenterol. 1997;35:109–12. [PubMed] [Google Scholar]