

Abstract
We report a fatal case of haemolytic crisis mimicking a pulmonary embolism in a previously healthy 42-year-old African man. The patient was admitted to hospital with fatigue, shortness of breath and jaundice lasting for 2 days. Laboratory tests were consistent with haemolysis and inflammation. The patient was treated as having a mycoplasma pneumonia. His condition deteriorated rapidly, with respiratory distress and circulatory failure. Echocardiography showed pulmonary hypertension and right heart dilation. Despite the fact that he was given fibrinolysis for suspected pulmonary embolism, he developed cardiac arrest and died after a long-lasting resuscitation attempt. Postmortem examinations revealed that the patient had a glucose-6-phosphate dehydrogenase deficiency and disseminated intravascular coagulation with pulmonary microthrombi. To the best of our knowledge, this is the first case of death caused by right heart failure due to microvascular obstruction resulting from multiple microvascular thrombosis in a patient with acute haemolysis due to glucose-6-phosphate dehydrogenase deficiency.
Background
Glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency) is the most prevalent of human enzyme deficiencies, affecting approximately 400 million people worldwide. Typically, the condition remains clinically silent, and only a small minority, with very low residual enzyme levels, exhibit signs of chronic haemolytic anaemia. However, all affected individuals are at risk of developing non-immune-mediated acute haemolytic attacks after exposure to oxidative stressors such as certain drugs, infections or classically after ingestion of fava beans. The latter explains why this deficiency is also known as favism. In otherwise healthy individuals, the typical course of a haemolytic attack is benign, and complete recovery is the rule.1 2 We describe a fatal case of acute haemolysis in a formerly healthy young male of Nigerian origin, who rapidly developed respiratory and circulatory failure, mimicking a pulmonary embolism. The suspicion of G6PD deficiency was confirmed postmortem, and autopsy showed pulmonary microvascular thrombotic obstruction.
Case presentation
A 42-year-old African man was admitted to a secondary care hospital after 2 days of malaise, shortness of breath and a mid-thoracic burning pain. On the day prior to admission, his wife had noticed that he had yellow sclera, and on the day of admission he experienced a syncopal episode. The man was of Nigerian origin, but had been living in Scandinavia for the past 8 years. During that period, he had visited Africa only once, which was 4 years ago. He had no medical history, and he did not take any medication.
Physical examination showed jaundice with yellow sclerae and dark urine. He was tender in the liver region, and appeared slightly confused with a Glasgow Coma Scale of 11. Vital signs included a blood pressure of 115/79 mm Hg, a regular pulse rate of 115 bpm, a peripheral saturation of 92% breathing room air, a respiratory rate of 24 breaths/min and a body temperature of 38°C. Laboratory tests were consistent with haemolytic anaemia, mild renal insufficiency and inflammation, as outlined in table 1. The ECG showed sinus tachycardia (figure 1A). A chest X-ray (figure 2A, B) and abdominal ultrasound revealed no abnormalities. The arterial blood sample showed hyperventilation due to hypoxia (pH in 7.44, PaO2 5.5 kPa, PaCO2 3.9 kPa, base excess −2.3 and lactate 2.5 mmol/L). The diagnosis was unclear, but the patient was suspected to have an undetected infection, and an antibiotic treatment with piperacillin/tazobactam and intravenous fluids was initiated. Owing to unresolved haemolysis, he was transferred to the haematology unit at the tertiary care university hospital. During interhospital transfer, his respiratory condition deteriorated. He became hypoxic with a peripheral saturation of 88% and the tachypnoea worsened, reaching 39 breaths/min. He had nasal oxygen in the ambulance.
Table 1.
Laboratory tests at admission, and after 11 h
| Laboratory test | Time=0 h | Time=11 h | Reference values |
|---|---|---|---|
| Haemoglobin (mmol/L) | 5.5 | 5.3 | 8.3–10.5 |
| Platelets (×109/L) | 128 | 101 | 145–350 |
| Leucocytes (×109/L) | 18.7 | 23.5 | 3.50–10.0 |
| C reactive protein (mg/L) | 65.2 | 75,2 | <8.0 |
| Creatine (μmol/L) | 154 | 226 | 60–105 |
| Lactate dehydrogenase (U/L) | * | * | 105–205 |
| Alanine aminotransferase (U/L) | * | 316 | 10–70 |
| Bilirubin (μmol/L) | * | 227 | 5–25 |
| Ferritin (μg/L) | – | 25 647 | 22–355 |
*Values not reported by the laboratory due to haemolysis in the samples.
Figure 1.

ECG (25 mm/s) (A) from the time of admission showing sinus tachycardia and (B) just before cardiac arrest showing right bundle branch block as a sign of right-sided load.
Figure 2.
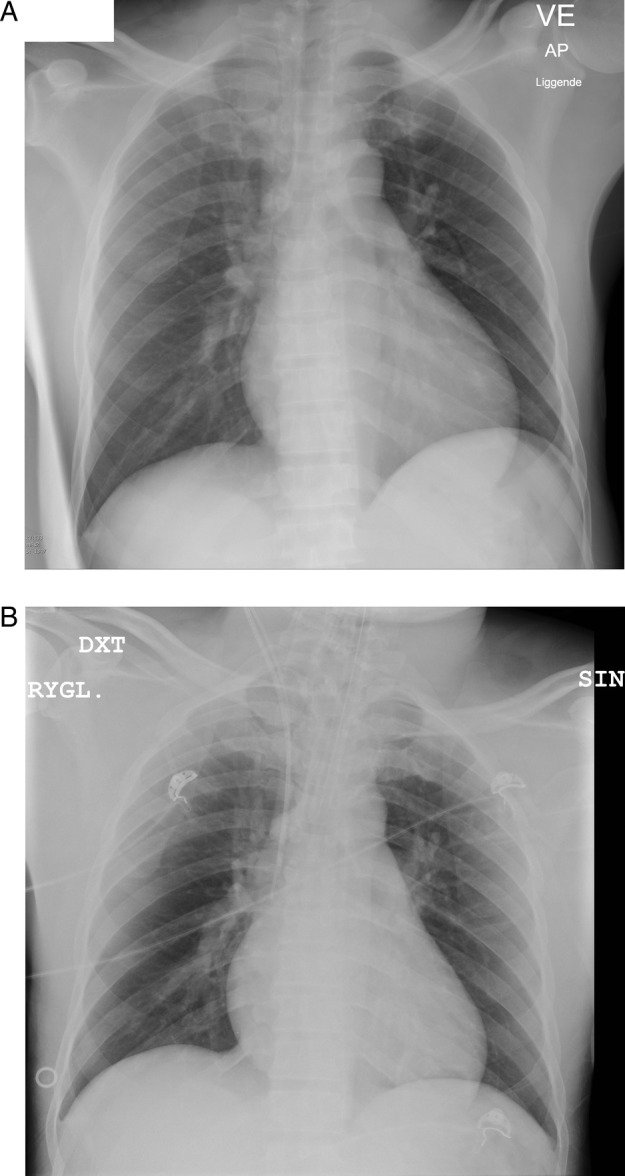
Chest X-ray (A) from the time of admission, where no pneumonic infiltrates are seen, and (B) after the patient's respiratory conditions have deteriorated, showing an endotracheal tube but still without pulmonary infiltrates or oedema.
Owing to the respiratory symptoms and the haemolysis, the tentative diagnosis was mycoplasma pneumonia with cold agglutinins. Antibiotic treatment was supplemented by macrolide antibiotic and glucocorticoids.
Despite this, his condition continued to deteriorate, and shortly after arrival at the tertiary hospital he was transferred to the intensive care unit due to respiratory failure. Even though he was tracheal-intubated and underwent mechanical ventilation with 90% oxygen, it was not possible to maintain sufficient saturation and the circulatory conditions worsened rapidly. Noradrenaline and dobutamine only transiently improved his circulation. At this stage, the arterial blood sample showed severe metabolic acidosis due to tissue hypoperfusion (pH 7.14, PaO2 9.3 kPa, PaCO2 6.8 kPa, base excess −12.3 and lactate 9.1 mmol/L). Distension of the jugular veins were observed and in contrast to earlier, the ECG showed right bundle branch block (figure 1B). Transthoracic echocardiography showed pulmonary hypertension with severe dilation of the right ventricle, a peak systolic tricuspid pressure gradient of 46 mm Hg and a distended inferior vena cava measuring 45 mm, corresponding to an estimated systolic pulmonary pressure exceeding 60 mm Hg (figure 3). The tricuspid annular plane systolic excursion was 10 mm, indicating severe right ventricular failure. There were no signs of deep vein thrombosis in the lower extremity or peripheral oedema. The left ventricular function was normal with an ejection fraction of 60%, but the cardiac output state was low due to a small left ventricular cavity compressed by the right ventricle. He developed cardiac arrest with ventricular asystole that converted to ventricular fibrillation. Cardiac defibrillation resulted in a short-lasting sinus rhythm. Owing to the suspicion of a pulmonary embolus, fibrinolysis with Alteplase was initiated on vital indication. After 60 min of cardiopulmonary resuscitation, the treatment was stopped and the patient died.
Figure 3.
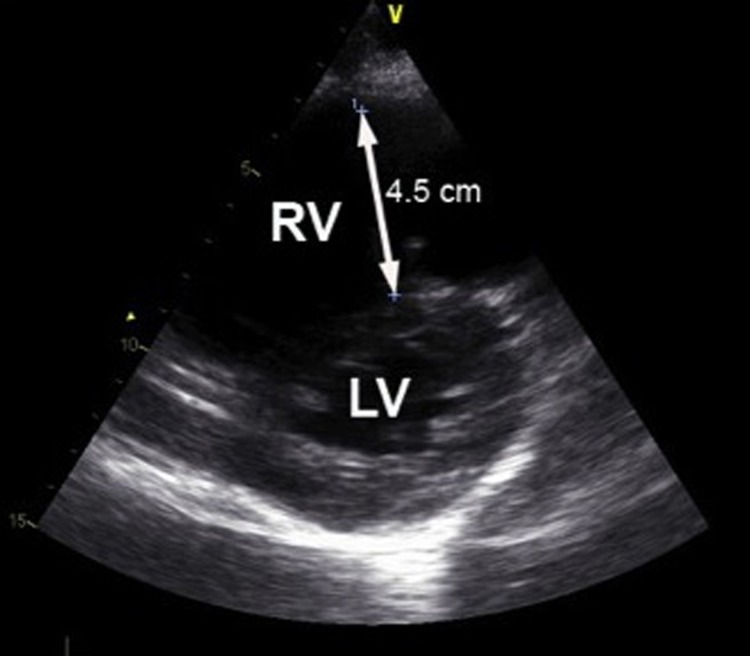
Echocardiography—a parasternal short axis view demonstrating severe dilation (45 mm) of the right ventricle (RV) causing compression of the left ventricle (LV) with flattening of the intraventricular septum resulting in a D-shaped LV.
Extended laboratory tests showed high D-dimer (>20 mg/L FEU), high International Normalised Ratio for blood clotting at 1.7, high cell-free haemoglobin (253 μmol/L) and normal activated partial thromboplastin time, P-antitrombin, P-fibrinogen. The haemoglobin level had decreased, although he received two portions of 260 mL erythrocyte pools. DAT (direct antiglobulin test) and extended DAT were negative and there were no schistocytes in the blood smear.
Autopsy showed right ventricular dilation without increased wall thickness. Surprisingly, there was no sign of a pulmonary embolism. However, histopathological examination showed extensive pulmonary microthrombi in the capillaries and midsized peripheral pulmonary vessels. These thrombi, which were considered to be a result of disseminated intravascular coagulation, consisted of fibrin, platelets and red blood cells (figures 4 and 5). There were no signs of infection in the lungs. No thrombus formation was found in other organs including the deep veins of the lower extremities. The spleen and the liver were not enlarged.
Figure 4.
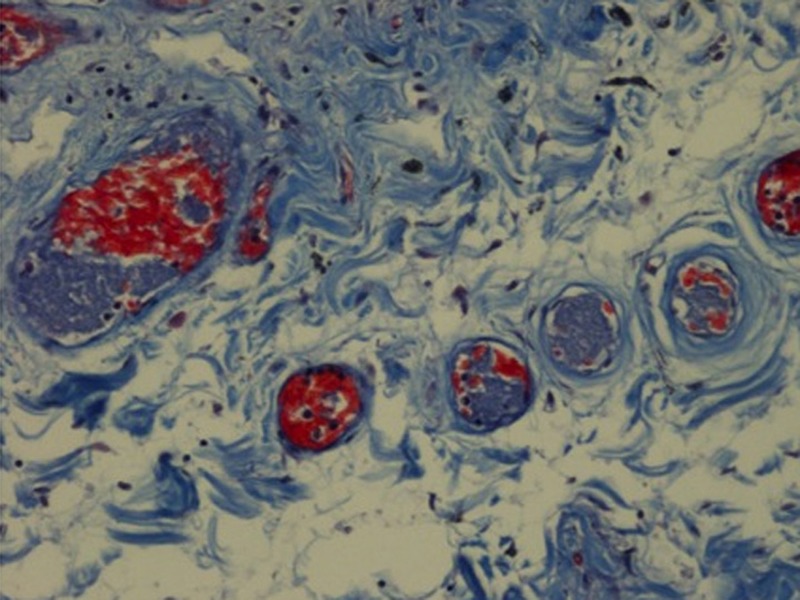
Picromallory, stain ×200. Small lung vessels with platelet microthrombi (blue) mixed with fibrin (red) and erythrocytes (orange) as a sign of intravascular coagulation.
Figure 5.
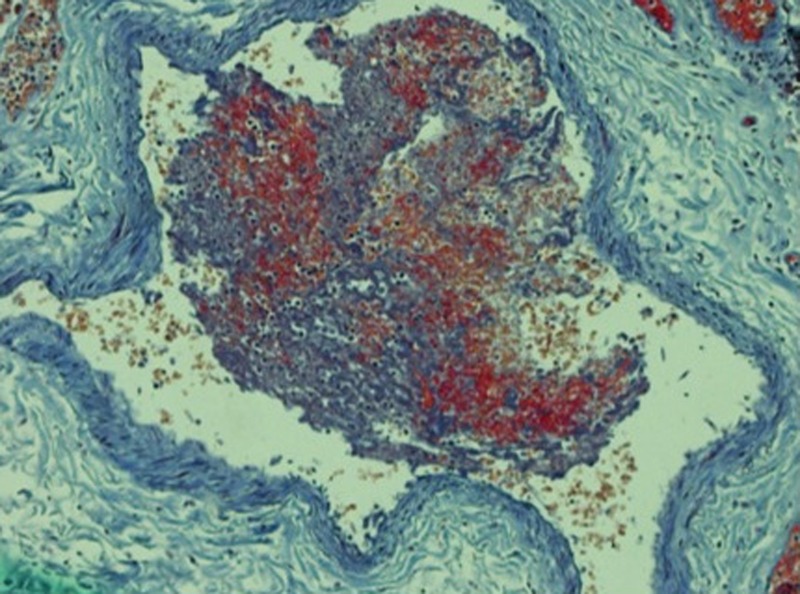
Picromallory, stain ×100. Lung vessel with a thrombus consisting of platelets (blue), fibrin (red) and erythrocytes (orange).
Viral tests were all negative, including tests for acute hepatitis A, B, C and cytomegalovirus. There were no mycoplasma antibodies. Blood and urine cultures were negative.
The peripheral blood smear showed three main findings which elucidated the pathophysiology of this case. First, poikilocytosis and reticulocytosis consistent with a rampant haemolysis were seen. Second, Heinz bodies were noted, which raised the suspicion of G6PD deficiency. This was confirmed by a gene test, which revealed that the patient was hemizygote for two mutations (202GTA and 376ATG) in the G6PD gene. Third, the blood smear contained macrothrombocytes, supporting the observation of an activated coagulation cascade (figure 6).
Figure 6.
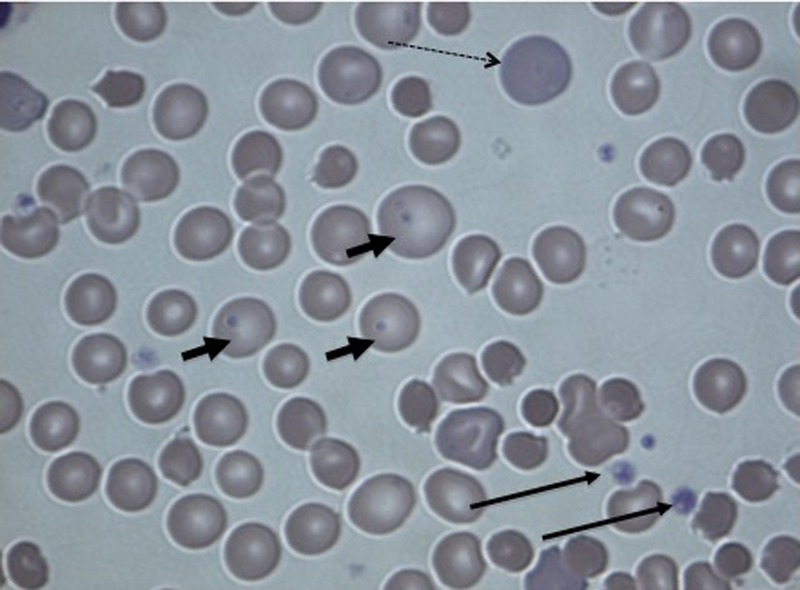
Peripheral blood smear, stain ×400. Dashed arrow: reticulocytes and poikilocytosis, small arrows: Heinz bodies and long arrows: macrothrombocytes.
After establishment of the G6PD deficiency diagnosis, his wife confirmed that, to the best of her knowledge, he had not been eating fava beans. The patient's brother confirmed once again that there was no family history of haemolytic anaemia.
Discussion
Typical symptoms of haemolytic crisis are a sudden increase in temperature, jaundice, dark urine, abdominal pain and weakness/malaise developing during the course of hours to a few days, as described in the present case. Besides these classical findings, fast and heavy breathing may be noticed, which may be caused by the combination of fever, anaemia and, of course, an accompanying/triggering pulmonary infection.1 In the present case, shortness of breath was present, and the arterial blood sample showed hypoxia, suggesting an affection of the lungs. The lack of a family history of haemolytic episodes, and the clinical picture with haemolysis, fever and respiratory symptoms in combination, suggested a mycoplasma pneumonia with cold agglutinins, even though the chest X-ray was without pathology. Thrombotic thrombocytopenic purpura/haemolytic uraemic syndrome was rejected as a differential diagnosis since the peripheral blood smear showed less than one schistocyte per high-power field. Owing to very high levels of S-ferritin, haemophagocytosis was considered as another possible explanation of haemolysis, but this was also rejected owing to a normal CD163 and the normal triglyceride level.
Despite antibiotics, the patient's condition deteriorated rapidly with respiratory and circulatory failure, and the suspicion of pulmonary embolism was supported by a very high D-dimer and echocardiography, which was consistent with acute cor pulmonale. There was no time for further image confirmation of pulmonary embolism. Even in the case of a negative pulmonary CT angiography test result, the clinical conditions were so severe that thrombolytic therapy seemed to be our patient's only chance. Tragically, the patient did not recover despite prolonged cardiac resuscitation after thrombolytic administration.
Theoretically, the acute pulmonary hypertension (PH) in the present case may have several potential causes. First, we cannot completely exclude the existence of a central pulmonary embolus that was lysed by fibrinolytic treatment. However, no embolus was found at the autopsy, so we find this explanation less likely. Second, our patient may have had an aggravation of a pre-existing PH. Chronic haemolysis ranks among the rare causes of PH, which is caused by mechanisms that may be related to an increased consumption of nitric oxide that acts as a potent pulmonary vasodilator and has antiproliferative properties.3 Only severe G6PD deficiency (WHO class I, less than 10% residual enzyme activity) is associated with a chronic haemolytic state, and our patient only had a mild G6PD deficiency (WHO class III, 10–60% residual enzyme activity).2 Additionally, the initial ECG and autopsy did not show signs of right ventricular hypertrophy, which would be expected in the presence of chronic PH. Third, PH may have been a result of the high cell-free haemoglobin seen in our patient. A study using a murine haemolysis model has demonstrated that platelet activation/aggregation plays a critical role in haemolysis-induced PH.4 By excessive intravascular haemolysis, the cell-free haemoglobin may reach a level where it saturates the capacity of protective haemoglobin-scavenging mechanisms. High cell-free haemoglobin contributes to clot formation and increased pulmonary vascular resistance by impairment of nitric oxide bioavailability, by stimulation of the endothelial adhesions molecule expression and by activation of the intrinsic coagulation cascade.5 6 The fourth explanation, which we consider to be the most plausible one, is that the PH may have been a consequence of the microthrombi identified in the pulmonary microvasculature. Signs of disseminated intravascular coagulation (DIC) were found in the small blood vessels of the lungs but there were no signs of bleeding, and blood tests were not fully consistent with DIC. DIC usually causes microthrombi in multiple organs,7 8 but the lungs are found to be one of the most frequented sites of microthombi, as found in this case.9
The DIC seen in our patient is suggested to be a consequence of the excessive inflammatory response due to haemolysis, as aforementioned. However, DIC is more often triggered by an infection.8–10 Since our patient had not taken any drugs or eaten any fava beans, he may most likely have had an undetected infection which triggered the haemolysis and DIC.
At present, no evidence-based guidelines exist on how to treat patients with acutely developed pulmonary hypertension and right heart failure in the absence of pulmonary embolism. Speculatively, it would have been reasonable to try to reduce the right ventricular afterload by pulmonary vasodilation. Prostacyclin has been demonstrated to improve survival in patients with PH, but like other pulmonary vasodilators such as calcium channel blockers and phosphodiesterase inhibitors, it also causes systemic vasodilation that would potentially aggravate the already threatening hypotension. Another option could have been inhaled nitric oxide which works locally by dilating the lung vessels.11 There is a theoretic possibility that this could have stabilised our patient.
To the best of our knowledge, death caused by acute right heart failure as a result of multiple pulmonary microthrombi in a patient with acute haemolysis and G6PD deficiency has not been described previously in the literature. This may be due to the rare incidence of fatal haemolytic attacks in patients with G6PD deficiency and, in continuance of this, a missing exploration of the real causes of death.
Learning points.
Not all cases of acute pulmonary hypertension and right heart failure are caused by pulmonary embolism. In patients with haemolysis, one should be cautious in attributing more than mild shortness of breath to anaemia.
In rare situations, as demonstrated by this fatal case, respiratory symptoms may reflect pulmonary hypertension secondary to chronic or acute haemolysis.
Although no answers exist on how our patient could have been saved, it is likely that hypoxia aggravated his pulmonary hypertension by pulmonary vasoconstriction. It remains speculative, but an early intensive oxygen therapy and pulmonary vasodilators might possibly have altered the course.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Luzzatto L. Hemolytic and anaemia due to acute blood loss. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson J, Losclzo J, eds. Harrison's principles of internal medicine. 18th edn New York: McGraw-Hill, 2012; chap106. http://accessmedicine.mhmedical.com/content.aspx?bookid=331&Sectionid=40726844 (accessed 5 Mar 2013). [Google Scholar]
- 2.Ronquist G, Theodorsson E. Inherited, non-spherocytic haemolysis due to deficiency of glucose-6-phosphate dehydrogenase. Scand J Clin Lab Invest 2007; 67:105–11 [DOI] [PubMed] [Google Scholar]
- 3.Galie N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493–537 [DOI] [PubMed] [Google Scholar]
- 4.Hu W, Jin R, Zhang J, et al. The critical roles of platelet activation and reduced NO bioavailability in fatal pulmonary arterial hypertension in a murine hemolysis model. Blood 2010;116:1613–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cappellini MD. Coagulation in the pathophysiology of hemolytic anemias. Hematology Am Soc Hematol Educ Program 2007:74–8. [DOI] [PubMed] [Google Scholar]
- 6.Rother RP, Bell L, Hillmen P, et al. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 2005;293:1653–62 [DOI] [PubMed] [Google Scholar]
- 7.Levi M. Current understanding of disseminated intravascular coagulation. Br J Haematol 2004;124:567–76 [DOI] [PubMed] [Google Scholar]
- 8.Levi M. Disseminated intravascular coagulation. Crit Care Med 2007; 35:2191–5 [DOI] [PubMed] [Google Scholar]
- 9.Wilde JT, Roberts KM, Greaves M, et al. Association between necropsy evidence of disseminated intravascular coagulation and coagulation variables before death in patients in intensive care units. J Clin Pathol 1988;41:138–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bleakly NT, Fontaine MJ, Pate LL, et al. Disseminated intravascular coagulation due to IgM-mediated autoimmune hemolytic anemia. Pediatr Blood Cancer 2011;57:329–31 [DOI] [PubMed] [Google Scholar]
- 11.Hoeper MM, Granton J. Intensive care unit management of patients with severe pulmonary hypertension and right heart failure. Am J Respir Crit Care Med 2011;184:1114–24 [DOI] [PubMed] [Google Scholar]