

Abstract
We present details of a man who was originally diagnosed with sarcoidosis, based on a combination of nodal granulomatous inflammation and radiology confirming bilateral hilar lymphadenopathy with pulmonary infiltrates. The patient subsequently developed splenomegaly and idiopathic thrombocytopenic purpura (ITP) and, latterly, a severe cavitating pneumonia. Serum immunoglobulins were checked, confirming panhypogammaglobulinaemia, and his diagnosis was revised to common variable immune deficiency (CVID). CVID is a heterogeneous condition, which can mimic sarcoidosis with granulomatous organ involvement and is commonly complicated by autoimmune disorders, including ITP. Prompt recognition is important to allow early introduction of immunoglobulin replacement therapy to decrease infection frequency, reduce development of secondary disease complications and retard progression of tissue damage. Given the potential for misdiagnosis and delay in recognition of CVID, serum immunoglobulin measurement should be a first-line investigation in patients with suspected sarcoidosis, even if the presentation is ‘typical’. Current international sarcoidosis guidelines should be revised accordingly.
Background
Sarcoidosis is a multisystem granulomatous disease of unknown cause, typically occurring in young adults, often under the age of 50. Commonly, it is associated with fatigue and general malaise (66%), and typically it affects the lungs (>90%), skin (24%), lymph nodes (15%) and eyes (12%). Around 3000 new cases of sarcoidosis are diagnosed each year in the UK.1
It is important to remember that sarcoidosis is essentially a diagnosis of exclusion and usually requires demonstration of granulomatous inflammation in an appropriate clinical context, that is, a typical clinical finding with rigorous exclusion of other disorders.2 3 Specifically, it is important to rule out malignancy (notably lymphoma), mycobacterial infection (especially tuberculosis), fungal infections and more obscure causes of granulomatous inflammation, such as immunodeficiency, response to foreign bodies, beryllium exposure and some drug therapies, for example, interferon α for hepatitis or highly active retroviral therapy for HIV infection.
We describe a case where clinicians confidently diagnosed ‘sarcoidosis’ only for an alternative diagnosis, requiring a significantly different therapeutic approach, to emerge some years later.
Case presentation
A 29-year-old Caucasian man presented in 1981 with axillary and cervical lymphadenopathy, splenomegaly and transient thrombocytopenia. An axillary lymph node biopsy was performed, demonstrating granulomatous inflammation supporting a clinical diagnosis of sarcoidosis. A lifelong non-smoker, working full time as an agricultural salesman and farmer, he was relatively well until 1998 when he developed conjunctivitis, cough, breathlessness and recurrent peripheral lymphadenopathy with nodular interstitial pulmonary shadowing and bilateral hilar lymphadenopathy. His serum ACE level was raised. Lymph node fine-needle aspiration once again showed granulomatous inflammation. Among a number of investigations at that time, he had serum immunoglobulins checked with low IgG 3.3 g/L (normal 6–16), IgA 0.5 g/L (0.8–2.8) and IgM 0.7 g/L (0.5–3) levels, although this was not recognised as being clinically important at that time. He was started on oral steroid therapy in 1999 and remained healthy over the next decade. In 2009, he presented again with increasing splenomegaly and thrombocytopenia, the latter thought to be due to idiopathic thrombocytopenic purpura (ITP). His marrow trephine showed normal megakaryocyte numbers along with the presence of granulomata (figure 1A). During the same year, he developed a left vocal cord palsy, the aetiology of which was uncertain but possibly related to his sarcoidosis. Over the next several years, his splenomegaly increased and he continued to have significant systemic malaise with treatment resistant ITP. In 2011, at the age of 42, he had a protracted right upper lobe cavitating pneumonia, growing Haemophilus influenzae on bronchoalveolar lavage, and he was now found to have agammaglobulinaemia (serum IgG, IgA and IgM all <0.3 g/L).
Figure 1.

(A) Histological appearance of granuloma in bone marrow trephine—magnification ×400. (B) Granulomas with a multinucleate giant cell in splenic parenchyma—magnification ×100.
The initial diagnosis of sarcoidosis was revised to common variable immune deficiency, complicated by disseminated granulomatous disease, splenomegaly and idiopathic (autoimmune) thrombocytopenia.
Treatment
Immunoglobulin replacement therapy was started in 2012, and initially the patient did well in terms of reduced infection frequency and improved general wellbeing. Subsequently, he developed worsening splenomegaly with severe thrombocytopenia unresponsive to high-dose prednisolone (50 mg daily) and deranged liver function tests of cholestatic pattern (thought likely to be due to granulomatous hepatitis). A platelet uptake scan demonstrated significant hepatic platelet sequestration only. Endoscopy revealed hiatus hernia, gastritis and varices in his oesophagus and stomach, which were not amenable to endoscopic banding. He was started on carvedilol. He underwent elective splenectomy in November 2012 mainly on the basis of symptomatic hypersplenism, but also in the hope of some partial improvement in his platelet count. Liver biopsy was performed at the time of his splenectomy but showed non-specific inflammatory change only. His splenic pathology included the presence of diffuse, non-caseating granulomata (figure 1B).
Outcome and follow-up
This man has now been on treatment for common variable immune deficiency (CVID) for 18 months with frequent shared-care immunology and respiratory follow-up. His general well-being is good with no systemic symptoms and with higher energy levels supporting his return to full-time working. He has had no further episodes of pneumonia or other significant infection and he remains on postsplenectomy antibiotic prophylaxis. His current maintenance therapy includes oral prednisolone, β-blocker and three-weekly intravenous immunoglobulin replacement therapy on which he has a satisfactory pre-infusion, trough IgG level maintained at 9-11 g/L.
From a respiratory perspective, he still has some exertional breathlessness on sustained effort, and although his chest radiograph is much improved (figure 2), he continues to have a degree of postpneumonic right lung scarring, albeit minor. His lung function remains impaired, with an FEV1 (forced expiratory volume in 1 s) of 62% predicted, FVC (forced vital capacity) of 75% predicted and PEF (peak expiratory flow) of 65% predicted. His resting oxygen saturation is 96%. He still requires a relatively high dose of prednisolone, 20 mg daily, which we hope to gradually wean.
Figure 2.
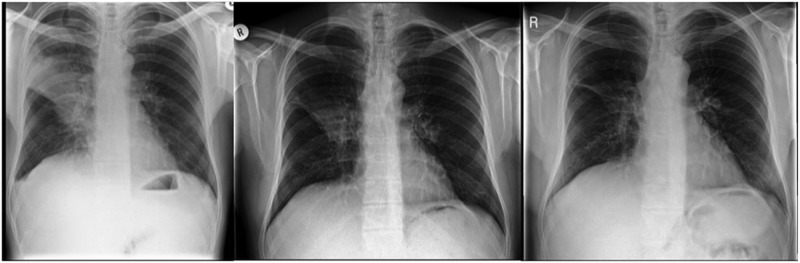
Chest radiographs noting right upper lobe pneumonia in October 2011 (left), April 2012 (middle) and June 2013 (right), noting gradual improvement with only minimal right mid-zone scarring remaining after initial antibiotics and approximately 18 months of immunoglobulin therapy.
With regard to extra-pulmonary manifestations of his CVID, his liver function tests remain elevated but are improving with, most recently, levels of alkaline phosphatase 265 U/L (normal 30–130 IU/L), alanine aminotransferase 84 IU/L (8–55 IU/L) and γ-glutamyltransferase 429 IU/L (4–35 IU/L) and with a normal albumin (41 g/L) and clotting profile. His platelet count has returned to normal postsplenectomy.
Discussion
CVID is the commonest symptomatic primary antibody deficiency, with an estimated population prevalence of 1–2.5/50 000,4 5 although awareness among clinicians remains generally low. It can present at any age, although in most patients it is first seen in childhood or early-to-mid adulthood. The condition may present with a heterogeneous combination of serious, persistent, opportunistic or recurrent (mainly bacterial) infections with immune dysregulatory complications such as autoimmune and hypersensitivity disorders or granulomatous inflammation. An increased relative risk of infection-associated cancers including lymphoma, gastric carcinoma and skin cancer is also seen. Multisystem granulomatous inflammation (commonly with a pulmonary component) occurs in 20% of cases.6 Chronic lung disease occurs in approximately a quarter of patients, with varying presentations including acute and chronic infection, bronchiectasis and granulomatous-lymphocytic interstitial lung disease.7
This case is typical, with CVID as a potential unifying diagnosis being considered years after the original presentation and following the evolution of other disease complications ultimately pointing towards a significant underlying complex defect of immune regulation. The relevance of his evolving hypogammaglobulinaemia in 1998 was not recognised at the time. With the later development of pneumonia, systemic granulomatous disease, autoimmune cytopenias and (recognised) hypogammaglobulinaemia, the ultimate diagnosis was more obvious. Often, these secondary complications do not present collectively or concurrently but evolve sequentially and subtly, which contributes in part to the frequent delay in the recognition and diagnosis of the underlying condition.
A key message for clinicians is that (simple and inexpensive) measurement of serum immunoglobulins should be considered routinely in the assessment of any patient with granulomatous disease, even if the clinical and radiological presentation appears typical for sarcoidosis. It is notable that current international sarcoidosis/interstitial lung disease guidelines do not highlight this issue despite similar existing reports of delayed diagnosis of CVID presenting initially with granulomatous disease.2 3 8
Learning points.
Sarcoidosis is a diagnosis of exclusion and the demonstration of granulomatous inflammation, while supportive in the appropriate clinical context, does not negate the possibility of alternative diagnoses.
It is typical to find a polyclonal gammopathy in sarcoidosis - hypogammaglobulinaemia should prompt immunology review and consideration of primary immunodeficiency disorders such as common variable immune deficiency (CVID).
Manifestations of CVID may develop over many years, and should be considered if a patient with apparent ‘sarcoidosis’ has an unusual clinical course including, for example, thrombocytopenia, other autoimmune manifestations or unusual infections.
Serum immunoglobulins (IgG, IgA, IgM) should be routinely checked in all patients with suspected sarcoidosis as part of initial assessment.
Footnotes
Contributors: RA was the clinician responsible for considering CVID as a diagnosis, and she asked for the immunoglobulin levels to be checked. RH was responsible for this case of CVID and the ongoing management. The manuscript was drawn together by A-MS and OD to present the key message of why immunoglobulins should be checked in those presenting with sarcoidosis, which is highlighted in this case.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Dempsey OJ, Paterson EW, Kerr KM, et al. Sarcoidosis. BMJ 2009;339:b3206. [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society. Statement on sarcoidosis. Am J Respir Crit Care Med 1999;160:736–55 [DOI] [PubMed] [Google Scholar]
- 3.Wells AU, Hirani N, on behalf of the British Thoracic Society Interstitial Lung Disease Guideline Group, a subgroup of the British Thoracic Society Standards of Care Committee, in collaboration with the Thoracic Society of Australia and New Zealand. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008;63:v1–58 [DOI] [PubMed] [Google Scholar]
- 4.Spickett GP. Current perspectives on common variable immunodeficiency. Clin Exp Allergy 2001;31:536–42 [DOI] [PubMed] [Google Scholar]
- 5.Cunningham-Rundles C. How I treat common variable immune deficiency. Blood 2010;116:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol 2009;133:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol 2010;134:97–103 [DOI] [PubMed] [Google Scholar]
- 8.Oppong P, Banik S, Derry D. Are cases of granulomatous common variable immunodeficiency misdiagnosed as sarcoidosis in routine clinical practice? Thorax 2011;6(4):91 [Google Scholar]