

Description
A man in his 50s had a history of a congenital atrial and ventricular septal defect requiring repair decades prior. He also had a history of heart block requiring pacemaker placement. During an evaluation for pacemaker battery replacement, he was noted to have nodules at the metacarpophalangeal joint of his fifth digit, bilaterally (figure 1). The nubbins were consistent with ulnar (also known as postaxial) polydactyly and, given the cardiac history, a diagnosis of Holt-Oram syndrome was considered highly likely. The patient tolerated his battery replacement without complication, and years later passed away from causes unrelated to his medical condition.
Figure 1.
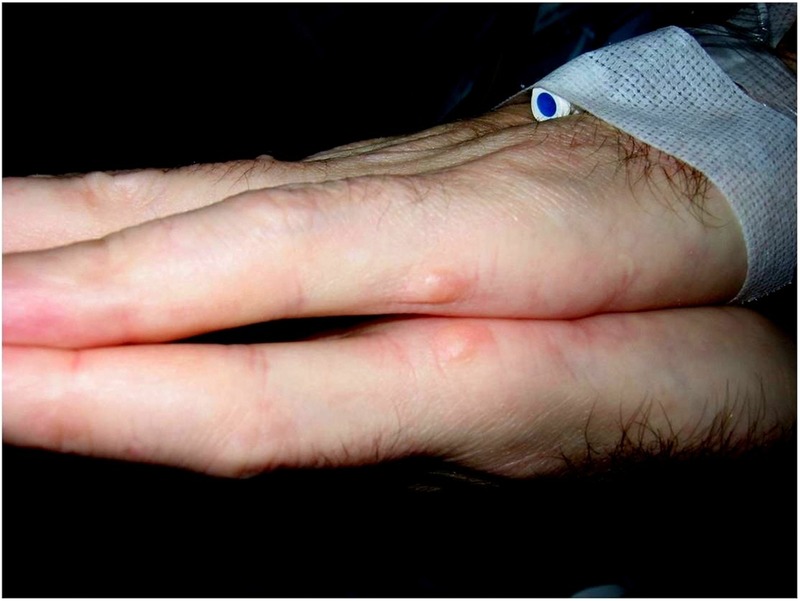
Nubbins present at the metacarpophalangeal joint the fifth digit are subtle manifestations of ulnar polydactyly.
Learning points.
Holt-Oram syndrome (HOS), otherwise known as heart–hand syndrome, affects approximately 1:100 000 live-births. It is an autosomal dominant syndrome caused by a mutation in the TBX5 transcription factor.1
TBX5 mutations manifest as cardiac defects, most commonly septal defects and radial ray upper limb abnormalities can range from preaxial (thumb) polydactyly triphalangeal or absent thumbs to phocomelia.1 Ulnar ray defects (postaxial) are a more rare manifestation of the syndrome.1
A wide variety of mutations can cause HOS, which may exhibit multitudinous phenotypes. Null allele mutations appear to cause significant limb and cardiac malformations, whereas certain missense mutations may cause more severe cardiac defects or significant limb abnormalities.2
The diagnosis can be confirmed by molecular genetic testing of TBX5 gene or with a posteroanterior hand X-ray as an abnormal carpal bone is present in all affected individuals.
Footnotes
Competing interests: None.
Patient consent: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Patel C, Silcock L, McMullan D, et al. TBX5 intragenic duplication: a family with an atypical Holt–Oram syndrome phenotype. Eur J Hum Genet 2012;20:863–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basson CT, Huang T, Lin R, et al. Different TBX5 interactions in heart and limb defined by Holt–Oram syndrome mutations. Proc Natl Acad Sci USA 1999;96:2919–24 [DOI] [PMC free article] [PubMed] [Google Scholar]