

Abstract
Intravascular lymphoma is a rare subtype of extranodal large cell non-Hodgkin's lymphoma that is usually seen in the elderly. It can occasionally present with neurological symptoms in the form of dementia, focal neurological deficit and seizure. Diagnosis is difficult because of non-specific clinical manifestation. We report a case of a 38-year-old woman presenting with rapidly progressive dementia and seizure. MRI of the brain showed bilateral diffuse involvement of cortex and subcortical white matter. Brain biopsy disclosed the aetiological confirmation of intravascular B-cell lymphoma. The patient was treated with monthly cyclophosphamide, doxorubicin, vincristine and prednisolone regimen, but unfortunately, she died after two chemotherapy cycles. So, high index of suspicion is warranted to diagnose and treat the condition early to have a better outcome.
Background
Intravascular lymphoma (IVL) is an uncommon variant of malignant extranodal large B-cell lymphoma. Pathologically, it is characterised by growth of lymphoma cells in the lumen of small vessels, especially capillaries. Initially, it was thought to have originated from the endothelium.1 Subsequently, lymphomatous origin was confirmed by Wick et al2 in 1986. Patients present with non-specific symptoms such as fever, fatigue and decreased appetite without lymphadenopathy. Two main variants are described in literatures: haemophagocytic and cutaneous variant. Neurological manifestations include seizure, dementia, sensory or motor deficits, altered sensorium, ataxia, vertigo and transient visual loss. The diagnosis is usually made by organ biopsy.
We report an unusual case of a 38-year-old woman who presented with rapidly progressive dementia (RPD) and seizure, later diagnosed as IVL. So, high index of suspicion is required to diagnose and initiate appropriate chemotherapy.
Case presentation
This case illustrates a 38-year-old woman, resident of Delhi (North India) from a low socioeconomic background who presented with a 2-month history of forgetfulness and behavioural abnormalities. At the onset of disease, she developed impaired recent memory (forgot what was heard or read before, misplaced things and sometimes forgot to add salt and other ingredients while cooking). Over time, her decision-making, judgement and problem-solving skills deteriorated. She also developed impaired organisational skills. She faced difficulty in learning new tasks. Gradually, she had abnormal remote memory as well as difficulty in recognising family members. Apart from this, she developed behavioural abnormalities in the form of aggressiveness, abusiveness, anger outbursts and disinhibition. She developed three episodes of generalised tonic–clonic seizure within 1 month. There was no history of fever, headache and vomiting. Her family members did not know of any history of focal neurological deficit, myoclonus or prior systemic illness. General physical examination was unremarkable. Neurological examination revealed that she was conscious but uncooperative for higher mental function examination. Overall cranial nerves examination was normal. Power, tone and deep tendon reflexes of upper and lower limbs were normal. Frontal release signs, including bilateral grasp and snout reflexes, were elicited. Extrapyramidal, cerebellar and autonomic examinations revealed no abnormalities.
Investigations
At the time of hospital admission, her complete blood count including haemoglobin (Hb 12 g%), total leucocyte count (TLC; 6670 cells/cumm), total platelet count (TPC; 2.35 lakhs/cumm), erythrocyte sedimentation rate (22 mm in first hour), liver function test and kidney function tests were normal. Cerebrospinal fluid (CSF) examination showed 3 cells/µL with normal glucose (68 mg/dL) and mildly raised protein (51 mg/dL) level. CSF acid-fast stain, Gram stain, India ink and cryptococcal antigens were negative. CSF PCR for herpes simplex virus was negative. During the hospital stay, her Hb, TLC and TPC deteriorated to 10 g%, 4000 cells/cumm and 1.2 lakhs/cumm, respectively. Liver function test showed increasing trend of enzymes (alanine aminotransferase 107 U/L, aspartate aminotransferase 186 IU/L, γ-glutamyl transpeptidase 1081 U/L). Blood and urine cultures were sterile. Thyroid function test, vitamin B12 and folic acid level were normal. Antinuclear antigens, antidouble-strandedDNA, extractable nuclear antigen profile and antithyroid peroxidase antibody were within normal range. ELISA for HIV, markers for hepatitis B and C virus, CSF Venereal Disease Research Laboratory test for neurosyphilis were negative. EEG showed generalised slow δ activity. Ultrasound of the abdomen revealed mild splenomegaly and chest X-ray was normal. MRI of the brain with T2 and fluid attenuated inversion recovery sequence showed bilateral asymmetrical metachronous, cortical and subcortical hyperintensities involving frontal, temporal and parietal lobes (figure 1A, B), and T1 hypointensity in the corresponding areas. Diffusion-weighted image revealed patchy diffusion restriction in the aforementioned lesion (figure 2A). Postcontrast image did not show enhancement (figure 2B). MR angiography of the head and neck was normal. Serum lactate dehydrogenase level was 400 IU/L (normal 100–190 U/L), serum ferritin level was 301 ng/mL (normal 12–300 ng/mL) and serum fibrinogen was 407 mg/dL (normal 200–400 mg/dL). The patient was subjected to brain biopsy. Histopathological examination showed B-cell IVL and increased perivascular lymphocytes. The small vessels were occluded with neoplastic cells. The atypical lymphocytes were found to be positive for CD20 (figure 3A–D). Luxol fast blue stain showed areas of demyelination. Random skin biopsy from right forearm was negative for malignant cells.
Figure 1.
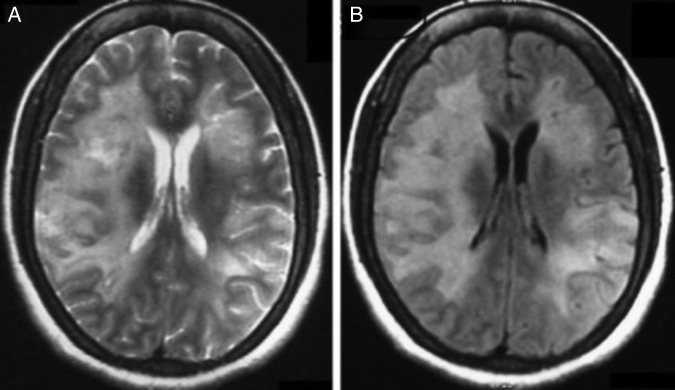
(A and B) MRI of the brain with axial T2 and fluid attenuated inversion recovery (FLAIR) sequence showing bilateral asymmetrical metachronous cortical and subcortical hyperintensities involving frontal, temporal and parietal lobes.
Figure 2.
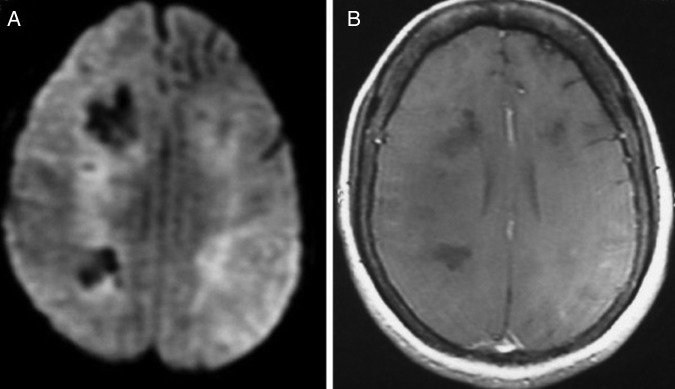
(A) Diffusion-weighted image revealing patchy diffusion restriction in frontal, temporal and parietal lobes. (B) Postcontrast image showing no enhancement.
Figure 3.

(A) Intravascular neoplastic cells and perivascular lymphocytes. (B) Vessel occluded with neoplastic cells. (C) CD20 cells in vascular lumen. (D) Luxol fast blue (LFB) showing area of demyelination.
Differential diagnosis
The diagnostic approach to RPD differs from chronic degenerative dementia. Although there is no clear definition of RPD, it refers to conditions that progress from the first symptom to dementia in less than 1–2 years.3 Multi-infarct dementia, cerebral amyloid angiopathy, central nervous system (CNS) vasculitis and IVL are among the major vascular causes of RPD. Other close mimics in the present case are Creutzfeldt-Jakob disease, Hashimoto's encephalopathy, autoimmune encephalitis, paraneoplastic limbic encephalitis, connective tissue disease such as systemic lupus erythematous and neuro Sjögren syndrome.3 Clinical spectrum, laboratory investigations and brain biopsy disclosed the aetiology.
Treatment and follow-up
During the hospital stay, the patient rapidly deteriorated and developed hospital-related infection. She was treated with antibiotics, intravenous fluid, antiepileptic (phenytoin 5 mg/kg) and ventilatory support. After histopathological confirmation, she was treated with intravenous methylprednisolone (MPS) (1 g daily) for 5 days followed by oral prednisolone (1 mg/kg). On the third day of IV MPS, she gradually improved. The Glasgow Coma Scale (GCS) score improved from E1V1M5 (GCS-7) to E4V2M5 (GCS-11), and she was weaned off the ventilatory support. However, she was dependent for all her daily activities. She was referred to the oncology department where she received two cycles of chemotherapy, with cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP) regimen. Rituximab could not be given due to financial constraints. Although there was transient improvement during the hospital stay, after two chemotherapy cycles, her clinical condition further declined and she died at home.
Discussion
IVL is a rare clinical entity with heterogeneous clinical manifestations.
It is characterised by selective growth of neoplastic and atypical cells in the small and intermediate-sized vessels. It involves all types of organs, including the CNS, lungs, skin, adrenal gland, liver, kidney, spleen, bone marrow, thyroid, pituitary and gastrointestinal tract.4 Two variants of IVL are described: haemophagocytic syndrome which is usually seen in Asian patients, and classical variant or cutaneous variant which is described in European patients. Patients with haemophagocytic syndrome present with haematological abnormalities in the form of anaemia, decreased red blood cell count, leucocytopenia, thrombocytopenia and hepatosplenomegaly.5 Early neurological manifestation is found in 25% of patients in the form of altered sensorium, focal neurological deficit, dementia and seizure.6 7 Organ biopsy is necessary for diagnosis of IVL. Random skin biopsy from the uninvolved site may help in diagnosis. Progressive decrease in blood cell count and raised lactate dehydrogenase level may support the possibility of IVL. The clinical outcome with 3-year survival rate with CHOP regimen was 33%.8 The response was increased after addition of rituximab (R-CHOP; 66% at 2 years).9 Some studies have shown good efficacy of autologous stem cell transplant in patients with IVL.
Learning points.
Intravascular lymphoma is a rare subtype of B-cell lymphoma in which the small vessels are occluded with neoplastic cells.
Timely and accurate diagnosis is required to initiate early treatment. Organ biopsy is mandatory to disclose the aetiological clue.
Early institution of chemotherapy including rituximab may halt the progression of the disease.
Footnotes
Contributors: AKP was involved in conception and design, analysis and interpretation of the data, and revising it critically for important intellectual content. SM was involved in acquisition, analysis and interpretation of the data, and drafting the article. AKP and SM gave final approval of the version published.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Pfleger L, Tappeiner J. [On the recognition of systematized endotheliomatosis of the cutaneous blood vessels (reticuloendotheliosis)]. Hautarzt 1959;10: 359–63 [PubMed] [Google Scholar]
- 2.Wick MR, Mills SE, Scheithauer BW, et al. Reassessment of malignant “angioendotheliomatosis”. Evidence in favor of its reclassification as “intravascular lymphomatosis”. Am J Surg Pathol 1986;10:112–23 [PubMed] [Google Scholar]
- 3.Geschwind MD. Rapidly progressive dementia: prion diseases and other rapid dementias. Continuum (Minneap Minn) 2010;16:31–56 [DOI] [PubMed] [Google Scholar]
- 4.Shimada K, Kinoshita T, Naoe T, et al. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol 2009;10:895–902 [DOI] [PubMed] [Google Scholar]
- 5.Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol 2007;25:3168–73 [DOI] [PubMed] [Google Scholar]
- 6.Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol 2004;127:173–83 [DOI] [PubMed] [Google Scholar]
- 7.Ferreri AJ, Campo E, Ambrosetti A, et al. Anthracycline-based chemotherapy as primary treatment for intravascular lymphoma. Ann Oncol 2004;15:1215–21 [DOI] [PubMed] [Google Scholar]
- 8.Ferreri AJ, Dognini GP, Govi S, et al. Can rituximab change the usually dismal prognosis of patients with intravascular large B-cell lymphoma? J Clin Oncol 2008;26:5134–6 [DOI] [PubMed] [Google Scholar]
- 9.Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol 2008;26:3189–95 [DOI] [PubMed] [Google Scholar]