

Abstract
EHD proteins have been implicated in intracellular trafficking, especially endocytic recycling, where they mediate receptor and lipid recycling back to the plasma membrane. Additionally, EHDs help regulate cytoskeletal reorganization and induce tubule formation. It was previously shown that EHD proteins bind directly to the C2 domains in myoferlin, a protein that regulates myoblast fusion. Loss of myoferlin impairs normal myoblast fusion leading to smaller muscles in vivo but the intracellular pathways perturbed by loss of myoferlin function are not well known. We now characterized muscle development in EHD1-null mice. EHD1-null myoblasts display defective receptor recycling and mislocalization of key muscle proteins, including caveolin-3 and Fer1L5, a related ferlin protein homologous to myoferlin. Additionally, EHD1-null myoblast fusion is reduced. We found that loss of EHD1 leads to smaller muscles and myofibers in vivo. In wildtype skeletal muscle EHD1 localizes to the transverse tubule (T-tubule), and loss of EHD1 results in overgrowth of T-tubules with excess vesicle accumulation in skeletal muscle. We provide evidence that tubule formation in myoblasts relies on a functional EHD1 ATPase domain. Moreover, we extended our studies to show EHD1 regulates BIN1 induced tubule formation. These data, taken together and with the known interaction between EHD and ferlin proteins, suggests that the EHD proteins coordinate growth and development likely through mediating vesicle recycling and the ability to reorganize the cytoskeleton.
Keywords: EHD proteins, Fer1L5, T-tubule, myoblast fusion, muscle growth
INTRODUCTION
Skeletal muscle is a complex tissue with inherent renewal properties that allow for ongoing repair and regeneration. The elongated nature of individual muscle cells, coupled with their functional role of contraction, renders muscle highly dependent on having a rapid and effective vesicle trafficking system for effective growth and repair (Grounds and Shavlakadze, 2011). Endocytic recycling, the internalization of membrane and membrane-associated proteins into the endosomal system and the return of sorted proteins back to the cell surface is essential for many cellular functions, including cell migration, cell division, and the regulation of signaling. Additionally, the ability to coordinate actin dynamics facilitating cytoskeletal rearrangements near the sarcolemma is also critical during myogenesis. Genes encoding proteins found in these vesicles, which also participate in trafficking pathways include caveolin-3, dynamin-2, Bin-1, Bin-3, and dysferlin. Loss-of-function mutations in many of these genes cause inherited myopathies including Limb Girdle Muscular Dystrophy (LGMD) and centronuclear myopathy (Bashir et al., 1998; McNally et al., 1998; Simionescu-Bankston et al., 2013).
The Eps15 homology-domain containing (EHD) family of proteins has been implicated in the regulation of multiple steps of endocytic recycling through binding partners such as caveolin-1 and BIN-1 (Grant and Caplan, 2008; Pant et al., 2009; Verma et al., 2010). Four highly homologous EHD proteins (EHD1-4) are expressed in mammals and are characterized by an amino-terminal ATPase domain thought to be required for the scission of vesicular membranes, a central coiled-coil region, and a single carboxyl-terminal Eps15 homology (EH) domain (Daumke et al., 2007; Mintz et al., 1999). The EH domain of EHD proteins binds proteins at an Asn-Pro-Phe (NPF) motif (de Beer et al., 1998). The founding member of the EHD family, the C. elegans RME-1 (Receptor-Mediated Endocytosis-1) protein, was identified in a genetic screen for mutants defective in endocytosis (Grant et al., 2001). Introduction of each human EHD protein in C. elegans rme-1 mutants rescued the recycling defect, demonstrating functional similarity between human and C. elegans EHD proteins (George et al., 2007). However, in mammals, the tissue-specific expression, non-complementation results, and distinct phenotypes from individual EHD gene deletions in mice are all consistent with non-redundant roles of individual mammalian EHD family members. Overexpression of EHD1 without the EH domain (EHD1ΔEH) in vitro did not rescue recycling and instead resulted in perinuclear transferrin accumulation (George et al., 2007). Ferlin proteins bind EHD proteins directly, and disruption of ferlin proteins also produces endocytic recycling defects (Doherty et al., 2008; Posey et al., 2011b).
There is additional support for the role of EHD proteins as regulators of vesicles sorting at critical nodes from the early endosome (EE) to the lysosome for degradation or to the endocytic recycling compartment (ERC) for recycling (Grant and Caplan, 2008). The cargo of these vesicles varies, but has been documented to include cell signaling receptors like insulin-like growth factor 1 receptor (IGF1R), and the glucose transporter GLUT4, both important for muscle growth (Guilherme et al., 2004; Naslavsky and Caplan, 2010; Rotem-Yehudar et al., 2001). EHD interacting proteins have been identified, which regulate the trafficking and recycling of cell-surface signaling receptors and/or rearrangement of the actin cytoskeleton (Braun et al., 2005; Grant and Caplan, 2008; Guilherme et al., 2004). Examples of EHD interacting proteins include vesicle membrane components like clathrin heavy chain and cytoskeletal rearrangement proteins like actin binding proteins EHBP1 and MICAL-L1 (Guilherme et al., 2004; Sharma et al., 2009). Ferlin proteins bind EHD proteins directly (Doherty et al., 2008; Posey et al., 2011b). The ferlin family members contain multiple C2 domains, and the second C2 domain, (C2B), of myoferlin contains an NPF motif that binds EHD1 and EHD2 (Doherty et al., 2008; Posey et al., 2011b). The related ferlin protein, Fer1L5, also contains an NPF motif in its second C2 domain and this region also directly binds to both EHD1 and EHD2 (Posey et al., 2011b).
EHD1-null mice display significant embryonic lethality but surviving mice exhibit reduced growth compared to wildtype mice; EHD1-null males are infertile due to defective spermatogenesis (Rainey et al., 2010). In another model of EHD1-deficiency, mouse embryonic fibroblasts from EHD1-null mice exhibited impaired transferrin recycling consistent with the role of EHDs in recycling (Rapaport et al., 2006). Analysis of EHD protein expression has shown a role for EHD2 in membrane repair and revealed that EHD3 and EHD4 were both highly upregulated after myocardial infarction in wildtype mice, suggesting a role for EHD proteins in damage response in both skeletal and cardiac muscle (Gudmundsson et al., 2010; Marg et al., 2012). In addition, the EHD proteins have been shown to directly associate with ankyrin, a protein required for membrane targeting and stability of ion channels, cell adhesion molecules, and signaling proteins, in cardiomyocytes (Gudmundsson et al., 2010).
We analyzed mice lacking EHD1 to investigate the in vivo role of EHD1 in muscle growth and development. Consistent with a recycling defect, myoblasts cultured from EHD1-null mice accumulate more intracellular transferrin than wildtype primary myoblasts. In addition, EHD1-null myoblasts have mislocalized caveolin-3 and Fer1L5 and are defective in the formation of large myotubes in vitro. EHD1-null skeletal muscle was found to lack the largest myofibers and have reduced muscle mass. We show tubule formation requires a functional ATPase domain of EHD1 and EHD1 regulates BIN1 induced tubule formation. These data extend the vesicle trafficking complex in muscle to include EHD1. The abnormal T-tubule structure seen in EHD1-null muscle supports that ferlin-EHD mediated vesicle trafficking regulates the biogenesis and maintenance of T-tubules, and this formation is essential for muscle growth.
MATERIALS AND METHODS
Animals
EHD1-null mice were previously generated by deleting exon 1 (Rainey et al., 2010). Heterozygous mice were interbred to generate EHD1-null mice. Mice were housed in specific pathogen free facility in accordance with the University of Chicago Institutional Animal Care and Use Committee regulations. Consistent with what was reported, less than 5% of EHD1 mice survived postnatally.
Muscle analysis
Muscles were examined from mice surviving until ten weeks of age. The quadriceps femoris and triceps brachii muscles were dissected from tendon to tendon from ten-week old male mice. The excised muscle was preserved in 10% formalin, bisected in the mid-belly, and embedded in paraffin. Sections from the center of the muscle were stained with hematoxylin and eosin. Images of entire muscle sections were taken at 1.5x and composite muscle sections were reconstructed using overlapping 10X images. Using ImageJ, the total number of fibers, mean fiber size, and total muscle area for both triceps and quadriceps muscles were analyzed. The mean fiber size was calculated from the area of 618 wildtype and 677 EHD1-null fibers (~ 200 fibers per animal, n=3 animals per genotype). Statistics were performed with Prism (Graphpad, La Jolla, CA) using an unpaired t-test.
Muscle preparation and immunostaining
Quadriceps muscles from wildtype and EHD1-null mice were dissected and frozen in liquid nitrogen-cooled isopentane and cut in 7 μm sections that were fixed in paraformaldehyde, blocked in 1X phosphate-buffered saline (PBS) containing 10% fetal bovine serum, and immunostained. Isolated myofibers were isolated from the flexor digitorum brevis (FDB). Muscle was removed and incubated in collagenase II (Invitrogen, 17101-015). After 2–3 hours, FDB bundles were moved to media containing 3% Bovine serum albumin and 0.1% gentamicin for trituration. Free fibers were incubated at 37 degrees overnight and plated on matrigel (BD Bioscience, 356234) coated coverslips. Fibers were fixed in 4% PFA, rinsed, and blocked in Super Block (Pierce, 37515) with 0.1% triton. Rabbit polyclonal anti-Fer1L5 antibody (Posey et al., 2011b) was used at 1:100, anti-caveolin3 (BD Transduction, 610420, Franklin Lakes, NJ) was used at 1:100, anti-BIN1 (Santa Cruz, sc-23918, Santa Cruz, CA) was used at 1:100, anti-DHPR (Pierce, MA3-920) was used at 1:100, and rabbit polyclonal anti-EHD1 (Rainey et al., 2010) was used at 1:100. Secondary antibodies goat anti-rabbit Alexa 488 antibody (Invitrogen, A11008, Eugene, OR) was used at 1:2500–1:5000, goat anti-mouse Alexa 594 (Invitrogen, A11005, Eugene, OR) was used at 1:2500–1:5000. Images were captured using a Zeiss Axiophot microscope and Axiovision software (Carl Zeiss, Maple Grove, MN). Single slice confocal images were captured on a Leica SP2 confocal or a Leica SP5 superresolution laser scanning confocal microscope.
Expression of wild type and T72A EHD1
The full-length EHD1 cDNA was amplified from the pSport EHD1 vector (Addgene) and ligated into the EcoR1 site of pcDNA3-mcherry. The T72A mutation was generated by site directed mutagenesis using the following primers: (A) GGGTACCATGTTCAGCTGGGTGAGCAAGG (B) GCACCGGCAAGGCCACCTTCATCCG (C) GCCGAAGGCGTTGAGCTTGCGGAAGG (D) CGGATGAAGGTGGCCTTGCCGGTGC. pEGFPC1-muscle Amphiphsin II (BIN1 variant 8) was purchased from Addgene. C2C12 cells were plated at 50,000 cells per well on glass coverslips in 6-well plates. Cells were cotransfected with equal amounts of BIN1, EHD1 and T72A EHD1 plasmid or in combination using Lipofectamine and Plus Reagent in Opti-MEM (Invitrogen). Twenty-four hours post transfection, cells were fixed, mounted in Vectashield with DAPI and imaged. Images were acquired on the Leica SP5 II STED-CW superresolution laser scanning confocal microscope. To analyze BIN1 tubule formation, a 20μm2 area extending from the perinuclear region to cell periphery was selected. A custom script was written and run in Image J. Briefly, fluorescent images were processed with a rolling ball filter. A threshold was applied to the filtered images to generate binary images, which were then skeletonized similar to (Beraud et al., 2009). Tubules were defined as individual line segments in the skeletonized images. The number and length tubules were determined in each image. n>1000 tubules from at least 5 cells per transfection condition.
Electron microscopy
Quadriceps muscles from 2-month old wildtype and EHD1-null mice were dissected and placed in 4% paraformaldehyde. The tissue was postfixed in 1% OsO4, rinsed, and processed for Epon embedding. Embedded samples were sectioned and stained with 1% uranyl acetate followed by lead citrate. Semithin (0.5 μm) toluidine blue stained sections were used to pre-screen the tissue and select areas for subsequent thin sectioning and electron microscopy (Philips CM10).
Isolation and culture of primary myoblasts
Primary myoblast cultures were isolated from embryos taken 17 days post fertilization (dpf) and cultured as described (Demonbreun et al., 2010b; Rando and Blau, 1994). Briefly, cells were plated in primary cell growth media (Ham’s F-10 with 20% fetal bovine serum and bFGF 2.5ng/ml (Promega) with 1% antibiotic-antimycotic (Invitrogen, 15240-062) and 1% penicillin/streptomycin (Invitrogen, 15070-063). Two hundred thousand cells per well were plated on ECL-treated NaOH treated coverslips in 6-well plates. Cells were induced to differentiate through serum withdrawal. After 72 hours, cells were fixed with PFA and stained with anti-desmin and visualized using a Zeiss Axiophot microscope and Ivision software. At least four random images were taken per culture each at an equivalent cell density. The number of nuclei per desmin positive cell was counted, binned, and an un-paired t-test was performed using Prism Graphpad.
Vesicle Analysis and Immunostaining
Primary myoblasts were isolated as above and plated at equal densities on NaOH-washed glass coverslips within 6-well plates at 200,000 cells per well. Cells were fixed in 4% paraformaldehyde for 10 minutes. Blocking and antibody incubations were performed in 1X phosphate-buffered saline (PBS) containing 5% fetal bovine serum. Rat polyclonal anti-lysosomal associated membrane protein (LAMP)-2 antibody (Abcam, ab13524) was used at 1:600 and goat anti-rat conjugated to Alexa 488 was used at 1:2000. Coverslips were mounted using Vectashield with DAPI (Vector Laboratories, Burlingame, CA). Images were captured using a Zeiss Axiophot microscope and Axiovision software (Carl Zeiss, Maple Grove, MN). ImageJ particle analysis software was used to analyze particle number and statistical analysis was performed with Prism (Graphpad) using an unpaired t-test.
Immunoblotting
Wildtype and EHD1-null quadriceps muscles were homogenized and 20μg of protein was separated on a 4–12% gel (Life Technologies). Protein was transferred to PDVF membrane and blocked with T-20 Starting Block (Pierce). Mouse polyclonal anti-Rab5 was used at 1:250 (BD Transduction Laboratories, 610281) and mouse anti Rab-11 was used at 1:250 (BD Transduction Laboratories, 610656). Secondary goat anti-mouse HRP was used at 1:2500. Images were acquired on a Bio Spectrum Imaging System (UVP) and processed in Adobe Photoshop.
Transferrin internalization and chase assay
Primary myoblasts were seeded in 6-well plates at 200,000 cells per well. Cells were incubated in binding media (DMEM, 0.1% BSA, 20mM HEPES, pH 7.2) for 30 minutes and then pulsed with Alexa-546 conjugated transferrin (25 μg/ml, Molecular Probes) in binding media for 15 minutes. Cells were liberated from wells with trypsin for 1 minute, washed in 1X PBS, and then fixed in 4% paraformaldehyde for 10 minutes. For chase assay, cells isolated and pulsed as described above, then were chased for 0, 20, or 40 mins with 50 mM deferoxamine, 250 μg/ml of holo-transferrin in 20% FBS, 20 mM HEPES, pH 7.2. For both assays, cells were resuspended in PBS for analysis in a FACS Aria (BD Biosciences). After gating to remove dead cells, aggregated cells and debris, a minimum of 10,000 events was scored per culture. Cultures were derived from three independent animals per genotype for the internalization assay and at least two independent animals per genotype for the chase assay. Median fluorescence values were determined using a histogram of Alexa-546 fluorescence and normalized to wildtype cultures. FACS data was analyzed using FlowJo software (Tree Star, Inc). Statistical analysis was performed using Prism (Graphpad) using an unpaired t-test.
RESULTS
Defective endosomal trafficking in EHD1-null myoblasts
EHD proteins as well as several ferlin family members have previously been implicated in endosomal trafficking (Austin et al., 2010; Demonbreun et al., 2010a; Demonbreun et al., 2010b; Naslavsky et al., 2007; Rotem-Yehudar et al., 2001). To determine whether endocytic recycling was altered in myoblasts, we studied EHD1-null myoblasts using the transferrin assay (Demonbreun et al., 2010a; Doherty et al., 2008). Labeled transferrin is endocytosed to the recycling compartment and then recycled from the cell within minutes. Primary myoblasts isolated from wildtype and EHD1-null embryos were incubated for 15 minutes with transferrin conjugated to Alexa-546. We then analyzed the pulsed myoblasts for fluorescent intensity and demonstrated that EHD1-null primary myoblast cultures consistently had more mean fluorescent intensity than wildtype myoblast cultures as shown by the rightward shift in the scatter plots in the EHD1-null graph (Figure 1A, right). When quantified, this difference was statistically significant (p<0.001) (Figure 1B). To determine if this increase was due to enhanced uptake of transferrin or delayed recycling, we pulsed the myoblasts for 15 mins as described above, chased the cultures for 0, 20 or 40 minutes with unlabelled transferrin, and then analyzed the fluorescent intensity of the individual myoblasts. After 0, 20, and 40 mins of chase, a greater number of EHD1-null myoblasts displayed transferrin fluorescence (p = 0.02, p=0.03 and p=0.08 respectively) compared to wildtype controls, confirming a defect in EHD1-null myoblast recycling (Figure 1C). Specifically, after 20 mins of chase, 2.3-fold more EHD1-null myoblasts retained labeled transferrin than wildtype myoblasts very similar to the 3.3-fold increase in EHD1-null fibroblasts reported by Rapapport et al 2006. The endocytic recycling defect of EHD1-null myoblasts was visualized further as an increased number of LAMP2+ vesicles was present within EHD1-null myoblasts (Figure 1D and 1E) (p = 0.002). To assess further the defects in the endocytic recycling pathway, immunblotting for Rab5 and Rab11 was performed. Rab5 and Rab11 are proteins known to regulate endocytic recycling from the plasma membrane to the endocytic recycling compartment and from the endocytic recycling compartment to the plasma membrane, respectively. EHD1-null skeletal muscle expressed similar amounts of Rab5 protein as WT muscle. However, EHD1-null muscle had increased levels of Rab11 protein. These data suggest that changes in the Rab proteins are not the cause of the defects in transferrin accumulation but rather Rab 11 is upregulated in response to the loss of EHD1 likely to compensate for the accumulation at the endocytic recycling compartment. Together these data demonstrate that EHD1 is involved in endocytic recycling in muscle, and that muscle like neurons and fibroblasts is dependent on EHD1 for influences on the recycling and lysosomal pathways (Lasiecka et al., 2010; Rapaport et al., 2006).
Figure 1.

Reduced recycling and lysosome accumulation in EHD1-null myoblasts. A) Wildtype and EHD1-null primary myoblasts were incubated with transferrin conjugated to Alexa-546 for 15 minutes and analyzed for fluorescence intensity by FACS. FACS profiles showed increased transferrin accumulation in EHD1-null myoblasts. B) EHD1-null myoblasts contained approximately 20% more transferrin fluorescence compared to wildtype myoblasts (n=3 replicates, p = 0.001). C) Wildtype and EHD1-null primary myoblasts were pulsed with transferrin conjugated to Alexa-546, chased with unlabelled transferrin for 0, 20, or 40 minutes and then analyzed by FACS for fluorescence intensity. A greater number of EHD1-null myoblasts retained Alexa-546 fluorescence at 0, 20, and 40 minutes consistent with a defect in recycling (* p< 0.03, + p=0.08). D) Lysosomes were identified in wildtype and EHD1-null myoblasts using anti-LAMP2 antibody (green). Nuclei are stained with DAPI (blue). Scale bar 10μm. E) The number of LAMP2-positive vesicles was increased in EHD1-null primary myoblasts (mean number 23 in wildtype myoblasts and 38 in EHD1-null myoblasts, n=13, p=0.002). F) Immunoblot of WT and EHD1-null muscle lysates. WT and EHD1-null muscle show similar Rab5 expression levels. Rab11 expression levels are increased in EHD1-null muscle. Actin is shown as a loading control.
EHD1-null myoblasts display abnormal Fer1L5 and caveolin-3 aggregation
We previously showed a direct interaction between Fer1L5 and EHD1 (Posey et al., 2011b). We immunostained myoblasts from EHD1-null mice and wildtype littermates with an antibody specific for Fer1L5 and also for caveolin-3, a protein also implicated in muscle vesicle trafficking and muscle disease. In the confocal images of wildtype myoblasts, Fer1L5 (red) and caveolin-3 (green) colocalize throughout the myoblast and are seen localized in discrete tubules (Figure 2, top panel, dotted arrow). However, in EHD1-null myoblasts, Fer1L5 and caveolin-3 primarily colocalize in the perinuclear region (Figure 2, bottom panel, white arrow). In EHD1-null myoblasts both Fer1L5 and caveolin-3 were more diffusely distributed in all cells analyzed, presumably due to the mislocalization of the proteins. DIC images are shown to outline the cells. This data suggests that EHD1 is necessary for proper trafficking and localization of Fer1L5 and caveolin-3 and suggests, in addition to general recycling defects, ferlin aggregation as a mechanism for developmental defects in EHD1-null muscle.
Figure 2.

Loss of EHD1 impairs protein trafficking and fusion in myoblasts. A) Representative images of wildtype and EHD1-null primary myoblasts immunostained with anti-Fer1L5 (red), anti-caveolin-3 (green), and DAPI (blue). In normal myoblasts, Fer1L5 colocalized with caveolin-3 throughout the cytoplasm in discrete tubules (top row, dotted arrow). In EHD1-null myoblasts (bottom row), the majority of Fer1L5 and caveolin-3 were found in the perinuclear region (white arrow) with only minimal diffuse staining in the cytoplasm (arrowhead). Scale 10μm. High magnification images are found to the far right. Scale 5μm. B) Wildtype and EHD1-null myoblasts were differentiated and then stained with an anti-desmin antibody (red). Desmin staining identifies cells that have entered into myogenic differentiation and, in combination with DAPI (blue nuclei) staining, highlights multinucleate versus single nucleate cells. Scale bar 50μm. C) EHD1-null myoblasts fused poorly with very few binucleate or multinucleate myotubes.
Impaired myoblast fusion without EHD1
We next examined whether loss of EHD1 perturbed myoblast fusion. EHD1-null mice exhibit a significant embryonic lethality (Mate et al., 2012; Rainey et al., 2010). Most EHD1-null mice die during gestation or shortly after birth (Mate et al., 2012; Rainey et al., 2010). To avoid this lethality, we utilized viable embryos (17 dpf) as the source for primary myoblasts. Primary myoblasts were isolated from wildtype and EHD1-null embryos (2 wildtype cultures, both performed in duplicate; 3 EHD1-null cultures, one performed in duplicate, all from independent embryos). Cells were allowed to differentiate for 72 hours via serum withdrawal, fixed, and then stained with anti-desmin, a myogenic marker. Representative images are shown in Figure 2B. The number of nuclei in each desmin positive cell was counted and binned. Results are expressed graphically as the percentage of fibers with n number of nuclei per fiber/cell (Figure 2C). Despite serum withdrawal EHD1-null cells remained singly-nucleated in over 95% of desmin positive cells, while wildtype cells had readily formed large multinucleate myotubes. EHD1-null cells rarely fused into microtubes containing 2–3 nuclei, 3.5%, compared to wildtype cultures 28.8%, and rarely fused into myotubes containing 4+ nuclei, 1.5%, versus wildtype 28.8%. Increasing the density of desmin positive cells per area within the culture did not increase the fusion potential of EHD1-null cultures, but did induce larger myotubes in wildtype cultures (data not shown). These results suggest that both recycling and fusion pathways are altered by the loss of EHD1 and ultimately contribute to muscle growth.
EHD1-null mice have smaller muscles
Surviving EHD1-null mice were previously noted to have significantly reduced body mass compared to wildtype littermates (Mate et al., 2012; Rainey et al., 2010). Because of the contribution of muscle mass to overall body mass, we examined muscle in EHD1-null mice. For these experiments, we used surviving EHD1-null mice and studied animals at 8–10 weeks of age. Representative images from wildtype and EHD1-null triceps and quadriceps muscle cross-sections are shown in Supplemental Figure 1A and 1D. The cross-sectional area (CSA) of whole muscles from EHD1-null was reduced in both the upper extremities (Supplemental Figure 1B) and lower extremities (Supplemental Figure 1E). In quadriceps muscle, the EHD1-null CSA was 16.80 mm2 and in WT it was 23.72 mm2, (n= 4 quadriceps from 3 mice, p=0.007). In the triceps muscle, the mean CSA was 12.30 mm2 in EHD1-null and 17.54 mm2 in WT (n=6 triceps from 3 mice, p=0.004.)
The reduced muscle size in EHD1-null mice derived from both reduction of myofiber number as well as myofiber size. EHD1-null quadriceps muscles contained approximately 20% fewer myofibers than wildtype littermates. In quadriceps muscle, the EHD1-null average fiber number was 5711 and in WT it was 7121, (n=3, p=0.0072). In triceps muscle, the average fiber number was 5095 in EHD1-null and 6193 in WT (n = 4, p=0.02.) (Supplemental Figure 1C,F). Individual myofibers also had reduced CSA in EHD1-null muscle relative to wildtype littermates (Figure 3A). EHD1-null quadriceps myofibers had a mean CSA of 1,589μm2 compared to the mean CSA of wildtype muscle fibers of 2,522μm2, which represents a 37% reduction in mean fiber size (n= at least two hundred fibers per mouse, three mice per genotype, p<0.0001). We examined the distribution of myofiber size in the quadriceps muscle and found that EHD1-null muscle specifically lacked the largest myofibers (Figure 3B, arrows). Myofibers larger than 3,500μm2 in area contribute nearly a quarter (23.1%) of wildtype muscle; these fibers only contribute 0.7% of EHD1-null muscle myofibers.
Figure 3.

Reduced muscle size in EHD1-null muscle. A) Multiple images of the quadriceps muscles were used to quantify fiber size as cross sectional area (CSA). EHD1-null myofibers had reduced CSA. The mean wildtype fiber CSA size was 2,522μm2 while the EHD1-null mean was 1,589μm2 (p<0.0001). B) EHD1-null muscle contained smaller muscle fibers and specifically the largest myofibers were absent (black arrows). C) Representative images from mature wildtype and EHD1-null quadriceps muscle. EHD1-null myofibers were smaller in cross sectional area and do not display obvious fibrosis as would be seen in muscular dystrophy. Scale bar 50μm. D) Electron micrographs of EHD1-null quadriceps muscle. White arrows depict lysosomal vesicles within EHD1-null muscle. Lysosomes were never seen in wildtype muscle. High magnification image (right) shows lysosomes filled with debris.
Lysosomal aggregation in EHD1-null skeletal muscle
Histopathology of EHD1-null muscle showed a largely intact muscle that was not characterized by fiber necrosis, fibrosis, fat, and immune infiltrate, features commonly found in muscular dystrophy (Figure 3C). At the ultrastructural level, sarcomere structure appeared normal. However, lysosomal accumulations were seen regularly in EHD1-null muscle and were never seen in WT tissue (Figure 3D). A higher magnification electron microscopy image shows lysosomes containing debris in EHD1-null muscle (Figure 3D, right) (Rodriguez et al., 1997). The accumulation of enlarged lysosomes is similar to what was seen in myoferlin-null skeletal muscle, suggesting a trafficking defect in EHD1-null muscle (Demonbreun et al., 2010b).
EHD1 localizes to the plasma membrane and T-tubules in healthy, wildtype skeletal muscle
BIN1 and DHPR are known markers of transverse tubules, a sarcolemmal invagination specialized for calcium delivery. BIN1 also colocalizes with other important muscle proteins at the tubule including caveolin-3 and Fer1L1, also known as Dysferlin (Klinge et al., 2007; Lee et al., 2002). In normal muscle imaged under high-resolution immunofluorescence microscopy, EHD1 localized to the T-tubule in skeletal muscle. In single slice confocal images of myofibers, EHD1 localizes partially with the T-tubule protein DHPR (Figure 4, DHPR red, EHD1 green, right merge). High magnification images are shown on the far right. Additionally, in single slice confocal images of myofibers, EHD1 localizes partially with BIN1, an additional T-tubule marker, at T-tubule structures and at the sarcolemma (Figure 4B; BIN1 red, EHD1 green, right merge). High magnification images are shown on the far right. The localization of EHD1 to the tubule region suggests a potential role for EHD1 at the T-tubule.
Figure 4.
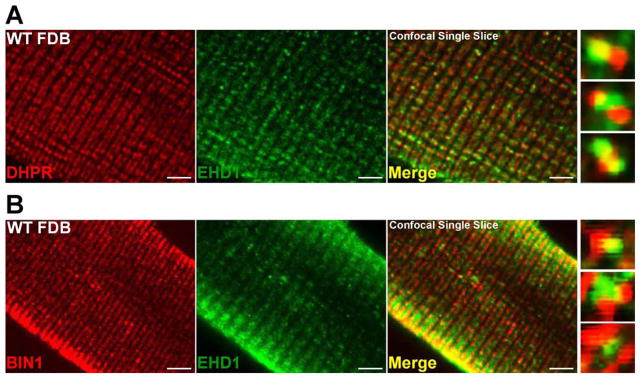
EHD1 localizes to the T-tubule in wildtype muscle. Representative single slice confocal images are shown with high magnification images on the right. A) Myofibers were stained with anti-DHPR (red) and anti-EHD1 (green) antibodies. EHD1 localizes partially with the T-tubule marker, DHPR. B) Additional myofibers were stained with anti-BIN1 (red) and anti-EHD1 (green) antibodies. EHD1 staining partially overlaps with BIN1 at the T-tubule and at the sarcolemma within wildtype myofibers. Scale bar 5μm.
T-tubular overgrowth in EHD1-null skeletal muscle
EHD1 has been implicated in tubule structure formation (Caplan et al., 2002). BIN1, also known as Amphiphysin 2, is involved in vesicle trafficking and in generating tubular invaginations implicated in T-tubule biogenesis, and mutations in BIN1 cause centronuclear myopathy (Nicot et al., 2007; Toussaint et al., 2007). Figure 5 shows mature muscle from wildtype (top row) and EHD1-null (bottom row) muscle with anti-Fer1L5 and anti-BIN1 processed and imaged identically. In longitudinal sections, EHD1-null myofibers contain elongated T-tubules with high levels of anti-BIN-1 expression in contrast to punctate structures viewed in wildtype controls (Figure 5, left panels). Fer1L5 localization mirrored what was seen for BIN1 in both the EHD1-null and wildtype muscle with elongated abnormal T-tubules within the EHD1-null muscle compared to the small punctate structures found in normal muscle (Figure 5, middle panels). Normal muscle rarely showed colocalization of Fer1L5 and BIN1 in T-tubule puncta, while the malformed EHD1-null T-tubules showed colocalization in the majority of T-tubules (Figure 5, right panels).
Figure 5.

T-tubule overgrowth in EHD1-null skeletal muscle. Longitudinal sections of wildtype and EHD1-null quadriceps muscle confirm increased anti-BIN1 (red) and anti-Fer1L5 (green) staining in malformed, elongated T-tubules of EHD1-null muscle compared to regularly shaped punctate tubules in wildtype controls. Scale bar 20μm.
Mislocalized caveolin-3 and Fer1L5 in EHD1-null muscle
We next studied the localization of both caveolin-3 and Fer1L5 in mature muscle (ten weeks of age). Immunofluorescence microscopy of wildtype muscle showed anti-Fer1L5 staining primarily within the fiber in small vesicles and tubules (Figure 6A top row, green) and caveolin-3 staining localized to the plasma membrane (Figure 6A, top row, red). Images of EHD1-null muscle, processed and imaged identically to the wildtype muscle, showed more intense staining of both Fer1L5 and caveolin-3 within the myofibers and caveolin-3 at the sarcolemma (Figure 6A bottom row, left and middle). Anti-dystrophin staining (green) is present at the sarcolemma in wildtype and EHD1-null myofibers at equal levels, demonstrating the integrity of both muscle sections despite the abnormal caveolin-3 membrane localization in the EHD1-null muscle (Figure 6B). Confocal imaging of wildtype fibers shows caveolin-3 was largely confined to the sarcolemma, while Fer1L5 was present at the sarcolemma and internal puncta (Figure 6C top row, merge right). In EHD1-null muscle, caveolin-3 was increased at the sarcolemma (Figure 6C bottom row, red) and internal caveolin-3, within the myofiber, was found to colocalize with internal Fer1L5 expression (Figure 6C bottom row, white arrow, right, merge). Fer1L5 was found in vesicles or tubular-like structures often adjacent to caveolin-3-rich areas at the plasma membrane (Figure 6C bottom row, right, merge). A low magnification image of internal tubules enriched in caveolin-3 and Fer1L5 within an EHD1-null myofiber is shown in Figure 6D.
Figure 6.

Abnormal localization of Fer1L5 and caveolin-3 in mature EHD1-null muscle. Mature muscle was stained with anti-Fer1L5 (green) and anti-caveolin-3 (red) antibodies. DAPI is shown in blue. A) Fer1L5 and caveolin-3 internal staining was increased in EHD1-null muscle (bottom row) compared to wildtype controls (top row). Scale bar 30μm. B) Wildtype and EHD1-null muscle were stained with anti-dystrophin (green) and DAPI (blue) to outline the myofibers, confirming the presence of a uniform sarcolemma in both samples. Scale bar 30μm. C) Fer1L5 and caveolin-3 colocalized in EHD1-null muscle at the membrane in vesicular patterns and within the muscle fiber on tubule structures (bottom row; merge, arrow). Scale bar 1μm. D) Low magnification image of an EHD1-null myofiber with excessive internal Fer1L5 and caveolin-3 tubules. Scale bar 10μm.
Ultrastructural overgrowth of T-tubules in the absence of EHD1
To confirm the immunofluorescence data, we analyzed the sarcotubule network by electron microscopy. At the ultrastructural level, elongated T-tubules (Figure 7, white arrowheads) were associated with multiple vesicles in the EHD1-null muscle (Figure 7, white arrows), distinct from what was seen in normal muscle. The majority of T-tubules in EHD1-null muscle stretched over 2μm in length, approximately 20 times longer than wildtype T-tubules. A magnified view of an elongated T-tubule surrounded by vesicular structures in EHD-null muscle is depicted in the right panel of Figure 7. Additionally, the sarcoplasmic reticulum was dilated in EHD1-null muscle compared to normal controls (Figure 7, black arrow). The abnormal T-tubules and sarcoplasmic reticulum are consistent with a defect in vesicle trafficking, which results in profound defects in the sarco-tubule system of mature muscle.
Figure 7.

Enlarged sarcoplasmic reticulum and T-tubular structures in the absence of EHD1. Representative electron microscopy images of swollen sarcoplasmic reticulum (black arrow), elongated T-tubules (white arrowhead), and vesicular structures (white arrow) juxtapose the T-tubules in EHD1-null quadriceps muscle. This was never seen in wildtype controls. A magnified view of the EHD1-null disrupted sarco-tubular system is shown in the right panel.
The EHD1 ATPase domain is necessary for EHD1 tubule formation
The EHD proteins are composed of an ATPase domain, a central coiled-coil region, and the EH domain necessary for many protein-protein interactions, including the EHD-ferlin interaction. Detailed structural and residue analysis of EHD1 is lacking. However, structural analysis of EHD2 is available including information on several key amino acid residues known to affect the ATPase domain and protein function (Daumke et al., 2007). The T72A mutation substitutes an alanine on a residue within the nucleotide binding P-loop of the ATPase domain and abolishes EHD2 ATPase activity (Daumke et al., 2007). ATPase activity is thought to be required for membrane scission of exocytic vesicles from the endocytic recycling compartment after EHD proteins oligomerize around the membrane (Daumke et al., 2007). Because EHD1 and EHD2 are over 70% homologous, and siRNA reduction of both proteins inhibited myoblast fusion myogenic in C2C12 cells (Posey et al., 2011b), we generated the T72A mutation within a carboxy-terminal EHD1-mcherry fusion protein and expressed it in undifferentiated C2C12 myoblasts. In the presence of endogenous EHD1, EHD1-mCherry localized to discrete tubules throughout the myoblast (Figure 8A, left). Upon EHD1T72A expression, EHD1T72A-mCherry was instead dispersed throughout the cytoplasm and EHD1T72A-mCherry positive tubules were not formed in C2C12 myoblasts (Figure 8A, right). A high resolution image of the boxed area within each myoblast is found in the lower right of each panel (Figure 8). The mis-localization of mutant EHD1 protein to the cytoplasm was reminiscent of what was seen when EHD2T72A was expressed in other cell types (George et al., 2007; Moren et al., 2012). Thus, the EHD1 ATPase activity is necessary for EHD1 tubule formation in myoblasts and tubule formation may relate to myoblast fusion.
Figure 8.

EHD1 regulates BIN1 mediated tubule formation. A) C2C12 myoblasts were transfected with wildtype EHD1-mCherry, EHD1T72A-mCherry, or BIN1-GFP. Cells were fixed twenty-four hours after transfection. A & B) EHD1 and BIN1 normally form tubules in C2C12 myoblasts. Expression of EHD1T72A results in lack of tubule formation and aggregation of EHD1T72A in the cytoplasm. High magnification images are shown in the lower right corner. C) BIN1-GFP was transfected into C2C12 cells in combination with EHD1-mCherry or EHD1T72A-mCherry. EHD1 and BIN1 together form an organized tubule network with discrete points of colocalization. The presence of EHD1T72A results in smaller (0.51μm compared to 0.662μm tubules, p=0.007) and more numerous BIN1-positive tubules (268 compared to 166 tubules per 20μm2 area, p=0.04). The white dotted box highlights the area of the cell depicted in the magnified image to the right. Scale bar 20μm, 1.5μm.
EHD1 regulates BIN1 tubule formation
Previous work established a molecular and functional interaction between EHD and amphiphysin/BIN orthologs RME-1 and AMPH-1 in C. elegans (Pant et al., 2009). BIN1 is known to be necessary for the formation of T-tubules in vivo (Lee et al., 2002). C2C12 cells were transfected with BIN1-GFP, EHD1-mCherry, and/or EHD1T72A alone or in combination. Twenty-four hours post transfection tubule formation within cells was monitored. BIN1-GFP or EHD1-mCherry individually can support the formation of intracellular tubules in C2C12 myoblasts (Figure 8A & B). The combination of EHD1-mCherry and BIN1-GFP generated large, organized tubules, extending from the perinuclear region to the periphery in C2C12 myoblasts (Figure 8C, top row). The combination of EHD1T72A-mCherry and BIN1-GFP produced tubules that were smaller (0.51μm compared to 0.662μm tubules, p=0.007), more numerous (268 compared to 166 tubules per 20μm2 area, p=0.04) and less organized (Figure 8C, bottom row). Comparison of the high magnification insets in Figure 8C highlights the less organized nature of the BIN1 tubules induced by EHD1T72A versus EHD1. These data are consistent with the observed in vivo ectopic tubule formation seen in EHD1-null muscle and suggest that this defect arises directly from misregulation of BIN1.
DISCUSSION
Defective recycling in myoblast from loss of EHD1
Mammalian models of loss-of-function mutations for EHD proteins have only recently become available, and the initial characterizations have focused on male fertility (George et al., 2010; Rainey et al., 2010; Rapaport et al., 2006). Although most EHD1-null mice die during in utero development, approximately 5–8% survive into adulthood (George et al., 2010; Rainey et al., 2010). Why some mice survive to adulthood is not known, but the high conservation between EHD proteins suggests that compensation by other EHD family members may, in some cases, be sufficient to allow survival. We now found that EHD1 is critically important for muscle development, growth and function. We previously linked EHD proteins to receptor recycling defects in muscle finding that myoferlin directly interacts with EHD1 and EHD2, and that loss of myoferlin leads to abnormal receptor trafficking (Demonbreun et al., 2010b; Doherty et al., 2008; Posey et al., 2011a; Posey et al., 2011b). IGF is a potent stimulator of prenatal growth. Normally IGF1 stimulation causes muscle fiber hypertrophy and in developing myofibers (Zanou and Gailly, 2013). Myoferlin-null mice do not respond to IGF1 stimulation (Demonbreun et al., 2010b) and notably after exposure to IGF, the IGF1 receptor was found in lysosomes in myoferlin null myoblasts. Dysferlin null mice, a model for limb girdle muscular dystrophy, also do not respond to IGF exposure (Demonbreun et al., 2010b). We now show that loss of EHD1 similarly disrupts recycling of transferrin and we expect loss of EHD1 similarly disrupts receptors that mediate growth in a variety of tissues including muscle. This may include the IGF1 receptor, but given the profound developmental phenotype in EHD1 mice, this molecular defect likely extends to implicate other receptors as well.
EHD1 is required for normal myogenesis
Muscle development can be modeled in cell culture where cultured myoblasts can be induced to differentiate and fuse in response to serum withdrawal. The formation of de novo myotubes is a process that requires membrane and vesicle trafficking (Posey et al., 2011a). Myoblast fusion requires the coalescence of two membranes. We found that cultured myoblasts from EHD1-null mice were highly defective in myoblast fusion. These cells expressed desmin, a marker of cell specification in the early stages of myogenesis, so loss of EHD1 does not impair the early phase of myoblast differentiation. Transcriptional control of myogenesis is well established where muscle specific transcription factors such as MyoD, Mef2, Myf5 and myogenin drive differentiation (Hasty et al., 1993; Molkentin et al., 1995; Olson et al., 1995; Rudnicki et al., 1993). We speculate that normal myoblast differentiation requires cues from the cytoplasm and matrix to be fully successful and this transmission requires normal vesicle trafficking.
The ferlins are found enriched at the plasma membrane, but also within internal vesicles that may be associated with the plasma membrane (Bansal et al., 2003; Doherty et al., 2005; Doherty et al., 2008; Posey et al., 2011b). Myoferlin is enriched at the sites of cell-cell fusion during myogenesis (Doherty et al., 2005). Intracellular aggregation of the ferlins is expected to be associated with an inhibition of normal function. Internalized ferlin staining patterns have been observed in muscle biopsies from patients with varying forms of muscle disease. However, the specificity and significance of this finding is not yet clear. In a form of centronuclear myopathy caused by a mutation in dynamin-2, dysferlin is sequestered from the plasma membrane and accumulates within the myofiber (Durieux et al., 2010). We suggest that ferlin mislocalization significantly contributes to the growth defect in EHD1-null muscle.
Vesicle trafficking in muscle
Loss of function mutations in dysferlin and caveolin-3, two other proteins involved in vesicle trafficking, cause muscular dystrophy (Bashir et al., 1998; McNally et al., 1998). EHD1-null muscle is not dystrophic, but rather is growth impaired, and this is associated with grossly malformed T-tubules. Loss of dysferlin, caveolin-3 or BIN1 causes vesicle accumulation and T-tubule abnormalities within muscle (Galbiati et al., 2001; Klinge et al., 2010; Toussaint et al., 2011). EHD1 lacks a transmembrane domain and is thought to interact with protein components via its EH domain. EHDs are associated with the cytoplasm and with actin binding proteins. We propose a model in which the EHDs, in an ATPase dependent manner, dynamically associate with ferlins, caveolin-3, and BIN-1 proteins as well as with other interacting partners during development at the T-tubule and at the sarcolemma allowing cytoplasmic rearrangements and vesicle membrane fusion. This is consistent with the role of the EHD family of proteins as an important regulator of intracellular trafficking at critical nodes of the endocytic recycling pathway like the Rab family of GTPases (Naslavsky and Caplan, 2010). Muscle, with its unique structure and development, is highly dependent on this intracellular trafficking.
Supplementary Material
Highlights.
Muscle without EHD1 lacks large myofibers.
Myoblasts without EHD are defective in myoblast fusion.
The ATPase domain of EHD1 is necessary for proper protein trafficking in muscle.
Muscle without EHD1 has overgrowth of transverse (T-) tubules.
EHD1 regulates BIN1 and T-tubule formation.
Acknowledgments
Supported by NIH NS047726.
Abbreviations
- CSA
cross sectional area
- T-tubule
Transverse tubule
- SR
Sarcoplasmic Reticulum
- EHD
Eps15 Homology Domain
Footnotes
CONFLICT OF INTEREST
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Austin P, Heller M, Williams DE, McIntosh LP, Vogl AW, Foster LJ, Andersen RJ, Roberge M, Roskelley CD. Release of membrane-bound vesicles and inhibition of tumor cell adhesion by the peptide Neopetrosiamide A. PLoS One. 2010;5:e10836. doi: 10.1371/journal.pone.0010836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–72. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I, Marconi G, Passos-Bueno MR, de Moreira ES, Zatz M, Beckmann JS, Bushby K. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, Saks V. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr. 2009;41:195–214. doi: 10.1007/s10863-009-9214-x. [DOI] [PubMed] [Google Scholar]
- Braun A, Pinyol R, Dahlhaus R, Koch D, Fonarev P, Grant BD, Kessels MM, Qualmann B. EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol Biol Cell. 2005;16:3642–58. doi: 10.1091/mbc.E05-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, Donaldson JG, Bonifacino JS. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. Embo J. 2002;21:2557–67. doi: 10.1093/emboj/21.11.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007 doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- de Beer T, Carter RE, Lobel-Rice KE, Sorkin A, Overduin M. Structure and Asn-Pro-Phe binding pocket of the Eps15 homology domain. Science. 1998;281:1357–60. doi: 10.1126/science.281.5381.1357. [DOI] [PubMed] [Google Scholar]
- Demonbreun AR, Fahrenbach JP, Deveaux K, Earley JU, Pytel P, McNally EM. Impaired muscle growth and response to insulin-like growth factor 1 in dysferlin-mediated muscular dystrophy. Hum Mol Genet. 2010a doi: 10.1093/hmg/ddq522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demonbreun AR, Posey AD, Heretis K, Swaggart KA, Earley JU, Pytel P, McNally EM. Myoferlin is required for insulin-like growth factor response and muscle growth. FASEB J. 2010b;24:1284–95. doi: 10.1096/fj.09-136309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–75. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty KR, Demonbreun AR, Wallace GQ, Cave A, Posey AD, Heretis K, Pytel P, McNally EM. The endocytic recycling protein EHD2 interacts with myoferlin to regulate myoblast fusion. J Biol Chem. 2008;283:20252–60. doi: 10.1074/jbc.M802306200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux AC, Vignaud A, Prudhon B, Viou MT, Beuvin M, Vassilopoulos S, Fraysse B, Ferry A, Laine J, Romero NB, Guicheney P, Bitoun M. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet. 2010;19:4820–36. doi: 10.1093/hmg/ddq413. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H, Jr, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–33. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- George M, Rainey MA, Naramura M, Ying G, Harms DW, Vitaterna MH, Doglio L, Crawford SE, Hess RA, Band V, Band H. Ehd4 is required to attain normal prepubertal testis size but dispensable for fertility in male mice. Genesis. 2010;48:328–42. doi: 10.1002/dvg.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George M, Ying G, Rainey MA, Solomon A, Parikh PT, Gao Q, Band V, Band H. Shared as well as distinct roles of EHD proteins revealed by biochemical and functional comparisons in mammalian cells and C. elegans. BMC Cell Biol. 2007;8:3. doi: 10.1186/1471-2121-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B, Zhang Y, Paupard MC, Lin SX, Hall DH, Hirsh D. Evidence that RME-1, a conserved C. elegans EH-domain protein, functions in endocytic recycling. Nat Cell Biol. 2001;3:573–9. doi: 10.1038/35078549. [DOI] [PubMed] [Google Scholar]
- Grant BD, Caplan S. Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic. 2008;9:2043–52. doi: 10.1111/j.1600-0854.2008.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grounds MD, Shavlakadze T. Growing muscle has different sarcolemmal properties from adult muscle: a proposal with scientific and clinical implications: reasons to reassess skeletal muscle molecular dynamics, cellular responses and suitability of experimental models of muscle disorders. Bioessays. 2011;33:458–68. doi: 10.1002/bies.201000136. [DOI] [PubMed] [Google Scholar]
- Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, Koval OM, Cunha SR, George M, Rainey MA, Kashef FE, Dun W, Boyden PA, Anderson ME, Band H, Mohler PJ. EH domain proteins regulate cardiac membrane protein targeting. Circ Res. 2010;107:84–95. doi: 10.1161/CIRCRESAHA.110.216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Soriano NA, Bose S, Holik J, Bose A, Pomerleau DP, Furcinitti P, Leszyk J, Corvera S, Czech MP. EHD2 and the novel EH domain binding protein EHBP1 couple endocytosis to the actin cytoskeleton. J Biol Chem. 2004;279:10593–605. doi: 10.1074/jbc.M307702200. [DOI] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–6. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Klinge L, Harris J, Sewry C, Charlton R, Anderson L, Laval S, Chiu YH, Hornsey M, Straub V, Barresi R, Lochmuller H, Bushby K. Dysferlin associates with the developing T-tubule system in rodent and human skeletal muscle. Muscle Nerve. 2010;41:166–73. doi: 10.1002/mus.21166. [DOI] [PubMed] [Google Scholar]
- Klinge L, Laval S, Keers S, Haldane F, Straub V, Barresi R, Bushby K. From T-tubule to sarcolemma: damage-induced dysferlin translocation in early myogenesis. FASEB J. 2007;21:1768–76. doi: 10.1096/fj.06-7659com. [DOI] [PubMed] [Google Scholar]
- Lasiecka ZM, Yap CC, Caplan S, Winckler B. Neuronal early endosomes require EHD1 for L1/NgCAM trafficking. J Neurosci. 2010;30:16485–97. doi: 10.1523/JNEUROSCI.3127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–6. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- Marg A, Schoewel V, Timmel T, Schulze A, Shah C, Daumke O, Spuler S. Sarcolemmal repair is a slow process and includes EHD2. Traffic. 2012;13:1286–94. doi: 10.1111/j.1600-0854.2012.01386.x. [DOI] [PubMed] [Google Scholar]
- Mate SE, Van Der Meulen JH, Arya P, Bhattacharyya S, Band H, Hoffman EP. Eps homology domain endosomal transport proteins differentially localize to the neuromuscular junction. Skelet Muscle. 2012;2:19. doi: 10.1186/2044-5040-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EM, de Sa Moreira E, Duggan DJ, Bonnemann CG, Lisanti MP, Lidov HG, Vainzof M, Passos-Bueno MR, Hoffman EP, Zatz M, Kunkel LM. Caveolin-3 in muscular dystrophy. Hum Mol Genet. 1998;7:871–7. doi: 10.1093/hmg/7.5.871. [DOI] [PubMed] [Google Scholar]
- Mintz L, Galperin E, Pasmanik-Chor M, Tulzinsky S, Bromberg Y, Kozak CA, Joyner A, Fein A, Horowitz M. EHD1--an EH-domain-containing protein with a specific expression pattern. Genomics. 1999;59:66–76. doi: 10.1006/geno.1999.5800. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 1995;83:1125–36. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- Moren B, Shah C, Howes MT, Schieber NL, McMahon HT, Parton RG, Daumke O, Lundmark R. EHD2 regulates caveolar dynamics via ATP-driven targeting and oligomerization. Mol Biol Cell. 2012;23:1316–29. doi: 10.1091/mbc.E11-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Caplan S. EHD proteins: key conductors of endocytic transport. Trends Cell Biol. 2010 doi: 10.1016/j.tcb.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Rahajeng J, Rapaport D, Horowitz M, Caplan S. EHD1 regulates cholesterol homeostasis and lipid droplet storage. Biochem Biophys Res Commun. 2007;357:792–9. doi: 10.1016/j.bbrc.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot AS, Toussaint A, Tosch V, Kretz C, Wallgren-Pettersson C, Iwarsson E, Kingston H, Garnier JM, Biancalana V, Oldfors A, Mandel JL, Laporte J. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134–9. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- Olson EN, Perry M, Schulz RA. Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Dev Biol. 1995;172:2–14. doi: 10.1006/dbio.1995.0002. [DOI] [PubMed] [Google Scholar]
- Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol. 2009;11:1399–410. doi: 10.1038/ncb1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey AD, Jr, Demonbreun A, McNally EM. Ferlin proteins in myoblast fusion and muscle growth. Curr Top Dev Biol. 2011a;96:203–30. doi: 10.1016/B978-0-12-385940-2.00008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey AD, Jr, Pytel P, Gardikiotes K, Demonbreun AR, Rainey M, George M, Band H, McNally EM. Endocytic recycling proteins EHD1 and EHD2 interact with fer-1-like-5 (Fer1L5) and mediate myoblast fusion. J Biol Chem. 2011b;286:7379–88. doi: 10.1074/jbc.M110.157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey MA, George M, Ying G, Akakura R, Burgess DJ, Siefker E, Bargar T, Doglio L, Crawford SE, Todd GL, Govindarajan V, Hess RA, Band V, Naramura M, Band H. The endocytic recycling regulator EHD1 is essential for spermatogenesis and male fertility in mice. BMC Dev Biol. 2010;10:37. doi: 10.1186/1471-213X-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–87. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport D, Auerbach W, Naslavsky N, Pasmanik-Chor M, Galperin E, Fein A, Caplan S, Joyner AL, Horowitz M. Recycling to the plasma membrane is delayed in EHD1 knockout mice. Traffic. 2006;7:52–60. doi: 10.1111/j.1600-0854.2005.00359.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem-Yehudar R, Galperin E, Horowitz M. Association of insulin-like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem. 2001;276:33054–60. doi: 10.1074/jbc.M009913200. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–9. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Sharma M, Giridharan SS, Rahajeng J, Naslavsky N, Caplan S. MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol Biol Cell. 2009;20:5181–94. doi: 10.1091/mbc.E09-06-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simionescu-Bankston A, Leoni G, Wang Y, Pham PP, Ramalingam A, Duhadaway JB, Faundez V, Nusrat A, Prendergast GC, Pavlath GK. The N-BAR domain protein, Bin3, regulates Rac1- and Cdc42-dependent processes in myogenesis. Dev Biol. 2013;382:160–71. doi: 10.1016/j.ydbio.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint A, Cowling BS, Hnia K, Mohr M, Oldfors A, Schwab Y, Yis U, Maisonobe T, Stojkovic T, Wallgren-Pettersson C, Laugel V, Echaniz-Laguna A, Mandel JL, Nishino I, Laporte J. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 2011;121:253–66. doi: 10.1007/s00401-010-0754-2. [DOI] [PubMed] [Google Scholar]
- Toussaint A, Nicot AS, Mandel JL, Laporte J. Mutations in amphiphysin 2 (BIN1) cause autosomal recessive centronuclear myopathy. Med Sci (Paris) 2007;23:1080–2. doi: 10.1051/medsci/200723121080. [DOI] [PubMed] [Google Scholar]
- Verma P, Ostermeyer-Fay AG, Brown DA. Caveolin-1 induces formation of membrane tubules that sense actomyosin tension and are inhibited by polymerase I and transcript release factor/cavin-1. Mol Biol Cell. 2010;21:2226–40. doi: 10.1091/mbc.E09-05-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanou N, Gailly P. Skeletal muscle hypertrophy and regeneration: interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.