

Abstract
Oncogenic mutations in components of the JAK/STAT pathway, including those in cytokine receptors and JAKs, lead to increased activity of downstream signaling and are frequently found in leukemia and other hematological disorders. Thus, small-molecule inhibitors of this pathway have been the focus of targeted therapy in these hematological diseases. We previously showed that t(8;21) fusion protein AML1-ETO and its alternatively spliced variant AML1-ETO9a (AE9a) enhance the JAK/STAT pathway via down-regulation of CD45, a negative regulator of this pathway. To investigate the therapeutic potential of targeting JAK/STAT in t(8;21) leukemia, we examined the effects of a JAK2-selective inhibitor TG101209 and a JAK1/2-selective inhibitor INCB18424 on t(8;21) leukemia cells. TG101209 and INCB18424 inhibited proliferation and promoted apoptosis of these cells. Furthermore, TG101209 treatment in AE9a leukemia mice reduced tumor burden and significantly prolonged survival. TG101209 also significantly impaired the leukemia-initiating potential of AE9a leukemia cells in secondary recipient mice. These results demonstrate the potential therapeutic efficacy of JAK inhibitors in treating t(8;21) AML.
Keywords: AML1-ETO, AML1-ETO9a, JAK/STAT, JAK2 inhibitors, t(8;21)
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease that is classified based on the presence of specific cytogenetic abnormalities [e.g. t(8;21)(q22;q22), inv(16)(p13q22) and t(15;17)(q22;q12)] and gene mutations (e.g. NPM1, CEBPA and FLT3 mutations), as well as the French-American-British (FAB) classification of the leukemia cells and immunophenotype.1–3 t(8;21) is one of the common cytogenetic abnormalities identified in AML,4 which is associated with nearly 40% of cases of the FAB-M2 subtype.5
t(8;21) disrupts the functions of the core binding factor (CBF), a heterodimeric transcription factor complex essential for hematopoiesis. CBF is composed of a DNA-binding CBFα subunit (AML1, 2, or 3, also called RUNX1, 3, or 2, respectively) and a non-DNA- binding CBFβ subunit. t(8;21) involves the AML1 (RUNX1) gene on chromosome 21 and the ETO (MTG8, RUNX1T1) gene on chromosome 8, generating the fusion transcription factor AML1-ETO.6 Studies in Runx1−/− and AML1-ETO knock-in mice indicate that AML1-ETO dominantly blocks AML1 function during early embryo development.7–10 AML1-ETO also modulates functions of several other transcription factors, thereby altering gene expression globally.11,12 Although AML1-ETO is critical for the pathogenesis of myeloid leukemia, it requires one or more additional mutations to cause leukemia in mice.6 A C-terminally truncated variant of AML1-ETO named AML1-ETO9a (AE9a), resulting from alternative splicing and found to co-exist with full-length AML1-ETO in most analyzed t(8;21) AML patients, causes rapid onset of leukemia in mice.13
Patients diagnosed with t(8;21) AML undergo conventional intensive chemotherapy and have a relatively favorable prognosis compared with other types of AMLs.14,15 About 90% of the patients achieve complete remission. However, despite this high remission rate, approximately half of them eventually relapse, which indicates the need for improved therapeutic strategies.12,16–18 We previously combined gene expression and promoter occupancy profiling assays using AE9a-induced primary murine leukemia cells to identify direct target genes of AE9a and explore potential therapeutic targets for treating t(8;21) AML. We showed that CD45, a negative regulator of JAK/STAT signaling, is significantly down-regulated in AE9a leukemia mice and human t(8;21) AML. Furthermore, we demonstrated that JAK/STAT signaling is hyper-activated in these leukemia cells.19 Thus JAK/STAT inhibitors may be effective in treating t(8;21) AML.
The JAK/STAT signaling pathway is frequently activated in leukemia and other hematological disorders. This may occur via activating mutations in upstream cytokine receptors including FLT3, cKIT and G-CSFR and constitutively active JAK kinases such as JAK2V617F and TEL-JAK2.20 These genetic aberrations are underlying causes of many hematological diseases. In particular, the JAK2-activating mutation JAK2V617F is found in a large proportion of myeloproliferative neoplasms such as polycythemia vera (PV; 81–99%), essential thrombocythemia (ET; 41–72%) and myelofibrosis (MF; 39–57%).21 Therefore, small-molecule inhibitors targeting JAK2 have been the focus in the development of targeted therapy.21,22 In addition to upstream activating mutations, down-regulation of a negative regulator of the JAK/STAT pathway could also contribute to activation of this pathway, as we showed previously in t(8;21) AML.19 In the current study, we test the therapeutic potential of JAK inhibition in AE9a-induced AML. We demonstrate that inhibition of JAK1 and/or JAK2 by shRNA or small-molecule inhibitors effectively suppresses the colony-forming ability of AML1-ETO and AE9a-transformed hematopoietic cells. A JAK2-selective inhibitor TG10120923 and a JAK1/2-selective inhibitor INCB1842424 inhibited proliferation and promote apoptosis of leukemia cells. Furthermore, TG101209 effectively reduced tumor burden in AE9a leukemia mice and prolonged survival. Importantly, TG101209 significantly impaired the leukemia-initiating potential of AE9a leukemia cells in secondary recipient mice. These results suggest a potential use of JAK/STAT signaling inhibitors in the treatment of t(8;21) AML.
Methods
Animals
MF-1 mice, as described previously,25 and C57BL/6 mice were used in this study. Animal housing and research were approved by the Institutional Animal Care and Use Committee of the University of California San Diego.
Generation of AE9a leukemia mice
Primary transplanted AE9a leukemia mice were generated as previously described.13 To generate secondary transplanted leukemia mice, AE9a leukemia cells from primary transplant were injected into sublethally irradiated (450 Rads) MF-1 mice via tail vein. Each mouse received 1 × 105 EGFP+ cells.
Plasmids
MSCV-IRES-EGFP (MigR1), MigR1-HA-AML1-ETO and MigR1-HA-AE9a have been described previously.13,26 MSCV-MLL-AF9-Flag-IRES-puromycin (MIP-MLL-AF9-Flag) was constructed by subcloning the MLL (EcoRI/SalI) and AF9-Flag-IRES (SalI/NcoI) fragments from MigR1-MLL-AF9-Flag (kindly provided by Dr. Nancy Zeleznik-Le) into MSCV-IRES-puromycin (EcoRI/NcoI). The siRNA sequences for the firefly luciferase gene and mouse JAK1 and JAK2 were designed using the RNAi Codex website (http://cancan.cshl.edu/cgi-bin/Codex/Codex.cgi) and cloned into the MSCV-LTRmiR30-PIG (LMP) retroviral vector (Thermo Scientific) following the manufacturer’s instructions. Firefly luciferase siRNA was used as a control. The sequences of the “sense” strands of the corresponding target genes are: (Luciferase) ACCGCTGAATTGGAATCGATAT; (JAK1) CCCAAAGCAATTGAAACCGATA; (JAK2#1) ACGTTAATGAGTGAAACCGAAA; (JAK2#2) CGCGAATGATTGGCAATGATAA.
JAK inhibitors
The JAK2-selective inhibitor TG101209 was provided by TargeGen/Sanofi. The JAK1/2-selective inhibitor INCB18424 (Ruxolitinib) was purchased from ChemieTek. Both inhibitors were dissolved in DMSO for in vitro studies. TG101209 was dissolved in 20% Tween 80 (Fisher Scientific) with pH of 4.0 for in vivo mouse treatment.
Cell culture
293T and EML cells were cultured as previously described.19 EML-AE and EML-AE9a cells were generated by retroviral transduction of the parental EML cells with MigR1-HA-AML1-ETO and MigR1-HA-AE9a, respectively. Then EGFP+ cells were sorted by fluorescence-activated cell sorting (FACS) with a FACSAria flow cytometer (BD Biosciences) and cultured as the parental EML cells.
Mouse fetal liver cells and bone marrow lineage negative (Lin-) cells were isolated and transduced as described.19 Briefly, fetal liver cells were cultured in Iscove’s DMEM (IMDM) supplemented with 20% FBS, 4% SCF conditioned media prepared from BHK/MKL cells stably expressing a cDNA encoding the secretory form of mouse SCF and 4% IL-3 conditioned media prepared from X63Ag-653 myeloma cells. Bone marrow Lin- cells were cultured in IMDM supplemented with 20% FBS, 10 ng/mL recombinant mouse (rm) SCF, 10 ng/mL rmIL-3 and 20 ng/mL recombinant human (rh) IL-6. Both fetal liver and bone marrow Lin- cells were cultured for 16 hours prior to retroviral transduction. The AEtr leukemia cell line was generated as described previously and cultured in IMDM supplemented with 20% horse serum, 10% SCF conditioned media, 10% IL-3 conditioned media and 20 ng/mL rhIL-6.19 All recombinant cytokines were purchased from Peprotech.
Retrovirus transduction
The retrovirus production and infection were performed as described previously.19
Colony assays
The procedure for colony-forming unit (CFU) assays was described previously.19 Fetal liver cells from MF-1 mice or bone marrow Lin- cells from MF-1 or C57BL/6 mice were transduced and transformed with MigR1-HA-AML1-ETO, MigR1-HA-AE9a or MIP-MLL-AF9-Flag by serial replating in methylcellulose (M3534) medium (StemCell Technologies), which contains rmSCF, rmIL-3 and rhIL-6. The transduced cells were selected by FACS (for MigR1 constructs) or puromycin (1.5 μg/mL, for MIP constructs). Then transformed cells from the third plating or beyond were replated in the presence of DMSO (control) or different concentrations of TG101209 or INCB18424 for CFU assays. For JAK1 and JAK2 knockdown experiments, AML1-ETO- and AE9a-transformed cells were infected with the LMP shRNA retroviruses, selected with puromycin (1.5 μg/mL) at 36 hours after infection in M3534 medium for 4 days and then collected for CFU assays.
JAK inhibitor treatment in AEtr leukemia cell line
The AEtr leukemia cell line was cultured in the presence of DMSO or different concentrations of TG101209 or INCB18424. Cell cycle analysis was done after 24 hours. Apoptosis and relative cell numbers were measured after 48 hours. Relative cell numbers were determined using the MTS assay (CellTiter96® AQueous One Solution Cell Proliferation Assay, Promega) following the manufacturer’s instructions. The absorbance (A490) of the DMSO control subtracting the background absorbance of culture medium was set as 100%.
Western blot analysis
Western blot was performed as previously described.19 Primary antibodies used for detection included rabbit antibodies against JAK1 (1:1000) or JAK2 (1:500) (Cell Signaling), and mouse antibody against α-tubulin (1:20,000) (Sigma). HRP-conjugated secondary antibodies (GE Healthcare) were used at 1:4000.
Flow cytometric analysis
Examination of EGFP and CD11b (PE-conjugated rat anti-mouse CD11b, Invitrogen) and cKit (PE-Cy7-conjugated rat anti-mouse CD117, BD Biosciences) surface expression, Annexin V-PE (BD Biosciences) staining for apoptosis analysis, propidium iodide (Sigma) staining for cell cycle analysis and intracellular phospho-specific flow cytometry were performed as described previously.19
In vivo TG101209 treatment and secondary bone marrow transplantation
The disease development of primary transplanted AE9a leukemia mice was assessed by peripheral white blood cell counts using the HEMAVET multispecies hematology analyzer (Drew Scientific) and percentages of circulating EGFP+ cells by flow cytometry on day 48 after transplantation. All mice developed leukocytosis and had at least 30% circulating EGFP+ cells prior to initiation of treatment on day 49. TG101209 was administered by oral gavage twice daily (b.i.d.) at 100 mg/kg for two weeks. The placebo group was treated with vehicle (20% Tween 80, pH 4.0) only. After two weeks of treatment, peripheral white blood cell counts and percentages of circulating EGFP+ cells were assessed again. The differentiation states (CD11b and cKit expression) of circulating EGFP+ cells after treatment were examined by flow cytometry. Mice transplanted with cells transduced with MigR1 control vector also received the same treatment and were analyzed as described above.
Secondary transplanted AE9a leukemia mice (see Generation of AE9a leukemia mice) were treated with TG101209 (100 mg/kg) or placebo at 3 weeks after transplantation twice daily for 5 days and then once daily for 9 days. The survival of mice was monitored.
To address whether TG101209 targets AE9a leukemia-initiating cells, primary transplanted AE9a leukemia mice were treated with TG101209 (100 mg/kg) or placebo at ~6 weeks after transplantation once daily for two weeks. The size of spleens was examined and their weight measured. Then EGFP+ spleen cells were sorted by FACS and transplanted into sublethally irradiated (450 Rads) MF-1 mice (1 × 104 EGFP+ cells per mouse). Leukemia development was assessed by monitoring percentages of circulating EGFP+ cells at 6 and 12 weeks after secondary transplantation.
Statistical analyses
The Log-rank test was used to analyze survival curves of TG101209 treatment and secondary bone marrow transplantation experiments, and two-tailed unpaired t-test (Student’s t-test) was used for the other statistical analyses (GraphPad Prism software version 5.0). A p-value of less than 0.05 was considered statistically significant.
Results
JAK1 and JAK2 inhibition impairs colony-forming ability of AML1-ETO- and AE9a- transformed cells
AML1-ETO and AE9a are capable of transforming primary murine hematopoietic stem/progenitor cells in serial replating assays.10,19 We assessed the effect of JAK1 or JAK2 knockdown by shRNA on the colony-forming ability of AML1-ETO- and AE9a-transformed murine hematopoietic cells, which had been replated at least three times in a serial replating assay (MigR1 vector-transduced cells have lost replating ability by this time). The effectiveness of JAK1 and JAK2 shRNA constructs was confirmed in EML cells, a murine hematopoietic stem/progenitor cell line, which express AML1-ETO or AE9a (supplemental Figure 1). Compared with control shRNA (firefly luciferase gene), JAK1 or JAK2 shRNA resulted in at least a 50% reduction of colony numbers from AML1-ETO- and AE9a-transformed cells (Figure 1A). To further investigate the role of JAKs in the transforming activity of AML1-ETO and AE9a, we performed colony formation assays in the presence of a JAK2-selective small molecule inhibitor, TG10120923 or a JAK1/2-selective inhibitor, INCB1842424 using AML1-ETO- and AE9a-transformed cells. Similar to the shRNA results, both inhibitors reduced the colony-forming ability of the transformed cells in a dose-dependent manner (Figure 1B and 1C; supplemental Figure 2 and data not shown). At low concentrations, INCB18424 showed a greater inhibitory effect than TG101209, suggesting that targeting both JAK1 and JAK2 has an additive effect. When used at high concentrations, TG101209 inhibited colony formation better than INCB18424, which could be due to its ability to inhibit FLT3 and RET in addition to JAK2.23 These results suggest that the JAK/STAT pathway contributes to leukemia transformation by AML1-ETO and AE9a. Both TG101209 and INCB18424 also decreased colony numbers of MLL-AF9-transformed cells (supplemental Figure 3). However, the effects of these two inhibitors were less severe on MLL-AF9-transformed cells than on AML1-ETO- or AE9a-transformed cells (supplemental Figure 3B and 3C). The colony size of MLL-AF9-transformed cells was also not as severely affected by the inhibitors as that of t(8;21) fusion protein-transformed cells (supplemental Figure 2 and data not shown).
Figure 1. Inhibition of JAK1 and JAK2 attenuates the colony-forming ability of AML1-ETO- and AE9a-transformed cells.

CFU assays of AML1-ETO- and AE9a-transformed mouse fetal liver or bone marrow cells after control, JAK1 or JAK2 shRNA expression (A). CFU assays of AML1-ETO (B) and AE9a (C) transformed mouse bone marrow cells in the presence of TG101209 or INCB18424 at the concentrations indicated. Colony numbers are presented as percentages of control shRNA (A) or DMSO controls (B) and (C). Averages with SEM of at least two independent experiments are shown. Statistical significance of each treatment compared with the control is indicated above each bar. Statistical significance between two different treatments is also indicated. The cells treated with 600 nM (C) and 1200 nM (B and C) TG101209 failed to generate any colonies and therefore a p-value cannot be calculated. ** p < 0.01, ‡ p < 0.001, # p < 0.0001
JAK inhibitors suppress growth and promote apoptosis of t(8;21) leukemia cells in vitro
To determine the importance of JAK/STAT signaling in t(8;21) leukemia cell proliferation and survival, we examined the effect of TG101209 and INCB18424 on leukemia cells in vitro. A murine t(8;21) leukemia cell line was previously established from an AEtr leukemia mouse.19 Like AE9a, AEtr lacks the NHR3 and NHR4 domains of the ETO protein due to a nucleotide insertion resulting in a premature stop codon.25 Both TG101209 and INCB18424 inhibited proliferation of the AEtr leukemia cell line in a dose-dependent manner (Figure 2A). Importantly, INCB18424 showed a significantly better inhibitory effect than TG101209 at concentrations below 1 μM. Both inhibitors also promoted apoptosis (Figure 2B) and delayed cell cycle progression (Figure 2C) of the AEtr leukemia cell line. When used at a high concentration (1.2 μM), TG101209 caused drastic growth inhibition (Figure 2A) accompanied by massive apoptosis (Figure 2B) and G2/M arrest (supplemental Figure 4). These data show that t(8;21) leukemia cells exhibit a dependency on JAK/STAT signaling for proliferation/survival, making this pathway a potential therapeutic target. Since targeting both JAK1 and JAK2 shows an additive effect, a JAK1/2 inhibitor may have a better therapeutic benefit than a JAK2-selective inhibitor.
Figure 2. In vitro effects of the JAK2 inhibitor TG101209 and JAK1/2 inhibitor INCB18424 on murine t(8;21) leukemia cells.

The AEtr leukemia cell line was treated with TG101209 or INCB18424 at the concentrations indicated for 48 hours. Relative cell numbers were determined by MTS assay (CellTiter96® AQueous One Solution Cell Proliferation Assay, Promega) and are presented as percentages of the DMSO control (A). Percentages of Annexin V-positive cells (B) and percentages of cells in the G1 phase upon treatment are also shown. Each bar represents the average with SEM of triplicate samples. Representative results from three independent experiments are shown. Statistical significance of each treatment compared with the control is indicated above each bar. Statistical significance between two different treatments is also indicated. * p < 0.05, ** p < 0.01, ‡ p < 0.001
JAK2 inhibitor TG101209 effectively targets AE9a leukemia cells in vivo
We next examined the in vivo effect of TG101209 in the primary transplanted AE9a leukemia mouse model. TG101209 and its derivative TG101348 (SAR302503) effectively target JAK2V617F and MPLW515L, mutations associated with myeloproliferative disorders, in several mouse models.23,27,28 They are also known to block wild-type JAK2 activity.23,27–29 On day 48 after transplantation, the white blood cell (WBC) count was above the normal range (> 10.7 K/μL) and at least 30% were EGFP+ cells in all the transplanted mice. TG101209 was administered by oral gavage twice daily at a dose of 100 mg/kg for two weeks. A 20–45% reduction in STAT5 or STAT3 phosphorylation was seen in AE9a leukemia cells isolated from mice treated with a single dose of TG101209 (100 mg/kg) compared with placebo (Figure 3A), demonstrating an inhibitory effect of the drug on the JAK/STAT pathway in vivo. After two weeks of treatment, all mice in the placebo group (n = 11) had an increased peripheral WBC count (1.44 – 5.39-fold increase), whereas eight out of the nine TG101209-treated mice had a 50 – 90% reduction (Figure 3B; supplemental Table 1). Consistent with a reduction in leukemia cell burden, the percentage of circulating EGFP+ cells significantly decreased (40 – 80%, Student’s t-test, p < 0.0001) in eight of the nine TG101209-treated mice (Figure 3C; supplemental Table 2).
Figure 3. In vivo efficacy of TG101209 on primary transplanted AE9a leukemia mice.

(A) The average mean fluorescence intensity (MFI) (± SEM) of pSTAT5 (Y694) and pSTAT3 (Y705) determined by flow cytometry in spleen cells of AE9a leukemia mice after a single-dose treatment of placebo (n = 3) or TG101209 (100 mg/kg) (n = 7). Cells were harvested 5 hours after treatment. EGFP+ cells were gated for the analysis. (B) Peripheral white blood cell (WBC) counts, and (C) percentages of circulating EGFP+ cells in placebo (n = 11) or TG101209-treated (100 mg/kg, twice daily) (n = 9) mice before (left panel) and after (right panel) two weeks of treatment. (D) Percentages of circulating CD11b+ cells (left panel) and cKit+ cells (right panel) within the EGFP+ population in placebo (n = 11) or TG101209-treated (100 mg/kg, twice daily) (n = 9) mice after two weeks of treatment. Horizontal bars indicate the mean values. (E) Spleens from one AE9a leukemia mouse treated with placebo (left) and two AE9a leukemia mice treated with TG101209 (100 mg/kg) (middle and right) once daily for two weeks. The scale bar is 1 cm. (F) The average spleen weight (± SEM) of AE9a leukemia mice treated with placebo (n = 10) or TG101209 (100 mg/kg) (n = 16) once daily for two weeks. The results are averages of two independent experiments. Each dot in (B), (C) and (D) represents one mouse. The p values were determined using Student’s t-test.
We have shown previously that the majority of AE9a (EGFP+) leukemia cells are Lin- (> 90%), among which most cells express cKit but are negative for Sca1.13 We have also shown that both this major population (Lin-/Sca1-/cKit+) and the Lin-/Sca1+/cKit+ population contain leukemia initiating cells and particularly the former caused faster leukemia development in secondary recipient mice.19 To address whether TG101209 promotes myeloid differentiation of AE9a leukemia cells, we examined CD11b and Gr1 expression. Within the EGFP+ population, 0.4 – 8.5% in the placebo group were CD11b+ cells, including CD11b+/Gr1- and CD11b+/Gr1+ cells, whereas the percentage of total CD11b+ cells within the EGFP+ population was significantly higher in five of the nine TG101209-treated mice (12.7 – 58.1%) (Figure 3D; supplemental Figure 5A; supplemental Table 3). Consistent with differentiation, the percentage of total cKit+ cells, including Sca1+/cKit+ and Sca1-/cKit+ cells, within the EGFP+ population decreased in the treated group compared to the placebo group (median 36.4% versus 58.6%) (Figure 3D; supplemental Figure 5B; supplemental Table 4). Importantly, the percentage of Sca1-/cKit+ cells, the population with a robust leukemia-initiating potential, decreased significantly after treatment (supplemental Figure 5B). These results indicate a significant in vivo effect of TG101209 on AE9a leukemia cells. Mice transplanted with cells transduced with the MigR1 control vector received the same treatment, but no significant differences in percentages of EGFP+ cells or WBC counts were observed between control and treated mice. There was no detectable health problem in the treated MigR1 mice other than a modest but significant decrease in hemoglobin and hematocrit levels (Figure 4 and data not shown), which is an expected on-target class effect of JAK2 inhibitors that is well established and seen in clinical practice.22 Furthermore, in a separate experiment where primary transplanted AE9a leukemia mice were treated once daily with TG101209 at a dose of 100 mg/kg for two weeks, splenomegaly was significantly reduced (mean spleen weight 0.37 g versus 0.62 g; Student’s t-test, p = 0.0002) (Figure 3E and 3F).
Figure 4. TG101209 caused a modest but significant decrease in hemoglobin and hematocrit levels in MigR1 control mice.

Percentages of EGFP+ cells (A) and white blood cell (WBC) counts (B) in the peripheral blood of MigR1 transplanted mice before and after treatment with placebo or TG101209 (100 mg/kg, twice daily) for one or two weeks. Horizontal bars indicate the mean values. (C) Comparisons of hemoglobin (Hb) concentrations and hematocrit (HCT) levels in the peripheral blood of MigR1 mice treated with placebo or TG101209 for one or two weeks. The p values were determined using Student’s t-test.
Survival time varies extensively among the primary transplanted AE9a leukemia mice, and prolonged TG101209 treatment resulted in severe anemia and eventual death in these mice (data not shown). On the other hand, secondary-transplanted mice receiving leukemia cells (1 × 10 5 EGFP+ cells) isolated from primary transplant generally develop leukemia more rapidly and succumb to the disease in a more synchronous way. We therefore examined whether TG101209 provides a survival benefit in this secondary AE9a leukemia. The treatment was initiated 3 weeks after transplantation and continued for 2 weeks. All the mice in the placebo group (n = 8) succumbed to leukemia within a week after treatment started. However, mice treated with TG101209 (n = 9) showed a longer survival time (Figure 5).
Figure 5. TG101209 prolongs lifespan of secondary transplanted AE9a leukemia mice.
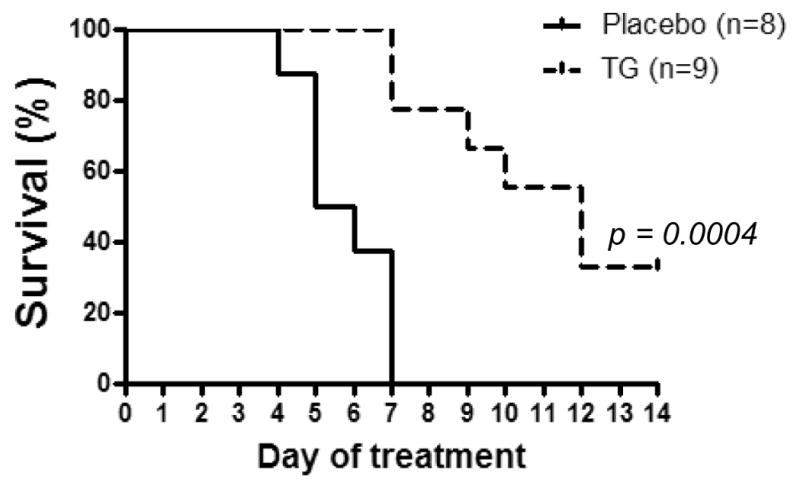
Survival curves of MF-1 mice transplanted with 1 × 105 EGFP+ leukemia cells from primary transplanted AE9a leukemia mice. Three weeks after secondary transplantation, mice were treated with placebo (n = 8) or TG101209 (100 mg/kg) (n = 9) twice daily for 5 days and then once daily for 9 days. The p value was determined by using the Log-rank test.
TG101209 impairs AE9a leukemia-initiating activity
Cancer-initiating cells are believed to have limitless self-renewal capability, so targeting them is a high priority.30–34 To examine whether TG101209 also affects AE9a leukemia-initiating potential, we performed secondary transplantation, as Wang et al35 reported previously, using sorted EGFP+ spleen cells from primary transplanted leukemia mice that were treated once daily with placebo or 100 mg/kg TG101209 for two weeks. TG101209 pretreatment resulted in a significant reduction of circulating EGFP+ cells at 6 and 12 weeks after secondary transplantation (Figure 6A) and significantly (p < 0.0001) delayed and prevented leukemia development (Figure 6B). On average, 50% of recipient mice in the treated group from two separate experiments (n = 8 and 10, respectively) lost EGFP+ cells and were free of leukemia at the end point of the study (Figure 6 and data not shown). Thus, the JAK2 inhibitor as a single agent has a suppressive effect on the leukemia-initiating potential of AE9a leukemia cells.
Figure 6. TG101209 impairs AE9a leukemia-initiating activity.

(A) Percentages of EGFP+ cells in peripheral blood of MF-1 mice receiving 1 × 104 sorted EGFP+ spleen cells from primary transplanted AE9a leukemia mice that had been treated with placebo or TG101209 (100 mg/kg) once daily for two weeks. Peripheral blood was examined at 6 and 12 weeks after secondary transplantation. At 12 weeks, there were only 5 live mice left in the placebo group (n = 8) and all of them had > 50% EGFP+ cells in blood. (B) Survival curves showing the lifespan of the secondary transplanted MF-1 mice in (A). *The remaining five mice in the TG101209 group (n = 8) had no detectable EGFP+ cells at the end point of the study and appeared healthy. The p values were determined by Student’s t-test (A) or Log-rank test (B). The results from one of two independent experiments are shown.
Discussion
In our previous study, we combined gene expression and promoter occupancy profiling approaches to identify potential therapeutic targets of t(8;21) AML. We discovered the down-regulation of CD45, a negative regulator of the JAK/STAT pathway, and the subsequent activation of this pathway by the t(8;21) fusion protein AML1-ETO and its alternatively spliced variant AE9a.19 In the current report, we demonstrate the in vitro and in vivo effects of JAK inhibition in AE9a-induced leukemia.
Chromosomal aberrations involving the JAK2 gene that generate a variety of chimeric fusion proteins with constitutively activated tyrosine kinase activity have been known for more than a decade.22 These JAK2 fusions often promote the development of leukemias of both myeloid and lymphoid origins. In addition, all activating point mutations, deletions and insertions in the JAK2 gene lead to myeloproliferative neoplasms that can progress to a myelodysplastic syndrome or acute leukemia following the acquisition of additional genetic lesions. These observations have inspired significant efforts towards the development of JAK2 inhibitors for the treatment of these hematological malignancies.22 Although JAK2 mutations are extremely rare in de novo AML, they do appear in approximately 0.5–8% of cases36–40 and are observed in some cases of therapy-related t(8;21) AML.41 The presence of the JAK2 V617F mutation was shown to be prognostically unfavorable in a few cases of t(8;21) leukemia affecting their disease-free survival rate.42 Furthermore, STAT3 and/or STAT5 are activated in the majority of AML samples.37 This activation could be caused by activating mutations in a cytokine receptor,20 increased expression of a cytokine receptor43,44 or down-regulation of a negative regulator of the JAK/STAT pathway as we showed previously in t(8;21) AML.19 In addition, genes encoding other negative regulators of the JAK/STAT signaling cascade such as the protein inhibitor of activated stats-2 (PIAS2),45 a direct negative regulator of Stats, and the suppressor of cytokine signaling-1 (SOCS1),46 a negative regulator of JAK kinases, are hypermethylated in approximately 70% of AML cases irrespective of the AML subtype. These observations suggest that there may be a potential use for JAK/STAT inhibitors for the treatment of AML.
In our previous microarray study of AE9a leukemia cells,19 we observed up-regulation of JAK1 (p = 0.000174) and JAK2 (p = 0.006515) transcripts, which was later confirmed by RT-qPCR (unpublished data). Notably, we also observed higher JAK1 and JAK2 mRNA levels in t(8;21) AML M2 patients compared with patients without the translocation (supplemental Figure 6). Thus, t(8;21) fusion proteins enhance JAK/STAT signaling in two ways: increasing the level of an activator (JAK1/2) while decreasing the level of an inhibitor (CD45) of this pathway. Therefore, targeting this pathway should significantly impair the growth advantage of AE9a leukemia cells. Here, we show that AE9a leukemia cells and AML1-ETO-transformed murine hematopoietic cells are sensitive to inhibition of JAK1 or JAK2. Furthermore, the JAK2 inhibitor TG101209 effectively reduced the leukemia cell burden and the leukemia-initiating potential in the AE9a AML mouse model. These results suggest that JAK/STAT signaling is critical in AE9a leukemogenesis and inhibition of this signaling pathway has therapeutic potential. Since enhanced activation of both JAK1 and JAK2 is detected in AE9a leukemia cells, inhibitors that more effectively target both JAK1 and JAK2, such as CYT387 and INCB18424, may have higher efficacy in treating t(8;21) AML. Our in vitro data of murine t(8;21) leukemia cells treated with TG101209 or INCB18424 further support this notion.
Clinically, aggressive chemotherapy is the standard treatment for t(8;21) AML. Although t(8;21) predicts favorable prognosis,14,15 other therapeutic options for t(8;21) AML patients have been investigated to further improve clinical outcome. Imatinib mesylate and dasatinib, which target BCR-ABL and the receptor tyrosine kinases cKIT and PDGFR, have been shown to inhibit proliferation and induce apoptosis in t(8;21)-positive leukemic cells47,48. When combined with cytarabine, these drugs prolong the survival of mice transformed by AML1-ETO and an activating cKit mutation.49 The histone deacetylase inhibitor Valproic acid was also reported to induce apoptosis and differentiation in the t(8;21)-positive Kasumi-1 cell line.50 In addition, Kasumi-1 cells are sensitive to arsenic-induced apoptosis.51 Furthermore, the diterpenoid Oridonin functions as an anti-tumor agent, promoting the degradation of AML1-ETO by caspase cleavage, and synergizing with Ara-C in the AEtr leukemia mouse model.52 Based on this preclinical work, clinical trials are investigating the efficacy of dasatinib in combination with standard chemotherapy in core binding factor leukemias [t(8;21) and inv(16)] (ClinicalTrials.gov identifiers NCT01238211 and NCT00850382).
More recently, the FDA-approved JAK inhibitor INCB18424/Ruxolitinib (Jakafi®; Incyte/Novartis) was used as a single agent in a clinical study in patients with refractory AML including postmyeloproliferative neoplasm AML.53 Although the response rate was very poor, 3 out of 18 patients did respond with 2 achieving complete remission (CR) while one achieved CR with incomplete blood recovery. Although the efficacy of INCB18424 was dismal as a single agent, it showed no significant toxicity in this study. Clinical trials with JAK inhibitors INCB18424 and TG101348 were also well tolerated in myelofibrosis patients and significantly reduced the disease burden, producing a durable clinical benefit.54,55 Furthermore, recent investigations suggest that JAK inhibitors may be useful in targeting chronic myeloid leukemia (CML) when combined with the conventionally used tyrosine kinase inhibitors.56–58 Therefore, targeting the JAK/STAT signaling in combination with other therapy could be a viable approach to achieve a greater clinical response. This proposition is further supported by observations that AML cells are sensitive to the combined treatment of dasatinib and JAK inhibitors in assays designed to mimic stromal-derived survival signals.59 Future studies are necessary to address the therapeutic potential of JAK/STAT inhibitors in t(8;21) AML and the possibility of combining them with conventional chemotherapies or other novel small molecule inhibitors for therapy.
Supplementary Material
Acknowledgments
Support for the work: National Institutes of Health R01CA104509 to DEZ Leukemia and Lymphoma Society Fellowship 5122-07 to MCL TargeGen/Sanofi providing the JAK2 inhibitor TG101209
We thank members of Zhang lab and Dr. Moshe Talpaz for valuable discussions, Jessica Mercer for editing the manuscript, Dr. Nancy Zeleznik-Le for providing the MigR1-MLL-AF9-Flag construct and TargeGen/Sanofi for providing TG101209.
Footnotes
Authorship
Contribution: MCL and LFP designed and performed the experiments, analyzed the data and wrote the manuscript. MY, XC, JHH, RCD and DN performed some experiments. DEZ supervised the research and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Supplementary information is available at Leukemia's website.
Reference List
- 1.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 2.Foran JM. New prognostic markers in acute myeloid leukemia: perspective from the clinic. Hematology Am Soc Hematol Educ Program. 2010;2010:47–55. doi: 10.1182/asheducation-2010.1.47. [DOI] [PubMed] [Google Scholar]
- 3.Lin TL, Smith BD. Prognostically important molecular markers in cytogenetically normal acute myeloid leukemia. Am J Med Sci. 2011;341(5):404–408. doi: 10.1097/MAJ.0b013e318201109d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973;16(2):109–112. [PubMed] [Google Scholar]
- 5.Groupe Français de Cytogénétique Hématologique. Acute myelogenous leukemia with an 8;21 translocation. A report on 148 cases from the Groupe Francais de Cytogenetique Hematologique. Cancer Genet Cytogenet. 1990;44(2):169–179. [PubMed] [Google Scholar]
- 6.Peterson LF, Boyapati A, Ahn EY, Biggs JR, Okumura AJ, Lo MC, et al. Acute myeloid leukemia with the 8q22;21q22 translocation: secondary mutational events and alternative t(8;21) transcripts. Blood. 2007;110(3):799–805. doi: 10.1182/blood-2006-11-019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. PNAS. 1996;93(8):3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yergeau DA, Hetherington CJ, Wang Q, Zhang P, Sharpe AH, Binder M, et al. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nat Genet. 1997;15(3):303–306. doi: 10.1038/ng0397-303. [DOI] [PubMed] [Google Scholar]
- 10.Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, van Deursen JM, et al. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood. 1998;91(9):3134–3143. [PubMed] [Google Scholar]
- 11.Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255–4262. doi: 10.1038/sj.onc.1207727. [DOI] [PubMed] [Google Scholar]
- 12.Goyama S, Mulloy JC. Molecular pathogenesis of core binding factor leukemia: current knowledge and future prospects. Int J Hematol. 2011;94(2):126–133. doi: 10.1007/s12185-011-0858-z. [DOI] [PubMed] [Google Scholar]
- 13.Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12(8):945–949. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 14.Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98(5):1312–1320. doi: 10.1182/blood.v98.5.1312. [DOI] [PubMed] [Google Scholar]
- 15.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29(5):487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 16.Marcucci G, Mrozek K, Ruppert AS, Maharry K, Kolitz JE, Moore JO, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23(24):5705–5717. doi: 10.1200/JCO.2005.15.610. [DOI] [PubMed] [Google Scholar]
- 17.Mrozek K, Bloomfield CD. Clinical significance of the most common chromosome translocations in adult acute myeloid leukemia. J Natl Cancer Inst Monogr. 2008;(39):52–57. doi: 10.1093/jncimonographs/lgn003. [DOI] [PubMed] [Google Scholar]
- 18.Reikvam H, Hatfield KJ, Kittang AO, Hovland R, Bruserud O. Acute myeloid leukemia with the t(8;21) translocation: clinical consequences and biological implications. J Biomed Biotechnol. 2011;2011:104631. doi: 10.1155/2011/104631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo MC, Peterson LF, Yan M, Cong X, Jin F, Shia WJ, et al. Combined gene expression and DNA occupancy profiling identifies potential therapeutic targets of t(8;21) AML. Blood. 2012;120(7):1473–1484. doi: 10.1182/blood-2011-12-395335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Basecke J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22(4):686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 21.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7(9):673–683. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 22.Jatiani SS, Baker SJ, Silverman LR, Reddy EP. JAK/STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders: Approaches for Targeted Therapies. Genes Cancer. 2010;1(10):979–993. doi: 10.1177/1947601910397187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21(8):1658–1668. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 24.Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan M, Burel SA, Peterson LF, Kanbe E, Iwasaki H, Boyapati A, et al. Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc Natl Acad Sci U S A. 2004;101(49):17186–17191. doi: 10.1073/pnas.0406702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson LF, Wang Y, Lo MC, Yan M, Kanbe E, Zhang DE. The multi-functional cellular adhesion molecule CD44 is regulated by the 8;21 chromosomal translocation. Leukemia. 2007;21(9):2010–2019. doi: 10.1038/sj.leu.2404849. [DOI] [PubMed] [Google Scholar]
- 27.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Geron I, Abrahamsson AE, Barroga CF, Kavalerchik E, Gotlib J, Hood JD, et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008;13(4):321–330. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Lasho TL, Tefferi A, Hood JD, Verstovsek S, Gilliland DG, Pardanani A. TG101348, a JAK2-selective antagonist, inhibits primary hematopoietic cells derived from myeloproliferative disorder patients with JAK2V617F, MPLW515K or JAK2 exon 12 mutations as well as mutation negative patients. Leukemia. 2008;22(9):1790–1792. doi: 10.1038/leu.2008.56. [DOI] [PubMed] [Google Scholar]
- 30.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 31.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23(43):7274–7282. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 32.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324(5935):1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13(6):483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lessard J, Faubert A, Sauvageau G. Genetic programs regulating HSC specification, maintenance and expansion. Oncogene. 2004;23(43):7199–7209. doi: 10.1038/sj.onc.1207940. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327(5973):1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JW, Kim YG, Soung YH, Han KJ, Kim SY, Rhim HS, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25(9):1434–1436. doi: 10.1038/sj.onc.1209163. [DOI] [PubMed] [Google Scholar]
- 37.Steensma DP, McClure RF, Karp JE, Tefferi A, Lasho TL, Powell HL, et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006;20(6):971–978. doi: 10.1038/sj.leu.2404206. [DOI] [PubMed] [Google Scholar]
- 38.Schnittger S, Bacher U, Kern W, Haferlach T, Haferlach C. JAK2V617F as progression marker in CMPD and as cooperative mutation in AML with trisomy 8 and t(8;21): a comparative study on 1103 CMPD and 269 AML cases. Leukemia. 2007;21(8):1843–1845. doi: 10.1038/sj.leu.2404707. [DOI] [PubMed] [Google Scholar]
- 39.Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106(10):3377–3379. doi: 10.1182/blood-2005-05-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwanaga E, Nanri T, Matsuno N, Kawakita T, Mitsuya H, Asou N. A JAK2-V617F activating mutation in addition to KIT and FLT3 mutations is associated with clinical outcome in patients with t(8;21)(q22;q22) acute myeloid leukemia. Haematologica. 2009;94(3):433–435. doi: 10.3324/haematol.13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schnittger S, Bacher U, Kern W, Haferlach C, Haferlach T. JAK2 seems to be a typical cooperating mutation in therapy-related t(8;21)/AML1-ETO-positive AML. Leukemia. 2007;21(1):183–184. doi: 10.1038/sj.leu.2404465. [DOI] [PubMed] [Google Scholar]
- 42.Illmer T, Schaich M, Ehninger G, Thiede C. Tyrosine kinase mutations of JAK2 are rare events in AML but influence prognosis of patients with CBF-leukemias. Haematologica. 2007;92(1):137–138. doi: 10.3324/haematol.10489. [DOI] [PubMed] [Google Scholar]
- 43.Chou FS, Griesinger A, Wunderlich M, Lin S, Link KA, Shrestha M, et al. The thrombopoietin/MPL/Bcl-xL pathway is essential for survival and self-renewal in human preleukemia induced by AML1-ETO. Blood. 2012;120(4):709–719. doi: 10.1182/blood-2012-01-403212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pulikkan JA, Madera D, Xue L, Bradley P, Landrette SF, Kuo YH, et al. Thrombopoietin/MPL participates in initiating and maintaining RUNX1-ETO acute myeloid leukemia via PI3K/AKT signaling. Blood. 2012;120(4):868–879. doi: 10.1182/blood-2012-03-414649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe D, Ezoe S, Fujimoto M, Kimura A, Saito Y, Nagai H, et al. Suppressor of cytokine signalling-1 gene silencing in acute myeloid leukaemia and human haematopoietic cell lines. Br J Haematol. 2004;126(5):726–735. doi: 10.1111/j.1365-2141.2004.05107.x. [DOI] [PubMed] [Google Scholar]
- 47.Grisolano JL, O'Neal J, Cain J, Tomasson MH. An activated receptor tyrosine kinase, TEL/PDGFbetaR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A. 2003;100(16):9506–9511. doi: 10.1073/pnas.1531730100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang YY, Zhou GB, Yin T, Chen B, Shi JY, Liang WX, et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci U S A. 2005;102(4):1104–1109. doi: 10.1073/pnas.0408831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang YY, Zhao LJ, Wu CF, Liu P, Shi L, Liang Y, et al. C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A. 2011;108(6):2450–2455. doi: 10.1073/pnas.1019625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S, Klisovic RB, Vukosavljevic T, Yu J, Paschka P, Huynh L, et al. Targeting AML1/ETO-histone deacetylase repressor complex: a novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J Pharmacol Exp Ther. 2007;321(3):953–960. doi: 10.1124/jpet.106.118406. [DOI] [PubMed] [Google Scholar]
- 51.Puccetti E, Beissert T, Guller S, Li JE, Hoelzer D, Ottmann OG, et al. Leukemia-associated translocation products able to activate RAS modify PML and render cells sensitive to arsenic-induced apoptosis. Oncogene. 2003;22(44):6900–6908. doi: 10.1038/sj.onc.1206747. [DOI] [PubMed] [Google Scholar]
- 52.Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie J, et al. Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo. Blood. 2007;109(8):3441–3450. doi: 10.1182/blood-2006-06-032250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119(20):4614–4618. doi: 10.1182/blood-2011-12-400051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Traer E, MacKenzie R, Snead J, Agarwal A, Eiring AM, O'Hare T, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26(5):1140–1143. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nair RR, Tolentino JH, Argilagos RF, Zhang L, Pinilla-Ibarz J, Hazlehurst LA. Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML. Leuk Res. 2012;36(6):756–763. doi: 10.1016/j.leukres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen M, Gallipoli P, Degeer D, Sloma I, Forrest DL, Chan M, et al. Targeting Primitive Chronic Myeloid Leukemia Cells by Effective Inhibition of a New AHI-1-BCR-ABL-JAK2 Complex. J Natl Cancer Inst. 2013;105(6):405–423. doi: 10.1093/jnci/djt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weisberg E, Liu Q, Nelson E, Kung AL, Christie AL, Bronson R, et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia. 2012;26(10):2233–2244. doi: 10.1038/leu.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.