

Abstract
Kidney ischemia-reperfusion (I/R) injury elicits cellular injury in the proximal tubule, and mitochondrial dysfunction is a pathological consequence of I/R. Promoting mitochondrial biogenesis (MB) as a repair mechanism after injury may offer a unique strategy to restore both mitochondrial and organ function. Rats subjected to bilateral renal pedicle ligation for 22 min were treated once daily with the SIRT1 activator SRT1720 (5 mg/kg) starting 24h after reperfusion until 72h–144h. SIRT1 expression was elevated in the renal cortex of rats after I/R + vehicle treatment (IRV), but was associated with less nuclear localization. SIRT1 expression was even further augmented and nuclear localization was restored in the kidneys of rats after I/R + SRT1720 treatment (IRS). PGC-1α was elevated at 72h–144h in IRV and IRS kidneys; however, SRT1720 treatment induced deacetylation of PGC-1α, a marker of activation. Mitochondrial proteins ATP synthase β, COX I, and NDUFB8, as well as mitochondrial respiration, were diminished 24h–144h in IRV rats, but were partially or fully restored in IRS rats. Urinary kidney injury molecule-1 (KIM-1) was persistently elevated in both IRV and IRS rats; however, KIM-1 tissue expression was attenuated in IRS rats. Additionally, sustained loss of Na+,K+-ATPase expression and basolateral localization and elevated vimentin in IRV rats was normalized in IRS rats, suggesting restoration of a differentiated, polarized tubule epithelium. The results suggest that SRT1720 treatment expedited recovery of mitochondrial protein expression and function by enhancing MB, which was associated with faster proximal tubule repair. Targeting MB may offer unique therapeutic strategy following ischemic injury.
Keywords: mitochondrial biogenesis, mitochondrial dysfunction, acute kidney injury, SRT1720, proximal tubule, ischemia-reperfusion
Introduction
Ischemia-reperfusion (I/R) is a primary cause of acute kidney injury (AKI), and the proximal tubule epithelium is particularly sensitive. Mitochondrial dysfunction is a hallmark of I/R injury and AKI in general, and we have recently shown that AKI results in early and persistent disruption of mitochondrial homeostasis in the outer cortex of the kidney, which correlated with sustained tubule damage (Funk and Schnellmann, 2012). In the outer cortex, a zone that is not typically susceptible to the overt necrotic injury seen in the outer medulla, mitochondrial electron transport chain mRNA and proteins were depleted within 24 h after I/R and did not recover by 144h after reperfusion. Our unpublished observations revealed that it took approximately two weeks for mitochondrial protein expression to be restored. Additionally, we showed that other markers of mitochondrial homeostasis were persistently disrupted, including increased expression of the mitochondrial fission protein Drp1, reduced expression of the fusion protein Mfn2, and up-regulation of proteins associated with mitochondrial biogenesis (MB) (Funk and Schnellmann, 2012).
We have previously characterized several pharmacological activators of MB in primary renal proximal tubule cells (RPTC), including a number of compounds which activate the NAD+-dependent class III histone and protein deacetylase sirtuin 1 (SIRT1) (Rasbach and Schnellmann, 2008; Funk et al., 2010; Rasbach et al., 2010). SRT1720 was initially reported as a potent SIRT1 activator, which could increase mitochondrial capacity through the MB regulator PPARγ coactivator-1α (PGC-1α) (Milne et al., 2007; Feige et al., 2008; Smith et al., 2009). In our hands, SRT1720 stimulated MB in primary cultures of RPTC within 24h of exposure, consistent with deacetylated PGC-1α, and elevated mitochondrial protein expression, basal and uncoupled oxygen consumption, and total cellular ATP (Funk et al., 2010). Additionally, SRT1720 restored mitochondrial and cellular function following oxidant-induced mitochondrial dysfunction in RPTC. More recently a protective role for SIRT1 in ischemic AKI has been reported (Fan et al., 2013). In the study, Fan, et al., demonstrated that younger mice, which were more resistant to AKI, expressed higher levels of SIRT1 compared to adult mice. Additionally, SRT1720 treatment in adult mice improved renal histopathology and function following I/R injury, whereas SIRT1 deficiency exacerbated injury. Treatment was associated with reduced p53 and p21, reduced apoptosis, and increased tubule cell proliferation (Fan et al., 2013). Longer SRT1720 dosing regimens in rodents have also been shown to mimic energy deprivation pathways typically stimulated with exercise, which can improve lifespan and pathological effects, such as reduced insulin sensitivity and glucose homeostasis, associated with high fat diet, obesity, and diabetes (Milne et al., 2007; Feige et al., 2008; Minor et al., 2011).
Although Fan, et al., demonstrated reduced cellular apoptosis, increased regeneration, and improved renal function with SRT1720 treatment after I/R, they did not examine the effects on recovery of mitochondrial function or tubule homeostasis. The aim of the current study was to examine MB as a repair component following AKI, and to examine whether SRT1720 treatment could expedite recovery of mitochondrial and tubule function in an I/R model. To do so, we examined recovery of mitochondrial and renal function in rats treated with SRT1720 starting 24h after I/R-induced AKI. The results indicated that SRT1720 treatment expedited recovery of mitochondrial proteins and function, as well as markers of tubule polarization and differentiation. Restoration of mitochondrial function via SIRT1/PGC-1α or other biogenic pathways may offer unique pharmacological targets for improved recovery from AKI.
Materials and Methods
Ischemia-reperfusion AKI model and SRT1720 dosing
Eight-week old male Sprague-Dawley rats weighing 180–200g were subjected to bilateral renal pedicle ligation as previously described (Zhuang et al., 2009). Briefly, renal artery and vein were isolated and blood flow was occluded with a microvascular clamp for 22 min. Sham rats underwent the same procedure, except the clamp was not applied. A heating pad against the dorsal side of the rats and heat lamps were turned on as needed to maintain body temperature at 37 °C, which was monitored by rectal probe. The rats were hydrated before, during, and after the procedure, including application of 1 ml of sterile, isotonic saline into the abdominal cavity during the clamped period. To maintain body temperature post-surgery, rats were placed in a warm recovery area for one hour. Twenty-four hours following I/R or sham procedure, rats were assigned to one of four treatment groups: sham + vehicle (V), sham + SRT1720 (S), I/R + vehicle (IRV), or I/R + SRT1720 (IRS). SRT1720 was synthesized as previously described (Milne et al., 2007; Funk et al., 2010). Dosing was initiated at 24 h after reperfusion, and rats were given either a daily injection of SRT1720 (5 mg/kg, i.p., 0.2 ml) or vehicle (10% DMSO in isotonic saline, i.p.) until euthanization at 72 h or 144 h. The dosing schedule was determined from preliminary studies in naïve rats and mice, which indicated that a single 5 mg/kg dose elevated mitochondrial gene expression within 24 h. All procedures involving animals were performed with approval from the Institutional Animal Care and Use Committee (IACUC) in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Assessing renal function
Tail vein blood was collected and serum was used to measure creatinine levels using a Quantichrom™ Creatinine Assay Kit (BioAssay Systems, Hayward, CA) according to manufacturer’s protocol. Urine was collected from rats housed in metabolic cages overnight (16 h collection time) at various time points throughout the study. Urine samples were used to determine kidney injury molecule-1 (Rat KIM-1 ELISA, Argutus Medical, Dublin, Ireland).
Immunoblot analysis
Surface regions of renal cortex, which are not generally within the zone of extensive necrosis, were dissected from flash frozen kidneys, and tissue was lysed in RIPA buffer containing cocktail protease and phosphatase inhibitors. Total protein content was measured by the BCA assay. Fifty μg total protein was loaded into SDS-PAGE gels and immunoblots were performed as previously described (Rasbach and Schnellmann, 2007). Antibodies used include phosphorylated AMPK, AMPK, acetylated-lysine, and histone H3 (Cell Signaling Technologies, Danvers, MA), SIRT1, PGC-1α, and Na+,K+-ATPase α1 subunit (EMD Millipore, Billerica, MA), GAPDH (Fitzgerald Antibodies, Acton, MA), ATP synthase β and VDAC (Abcam, Cambridge, MA), cytochrome c oxidase subunit I and NDUFB8 (Invitrogen, Grand Island, NY), Mfn2 and vimentin (Sigma-Aldrich, St. Louis, MO), Drp1 (Santa Cruz Biotechnology, Dallas, TX), and KIM-1 (R&D Systems, Minneapolis, MN). Nuclear lysates were prepared as previously described [Funk 2010]. Briefly, a piece of kidney cortex was homogenized in sucrose buffer (50 mM Tris-HCl, 1 mM β-mercaptoethanol, 1 mM EDTA, and 320 mM sucrose) with a pestle and sonication (10 s). Lysates were centrifuged at 900 X g for 10 min, and nuclear pellets were resuspended in RIPA buffer. Histone H3 was used as a load control for nuclear lysate immunoblots.
Immunoprecipitation and PGC-1α acetylation
Immunoprecipitation was performed as previously described according to manufacturer’s protocol (Roche Applied Science, Indianapolis, IN) (Funk et al., 2010). Briefly, 500 μg total protein from whole tissue lysates or nuclear lysates was incubated with protein A-agarose beads (Roche Applied Science, Indianapolis, IN) for 1 h to clear samples. After spinning down the samples, the supernatant was extracted and incubated with Rabbit anti-acetylated lysine antibody (5 μg, Cell Signaling Technologies) overnight at 4 °C with gentle agitation. Lysates were incubated with protein A-agarose beads for 3 h at 4 °C, and after washing, the beads were boiled to release the product, which was subsequently run on SDS-PAGE and immunoblotted for PGC-1α expression.
Immunohistochemistry
At euthanasia, the right kidney was sectioned coronally at the midline, and one half was immersion fixed for 24 h at 4 °C in 10% phosphate-buffered formalin solution. Twenty-four hours after immersion fixation, the sections were rinsed in water and then transferred to 70% EtOH until paraffin-embedded. Paraffin-embedded sections were cleared in xylenes, and rehydrated in a graded ethanol wash. Antigen unmasking was performed by boiling sections in citrate buffer for 10 min followed by cooling at room temperature for 30m. Endogenous peroxidase activity was quenched by incubating sections in 3% H2O2 for 10 min. Sections were then blocked in 10% normal goat serum for 1h, followed by primary antibody (KIM-1, Na+,K+-ATPase α1, or SIRT1) overnight at 4°C. Sections were then incubated in biotinylated anti-rabbit secondary antibody for 30 min followed by HRP-linked avidin-biotin complex reagent (Vector Labs, Burlingame, CA) for 30 min. Finally, antibody detection was visualized by DAB peroxidase substrate developer (Vector Labs), counterstained with hematoxylin, mounted and cover slips applied. Images were acquired with a Nikon microscope under control of QCapture imaging software. Low magnification images were captured at 10X and high magnification images were captured at 40X.
SIRT1 Nuclear Localization
SIRT1 protein was detected in paraffin-embedded kidney sections by IHC as described above. SIRT1-positive (SIRT1+) nuclei were counted in tubules and the data were presented as a percentage of SIRT1+ nuclei out of the total number of tubule nuclei counted in sections from each of the four groups listed above (V, S, IRV, IRS). At least four sections from each group were stained for SIRT1 immunoreactivity, and six images were randomly captured throughout the renal cortex from each section.
Mitochondrial isolation and oxygen consumption
At the time of euthanasia, kidneys were excised from rats and submerged in an ice-cold mitochondrial isolation buffer on ice. Mitochondria were isolated by differential centrifugation as previously described (Covington and Schnellmann, 2012). The mitochondrial pellet was resuspended in an incubation buffer and oxygen consumption (QO2) was measured using a Clark oxygen electrode. Briefly, 1.5 ml of mitochondrial suspension was added to the chamber and state 2 respiration was measured. Uncoupled oxygen consumption was determined by injecting FCCP for a final concentration of 1 μM. An aliquot of the mitochondrial suspension was saved for protein measurement, and rates were calculated as nmol O2 consumed per minute, and were normalized to the amount of total protein in each sample.
Statistical Analysis
Graphs represent a sample size of 3 to 6 for each group. Data were analyzed by ANOVA followed by Newman-Keuls post-hoc analysis for individual group comparisons. In each of the graphs presented, an asterisk (*) indicated data were statistically different from the sham + vehicle group (V), and a pound (#) sign was used to indicate the IRS group was different from the IRV group.
Results
Approximately 8-week old Sprague-Dawley rats were divided into four groups that underwent either a sham or renal I/R procedure, followed by treatment with either SRT1720 (5 mg/kg, i.p.) or vehicle (DMSO) starting at 24 h after the procedure. The four groups were designated as: sham + vehicle (V), sham + SRT1720 (S), I/R + vehicle (IRV), and I/R + SRT1720 (IRS), and the rats were euthanized at 72h or 144h after the sham or I/R procedure.
SIRT1 expression and localization
Because it has been reported that SIRT1 activators may act on AMPK instead of or in conjunction with SIRT1 (Baur et al., 2006; Dasgupta and Milbrandt, 2007), we examined expression levels of phosphorylated AMPK (pAMPK, Thr172), AMPK, and SIRT1. pAMPK increased in both IRV and IRS rats; however, there was no effect of SRT1720 treatment on pAMPK expression (Fig 1A). Total AMPK did not change with treatment or after injury. Tong, et al., recently demonstrated impaired nuclear SIRT1 shuttling in aged animals following myocardial infarction, and increased nuclear SIRT1 accumulation in cardiomyocytes treated with SRT1720 (Tong et al., 2012). To determine the effects of I/R injury and SRT1720 treatment on SIRT1 expression and localization in the kidney, SIRT1 protein was examined by immunoblot and immunohistochemistry in kidneys from rats at 144 h after reperfusion (Fig 1B–E). SIRT1 was minimally expressed in sham animals, but was induced after I/R, and further augmented with SRT1720 treatment after I/R (Fig 1B and 1C). In sham animals, regardless of treatment, SIRT1 was predominantly located within the nucleus of proximal tubule epithelial cells (Fig 1D, arrowheads), although some distal tubules also showed cytoplasmic staining. In kidneys from IRV rats, there was a noticeable loss of nuclear SIRT1 (Fig 1D arrows), which was replaced by more intense cytoplasmic staining that was not seen in sham animals (Fig 1D, asterisks). In kidneys from IRS animals, nuclear SIRT1 immunoreactivity was restored similar to what was observed in sham animals (Fig 1D, arrowheads). The number of SIRT1-positive (SIRT1+) nuclei were counted and were reduced in IRV kidneys compared to sham animals, and SRT1720 treatment restored SIRT1 nuclear localization (Fig 1E).
Figure 1. SIRT1 expression and localization after I/R and SRT1720 treatment.

pAMPK and total AMPK (A) and SIRT1 (B) were examined in the renal cortex at 144 h by immunoblot analysis. (C) SIRT1 expression was quantified from immunoblot images, and normalized to GAPDH (n = 3). (D) SIRT1 expression was examined by IHC (40X objective, DAB chromogen, hematoxylin counterstain). In sham animals, nuclear SIRT1 staining was noticeable in tubule epithelial cells (arrowheads). Nuclear localization was lost after I/R, and many tubules exhibited intense cytoplasmic staining with less nuclear staining (asterisks) or complete loss of SIRT1 staining (arrows). Nuclear localization was restored with SRT1720 treatment after I/R (arrowheads). (E) The percentage of SIRT1-positive nuclei per field for each group was quantified (n = 3 – 6, 8 fields per n, Mean % +/− SEM). In (C) and (E), asterisks (*) indicate data are significantly different from V group, and pound signs (#) indicate IRS was significantly different from the IRV group (ANOVA followed by Newman-Keuls, p<0.05).
PGC-1α expression and acetylation state
PGC-1α protein was up-regulated at 72 h and 144 h in both IRV and IRS kidneys (Fig 2A). Differences in nuclear localization of PGC-1α may also affect its activity, and we have previously shown increased nuclear PGC-1α with SRT1720 treatment (Funk et al., 2010). Similar to the cortical tissue lysates, PGC-1α expression was increased in nuclear lysates after I/R and there were no differences between the IRV and IRS groups (Fig 2B). Because SIRT1 is a protein deacetylase, and PGC-1α is a SIRT1 target (Nemoto et al., 2005), and because we have previously shown that SRT1720 treatment results in deacetylated PGC-1α in RPTC (Funk et al., 2010), PGC-1α acetylation (Ac-PGC-1α) was examined in lysates after I/R and SRT1720 treatment (Fig 2C). Renal cortical lysates were subjected to immunoprecipitation with an acetylated-lysine antibody, and then immunoblot analysis with an antibody to PGC-1α. There was increased Ac-PGC-1α in IRV rats at 72 h and 144 h after injury that was reduced to control levels in IRS rats (Fig 2C, representative immunoblots and graph), demonstrating that the majority of PGC-1α which is increased in IRS rats after injury was deacetylated.
Figure 2. SRT1720-induced deacetylation of PGC-1α after I/R.
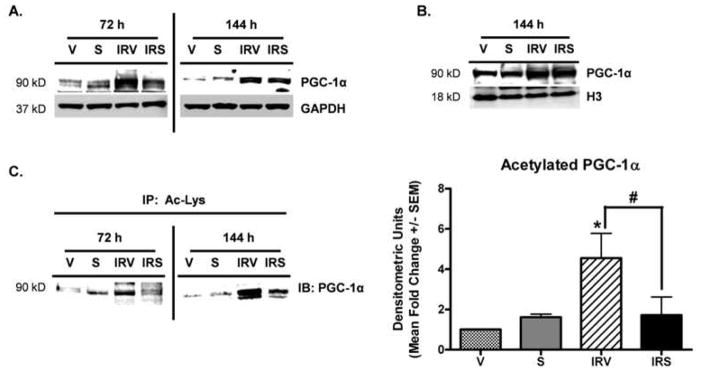
(A) PGC-1α protein in the renal cortex at 72 h and 144 h after I/R by immunoblot analysis. (B) Nuclear PGC-1α from the renal cortex at 144 h after I/R by immunoblot analysis. (C) Acetylation of PGC-1α at 72 h and 144 h after I/R by immunoprecipitation with an anti-acetylated-lysine antibody, followed by immunoblot analysis of PGC-1α, with densitometric quantification of (C) at the 144 h time point in the graph to the right (n = 3). Asterisk (*) indicates data are significantly different from V group, and pound sign (#) indicates IRS was significantly different from the IRV group (ANOVA followed by Newman-Keuls, p<0.05).
Mitochondrial electron transport chain (ETC) and fission/fusion protein expression
From mouse renal I/R studies, we have shown that mitochondrial proteins are depleted early after injury and are persistently down-regulated until at least 144 h after injury (Funk and Schnellmann, 2012). Because SRT1720 treatment activated PGC-1α, we examined recovery of mitochondrial ETC protein expression in the rat kidneys after I/R injury (Fig 3). Twenty-four hours after ischemia, prior to initial SRT1720 dosing, the nuclear-encoded proteins ATP synthase β and NDUFB8, and the mitochondrial-encoded protein COX I were decreased (Fig 3A). As expected, mitochondrial ETC proteins were still diminished at 72 h and 144 h in IRV rats (Fig 3A). Although mitochondrial ETC proteins were also reduced in the kidneys of IRS rats at 72 h, all were at least partially restored by 144 h (Fig 3A).
Figure 3. Mitochondrial protein expression after I/R and SRT1720 treatment.

Mitochondrial proteins (A) ATP synthase β, cytochrome c oxidase subunit I, NDUFB8 and (B) Drp1 And Mfn2 were examined in the renal cortex at 24 h, 72 h and 144 h by immunoblot analysis, with densitometric quantification (normalized to GAPDH) of the 144 h time point in the graph to the right of each representative blot (n = 3 – 6, Mean fold change +/− SEM). Asterisks (*) indicate data are significantly different from V group, and pound signs (#) indicate IRS was significantly different from the IRV group (ANOVA followed by Newman-Keuls, p<0.05).
In our previous study, proteins associated with mitochondrial fusion/fission were also persistently altered after I/R, including dynamin-related protein (Drp1), which mediates mitochondrial fission, and mitofusin 2 (Mfn2), which is involved in fusion of mitochondria (Funk and Schnellmann, 2012). Examination of cortical tissue lysates revealed that both of these proteins were similarly altered after I/R in rats. Mfn2 expression was decreased at 24 h, prior to initial dosing, and was still reduced in both IRV and IRS kidneys at 72 h and 144 h after injury (Fig 3B). Drp1 expression was not changed at 24 h, but was elevated at both 72 h and 144 h in IRV and IRS rats (Fig 3B).
Respiration and protein expression in isolated mitochondria
Because reduced expression of ETC proteins and altered mitochondrial fission/fusion can affect mitochondrial function, we examined mitochondrial respiratory capacity in each of the groups at 144 h. Basal respiration was not significantly altered in any of the groups (Fig 4A, shaded bars). However, FCCP-uncoupled respiration, indicative of maximal respiratory capacity, was reduced approximately 40% in IRV kidneys and completely restored in IRS rats (Fig 4A, filled bars). Mitochondrial ETC and fusion/fission proteins were also examined in isolated mitochondria (Fig 4B–D). In mitochondrial lysates, only COX I was reduced in IRV kidneys. Interestingly, whereas COX I was reduced approximately 60% in IRV whole tissue kidney lysates (Fig 3A), it was only reduced approximately 25% in mitochondrial lysates, and was completely restored in IRS kidneys (Fig 4B and 4C).
Figure 4. Oxygen consumption and protein expression in isolated mitochondria.
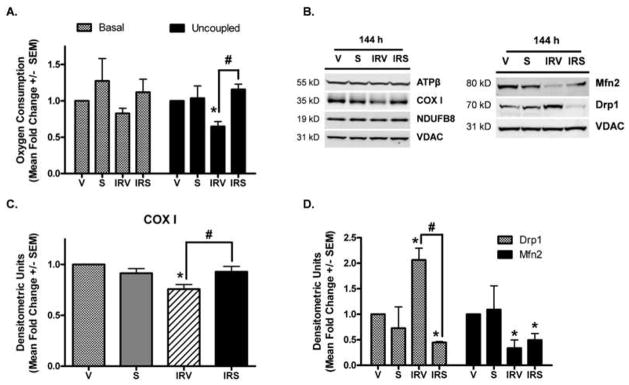
(A) Basal and FCCP-uncoupled respiration were measured in mitochondria isolated from the renal cortex at 144 h (n = 4, Mean fold change +/− SEM). (B) ATP synthase β (ATPβ), cytochrome c oxidase subunit I (COX I), and NDUFB8, and the fusion/fission proteins Mfn2 and Drp1 were examined in mitochondria isolated from the renal cortex at 144 h, with densitometric quantification (normalized to VDAC) in (C) and (D) (n = 3). Asterisks (*) indicate data are significantly different from V group, and pound signs (#) indicate IRS was significantly different from the IRV group (ANOVA followed by Newman-Keuls, p<0.05).
Drp1 is expressed in the cytosol and is recruited to mitochondria undergoing fragmentation by the outer membrane protein Fis1. Therefore, examination of renal cortical expression may not reflect the level of protein actually associated with mitochondrial fission. When Drp1 was examined in isolated mitochondria, there was increased mitochondrial-associated Drp1 in IRV rats, which was attenuated in IRS rats (Fig 4B and 4D). Mfn2 remained down-regulated in mitochondria from IRV rats and was not restored with SRT1720 treatment.
Renal function analysis
Renal histopathology was assessed at 144 h post-reperfusion in IRV and IRS rats by PAS and H&E staining (Fig 5A). Both groups displayed similar levels of brush border loss, presence of casts, inflammatory cells, tubular dilation, degeneration, and interstitial edema, which was reflected in similar composite severity scores (Fig 5B). Additionally, serum creatinine (sCr) measurements followed a similar pattern after injury in both IRV and IRS rats (Fig 5C). sCr was increased from approximately 0.5 mg/dl pre-injury to approximately 2.0–2.3 mg/dl at 24 h after reperfusion in both I/R groups (Fig 5C). sCr recovered consistently and equally in both IRV and IRS rats between 24 h and 144 h to control levels.
Figure 5. Histopathology and renal function after I/R.
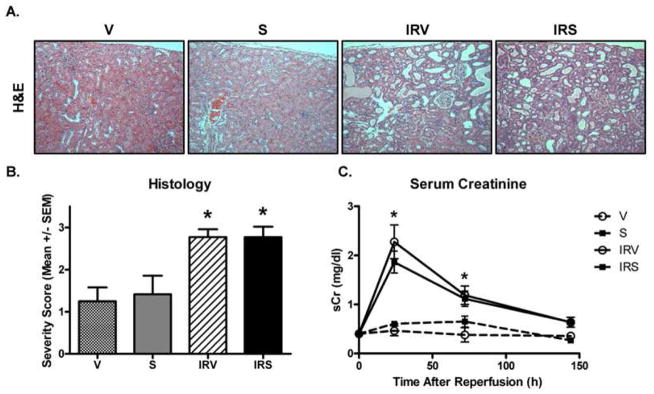
(A) Renal histology was examined by hematoxylin and eosin (H&E) staining at 144 h (10X objective). (B) Histological grades were determined by evaluating H&E and PAS sections to generate a composite severity score from individual scores (0 – 4) for factors including: loss of brush border, casts, inflammatory cells, tubular dilation, degeneration, interstitial edema, and wide interstitium (n = 5, Mean +/− SEM). (C) Serum creatinine was monitored in vehicle and SRT1720-treated I/R and sham rats from 24h to 144h after injury (n = 6, Mean mg/dl +/− SEM). Asterisks (*) indicate data are significantly different from V group (ANOVA followed by Newman-Keuls, p<0.05).
Proximal tubule analysis
Kidney injury molecule-1 (KIM-1) is a sensitive and specific marker of kidney injury with tubular damage (Han et al., 2009). It is up-regulated early after injury, and is expressed until the tubular epithelium has recovered (Ichimura et al., 1998). Urinary KIM-1 increased to maximal levels 24 h after I/R and did not decrease in either group during the 144h study period (Fig 6A), suggesting that there was persistent tubular damage even though glomerular filtration improved. KIM-1 protein was not expressed in kidneys of uninjured rats; however, it was robustly expressed in the renal cortex of IRV rats at 72 and 144 h (Fig 6B and 6D). Although it was still expressed in the IRS rats, KIM-1 was attenuated with SRT1720 treatment at 144 h (Fig 6B and 6D). Following injury, depolarized and dedifferentiated proximal tubule cells undergo numerous morphological and biochemical changes, which are evident by alterations in the expression of various epithelial and mesenchymal markers (Molitoris, 1991; Witzgall et al., 1994). One such alteration is the redistribution and loss of Na+,K+-ATPase expression early after injury in conjunction with the loss of apical-basolateral polarity (Brenner, 1977; Molitoris et al., 1989; Molitoris, 1991). Additionally, the developmental marker vimentin, which is not typically expressed in the normal differentiated tubular epithelium, is increased after injury in dedifferentiated proliferating cells (Witzgall et al., 1994). By immunoblot analysis, Na+,K+-ATPase expression was reduced in IRV rats at 72 and 144 h, but was restored in IRS kidneys at 144 h after injury (Fig 6C and 6E). Vimentin was minimally expressed in uninjured rats, but was markedly elevated in IRV rats at 72 h and 144 h after injury (Fig 6C and 6E). Although it was still elevated compared to uninjured rats, vimentin expression was attenuated in IRS rats compared to IRV rats at 144 h (Fig 6C and 6E). These data suggest that SRT1720 treatment after I/R expedited recovery of a polarized, differentiated tubule epithelium.
Figure 6. Markers of proximal tubule injury and homeostasis.

(A) Urinary kidney injury molecule-1 (KIM-1) measured by ELISA in urine collected from IRV and IRS rats (n = 6, Mean ng/ml +/− SEM). (B) KIM-1 in the renal corex at 72 h and 144 h after injury by immunoblot analysis, with (D) densitometric quantification (normalized to GAPDH) at the 144 h time point (n = 5, Mean +/− SEM). (C) Na+,K+-ATPase and vimentin were examined in the renal cortex at 72 h and 144 h after injury by immunoblot analysis, with (E) densitometric quantification (normalized to GAPDH) at the 144 h time point (n = 4, Mean +/− SEM). Asterisks (*) indicate data are significantly different from V group, and pound signs (#) indicate IRS was significantly different from the IRV group (ANOVA followed by Newman-Keuls, p<0.05).
Because there was some variability in the initial injury (24 h post-reperfusion, prior to treatment intervention), it was anticipated that the kidney might be at different stages of injury/recovery at 144 h post-reperfusion. Therefore, kidneys were stratified into either moderate or severe injury groups. The moderate injury group exhibited a sCr between 1.3 – 2.0 mg/dl at 24 h and the severe injury group exhibited a sCr between 2.5 – 3.0 mg/dl. In sham animals, KIM-1 protein was not detected, PCNA was sporadically dispersed and minimally detected throughout the renal cortex, and Na+,K+-ATPase was ubiquitously expressed at the basolateral membrane of renal tubules (Fig 7A). In kidneys from IRV animals, regardless of severity of initial injury, KIM-1 protein was localized to the apical membranes and cellular debris within the luminal space of injured tubules throughout the renal cortex (Fig 7A). Similar to the results obtained by immunoblot analysis (Fig 6), the mean number of KIM-1 positive (KIM-1+) tubules per field in the renal cortex of IRS rats was reduced in both the moderate and the severe injury groups (Fig 7A and 7B) compared to IRV rats. Na+,K+-ATPase expression and basolateral localization were lost in IRV kidneys in both the moderate and the severe injury groups (Fig 7A). Consistent with the results obtained by immunoblot analysis (Fig 6), Na+,K+-ATPase expression was restored in IRS kidneys from both injury groups; however, whereas localization was predominantly restored to the basolateral membrane in the moderate injury group, expression was dispersed throughout the cytosol in a number of tubules in the severe injury group (Fig 7A).
Figure 7. Immunohistochemical analysis of proximal tubule injury/recovery after I/R.

(A) KIM-1, Na+,K+-ATPase, and PCNA were examined by IHC after I/R with SRT1720 or vehicle treatment (DAB chromogen, hematoxylin counterstain). Injury groups were evaluated separately (moderate or severe) based on extent of initial injury at 24 h post-reperfusion. KIM-1 was not detected in sham animals (40X objective). In IRV and IRS kidneys, KIM-1 was localized to the apical membrane of injured tubules (asterisks), and was associated with cellular debris in the luminal space (arrows). Na+,K+-ATPase was ubiquitously expressed at the basolateral membrane of tubules throughout the renal cortex in sham animals (20X objective). In IRV kidneys, there was an overall loss of Na+,K+-ATPase localization and expression. In IRS kidneys, Na+,K+-ATPase expression was mostly restored in both the moderate and the severe injury groups, and localization to the basolateral membrane was more evident in the moderate injury group, but was also present in the severe injury group (arrows). In sham kidneys, PCNA was sporadically detected in the nucleus of isolated tubule cells throughout the renal cortex (arrow, 40X objective). After injury, the number of PCNA-positive (PCNA+) cells increased, and could be detected in groups of tubule cells throughout the renal cortex (arrows). (B) The number of KIM-1 positive tubules per field was quantified in IRV and IRS kidneys from the moderate and the severe injury groups (n = 3, 8 fields per n, Mean +/− SEM). (C) The number of PCNA+ cells per field from IRV and IRS kidneys in the moderate and the severe injury groups was quantified (n = 3, 8 fields per n, Mean +/− SEM). Pound signs (#) indicate IRS was significantly different from the IRV group (ANOVA followed by Newman-Keuls, p<0.05).
The number of PCNA-positive (PCNA+) nuclei was similar in kidneys from IRV rats from both the moderate and the severe injury groups (Fig 7A and 7C). Interestingly, PCNA staining was patchy, and only sporadically expressed in kidneys from IRS rats in the moderate injury group (Fig 7A). Compared with kidneys from IRV rats with moderate initial injury, IRS kidneys displayed fewer PCNA+ cells, which suggested that proliferation was mostly complete by 144 h in these animals (Fig 7C). In contrast, PCNA was ubiquitously expressed throughout the renal cortex in IRS kidneys from the severe injury group (Fig 7A). Compared to IRV kidneys with similar initial injury, IRS kidneys displayed significantly greater numbers of PCNA+ cells, indicating that proliferation and regeneration was more active in these animals (Fig 7C).
Discussion
In this study we showed that SRT1720 stimulated renal MB in vivo after AKI, and treating rats with SRT1720 after renal ischemia-reperfusion (I/R) expedited recovery of mitochondrial and tubular homeostasis. The effects of SRT1720 were associated with increased nuclear SIRT1 expression and PGC-1α deacetylation, suggesting that the mechanism of action is through a SIRT1-mediated pathway. Overall, we think that using agents that improve mitochondrial function after AKI may offer unique therapeutic benefits to aid in restoration of kidney function. These agents may also offer new therapeutic strategies for treating injuries and/or diseases of other organs, which are plagued by mitochondrial dysfunction.
Following renal I/R, loss of nuclear SIRT1 immunoreactivity was observed in renal cortical proximal tubules and more intense cytoplasmic staining was detected. SRT1720 treatment restored nuclear SIRT1 immunoreactivity in proximal tubules. This finding is similar to what has been shown with SRT1720 treatment after myocardial infarction (Tong et al., 2012). Tong, et al., also demonstrated that SRT1720 treatment conferred PGC-1α deacetylation, which was also seen in the current study, and was cardioprotective. Because PGC-1α functionally interacts with SIRT1, promoting deacetylation of PGC-1α (Nemoto et al., 2005; Gerhart-Hines et al., 2007; Canto and Auwerx, 2009), subcellular localization of SIRT1 is an important determinant of its activity and substrate accessibility. SIRT1-dependent deacetylation of PGC-1α, particularly under stress conditions or energy deprivation, promotes its nuclear accumulation and transcriptional activity (Anderson et al., 2008; Funk et al., 2010). As a transcriptional co-activator, deacetylated PGC-1α more effectively recruits transcription factors to stimulate synthesis of its target genes, which are associated with gluconeogenesis, fatty acid oxidation, and mitochondrial biogenesis (Gerhart-Hines et al., 2007; Rohas et al., 2007; Rodgers et al., 2008).
In addition to the effects on PGC-1α, nuclear versus cytosolic SIRT1 may also influence other cellular fates, such as differentiation and cell death. Translocation of SIRT1 from the cytoplasm into the nucleus promotes differentiation of embryonic neuronal precursor cells to neurons via repression of Notch-Hes1 signaling (Hisahara et al., 2008). Notch-Hes1 signaling is also important in kidney development, and renal cell regeneration following AKI (Kobayashi et al., 2008; Sirin and Susztak, 2012). It is possible that, after injury, SRT1720-mediated SIRT1 nuclear translocation promoted differentiation of renal tubule cells by modulating similar signaling pathways. Additionally, cytoplasmic SIRT1 may make some cells more susceptible to apoptosis, likely the result of reduced interaction with nuclear apoptotic mediators, including p53, FOXO, and Ku70 (Jin et al., 2007). Consistent with these findings, Fan, et al., found that SRT1720 treatment after renal I/R was associated with SIRT1-dependent deacetylation and degradation of p53 and reduced apoptosis (Fan et al., 2013). Similarly, in a mouse model of myocardial infarction, Tong, et al., demonstrated that age-related impaired SIRT1 nuclear translocation, which was associated with higher levels of p53 acetylation and apoptosis, could be reversed with SRT1720 treatment (Tong et al., 2012). Although Fan, et al., did not specifically address SIRT1 localization in their studies, it is likely that nuclear translocation of SIRT1 after injury was a component of SRT1720 treatment in light of the findings presented here and in the heart.
As expected, sCr was elevated 24 h after injury and recovered by 144 h. SRT1720 treatment did not alter recovery of sCr compared to vehicle treatment after injury. This was not entirely unexpected as sCr has been shown to recover quickly, though not completely, in similar models (Funk and Schnellmann, 2012). Fan et al., reported improved sCr and BUN in mice treated with SRT1720 following I/R injury (Fan et al., 2013). Differential injury response and recovery in rats compared to mice may account for the inconsistent results. Whereas Fan, et al., subjected mice to 45 min ischemia followed by 48 h reperfusion, our study subjected rats to 22 min ischemia and 144 h reperfusion to produce an injury comparable to our previously published mouse model (Funk and Schnellmann, 2012). In our previous study, mice had persistent renal dysfunction based on sCr at 144 h; however, rats demonstrated complete recovery of sCr in the current study, which may suggest differential degrees of initial injury and/or recovery. Additionally, Fan et al., gave a daily oral dose of 100 mg/kg, and a dose of 5 mg/kg (ip) was used in this study. Although much lower, this dose was selected after preliminary studies in mice and rats indicated that a single injection of SRT1720 at 5 mg/kg increased PGC-1α and mitochondrial gene expression within 24 h. Even though sCr and histopathology did not improve with SRT1720 treatment, other markers of AKI and tubule homeostasis, including renal KIM-1 expression, vimentin expression, and Na+,K+-ATPase expression and localization, consistent with an overall improvement in renal function.
Interestingly, there were some distinct differences in protein expression and localization when the rats were evaluated based on severity of initial injury. In rats that initially exhibited a moderate injury (i.e., sCr < 2 mg/dl at 24 h), the combined results of reduced KIM-1 and vimentin expression, restoration of Na+,K+-ATPase expression and localization, and reduced PCNA immunoreactivity suggests that, at the time point examined, proliferation and regeneration of the tubule epithelium was mostly complete, and a polarized, differentiated epithelium was restored with SRT1720 treatment. In rats that exhibited a more severe initial injury (i.e. sCr > 2.5 mg/dl at 24 h), KIM-1 and vimentin were still reduced, Na+,K+-ATPase expression was restored, but Na+,K+-ATPase basolateral localization was not, and PCNA was further increased in rats treated with SRT1720 compared to vehicle. This finding suggests that proliferation was more active in these animals, similar to what has been reported in mice treated with SRT1720 at 48 h post-reperfusion (Fan et al., 2013). Because our evaluation was at a time point (144 h post-reperfusion) later than most comparable studies, the discrepancies in PCNA reactivity and Na+,K+-ATPase localization were not surprising. We suggest that recovery from tubule damage is dependent on the extent of the initial insult, and that more severe injury correlated with a longer time course of recovery, and thus the animals were at different stages on the renal injury-recovery continuum.
Yuan et al., have also demonstrated SIRT1-mediated protection of podocytes from aldosterone toxicity with the SIRT1 activator resveratrol (Yuan et al., 2012). In the study, the authors found that aldosterone exposure reduced PGC-1α expression and induced mitochondrial and podocyte injury, and that this could be reversed with SIRT1 overexpression or resveratrol treatment. Additionally, Tran, et al., provided evidence that PGC-1α expression was essential for recovery of renal function following acute endotoxemia (Tran et al., 2011). The authors found that PGC-1α and genes associated with oxidative phosphorylation were suppressed after AKI but recovered concurrently with organ function. Furthermore, PGC-1α deficiency led to persistent renal dysfunction. Overall, our findings as well as others point to SIRT1 and PGC-1α as potential targets to improve renal recovery following acute ischemic or toxic injury.
We previously showed that mitochondrial proteins such as ATP synthase β, NDUFB8, and COX I were depleted early after AKI (i.e. I/R-mediated or glycerol-induced myoglobinuric AKI) and were not restored within at least 144h after injury (Funk and Schnellmann, 2012). Our unpublished experiments revealed that these proteins took approximately two weeks to return to normal expression. In the current study, we again report persistent depletion of kidney cortex mitochondrial proteins in rats subjected to I/R. In rats treated with SRT1720, however, mitochondrial proteins were mostly restored by 144h after injury. In conjunction with the return of mitochondrial proteins, mitochondrial function returned as determined in isolated mitochondria. The increased SIRT1 expression and nuclear localization, increased active nuclear PGC-1α, and return of mitochondrial proteins and function, provides strong evidence that SRT1720 treatment stimulated MB to overcome the persistent mitochondrial injury/dysfunction.
Not only did SRT1720 treatment restore mitochondrial protein expression and function, but also reduced mitochondrial-associated Drp1, suggesting that mitochondrial fragmentation was diminished by SRT1720 in rats after I/R. The influence of Drp1, Mfn, and mitochondrial fragmentation on apoptosis and exacerbation of injury has been documented in several studies from Dr. Zheng Dong’s group (Brooks et al., 2007; Brooks et al., 2009; Brooks et al., 2011), and inhibiting this response protects the kidney from further injury after I/R. The specific mechanism by which SRT1720 decreased mitochondrial-associated Drp1 is not known; however, activation of the MB program may also influence mitochondrial dynamics after I/R. It should be pointed out that the method used to examine mitochondrial-associated Drp1 is not without limitations, as the Drp1-mitochondrial association could be lost during the isolation process.
Following I/R injury, loss of Na+,K+-ATPase localization and expression, and increased expression of vimentin, correlated with tubule histopathology. This was not surprising as changes in Na+,K+-ATPase and vimentin expression/localization and disrupted tubule epithelial cell morphology, including cytoskeletal disruptions, loss of brush border integrity, and flattened elongated morphology, have been previously characterized (Molitoris, 1991; Witzgall et al., 1994; Bonventre and Yang, 2011). Restoration of normal Na+,K+-ATPase and vimentin expression with SRT1720 treatment correlated with recovery of renal KIM-1 expression. KIM-1 also co-localizes with α-smooth muscle actin (α-SMA), aquaporin, and vimentin, which suggests that it is expressed in dedifferentiated proximal tubule cells (van Timmeren et al., 2007), and the results obtained in the current study are consistent with that conclusion.
It is possible that SIRT1 has a direct effect on Na+,K+-ATPase expression and/or localization in I/R injury. Seo-Mayer, et al., demonstrated that basolateral expression of the Na+,K+-ATPase may be preserved by activating AMPK prior to I/R (Seo-Mayer et al., 2011). The authors suggested that, in the face of energy-deprivation, stimulation of AMPK may alleviate some of the epithelial cell damage by activating energy-conserving pathways. Indeed, Na+,K+-ATPase expression and function may be regulated by AMPK and AMPK family members, such as the salt-inducible kinases (Taub et al., 2010). It is possible that similar energy-salvaging mechanisms are activated upon SIRT1 activation which restore Na+,K+-ATPase expression and localization after I/R, although this has yet to be shown. Several reports have also indicated that SRT1720 and other reported SIRT1 activators target AMPK, but we have not observed this effect in the current study or previously (Funk et al., 2010). AMPK and SIRT1 pathways seem to converge quite often and have been suggested to work synergistically on PGC-1α activation (Canto and Auwerx, 2009; Canto et al., 2009; Canto et al., 2010). As a modulator of PGC-1α activity/expression after injury, SRT1720 treatment may be manipulating one or a combination of these pathways to preserve Na+,K+-ATPase expression after I/R. Although we did not observe any evidence of direct AMPK activation with SRT1720 treatment in our model, it is possible that the combined effects of AMPK activation after injury with SRT1720-induced SIRT1 activation modulated Na+,K+-ATPase expression via PGC-1α or another unidentified mechanism.
Finally, SIRT1 may regulate many functions in the cell and it is possible that SRT1720 treatment activated recovery mechanisms not related to its effects on mitochondrial function. Mice overexpressing SIRT1 specifically within the kidney were protected against cisplatin-induced nephrotoxicity (Hasegawa et al., 2010). Although they observed sufficient protection of mitochondrial function, but not mitochondrial numbers, in the SIRT1 transgenic mice (SIRT1-Tg), the authors attributed the protection to a preservation of peroxisome function after injury. Retaining peroxisome numbers increased levels of catalase, thus reducing ROS and apoptosis and retaining kidney function (Hasegawa et al., 2010). The authors also examined I/R injury in the SIRT1-Tg mice, but did not see any protection in this model. It should be noted that in our model, SRT1720-mediated SIRT1 stimulation was not initiated until 24h after reperfusion when tissue injury is extensive and when functional markers such as SCr have reached peak levels. In the SIRT1-Tg mice, there was significant protection from tissue damage, thus there are inherent differences in how the injured cells may respond to SIRT1 activation under different conditions.
In conclusion, treatment with the SIRT1 activator SRT1720 improved renal cortical mitochondrial function following I/R within 6 days after injury. Restoration of mitochondrial function correlated with normalization of proximal tubule polarization and differentiation. Recovery of mitochondrial function following AKI appears to be an essential component of the recovery process, in particular for recovery of normal proximal tubule homeostasis. SIRT1 activation and other compounds that target PGC-1α activity and/or expression offer unique therapeutic options to improve tubule repair after injury.
Highlights.
We examined recovery of mitochondrial and renal function after ischemia-reperfusion
SRT1720 treatment after I/R induced mitochondrial biogenesis via SIRT1/PGC-1α
Recovery of mitochondrial function was expedited with SRT1720 treatment
Restoration of tubule homeostasis was expedited with SRT1720 treatment
Activators of SIRT1 and PGC-1α may offer new therapeutic targets for AKI
Acknowledgments
This work was supported by NIH/NIGMS Grant 084147 and by the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs Merit grant BX000851. Animal facilities were funded by NIH grant C06 RR-015455.
Abbreviations
- SIRT1
sirtuin 1
- PGC-1α
peroxisome proliferator activated receptor γ coactivator-1α
- ATPβ
ATP synthase subunit β
- COX I
cytochrome c oxidase subunit I
- NDUFB8
NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8
- Drp1
dynamin related protein 1
- Mfn2
mitofusin-2
- sCr
serum creatinine
- KIM-1
kidney injury molecule 1
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner BM. Functional and structural determinants of glomerular filtration. A brief historical perspective. Fed Proc. 1977;36:2599–2601. [PubMed] [Google Scholar]
- Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 2011;300:C447–455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. The Journal of clinical investigation. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington MD, Schnellmann RG. Chronic high glucose downregulates mitochondrial calpain 10 and contributes to renal cell death and diabetes-induced renal injury. Kidney Int. 2012;81:391–400. doi: 10.1038/ki.2011.356. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Yang HC, You L, Wang YY, He WJ, Hao CM. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2013;83:404–413. doi: 10.1038/ki.2012.394. [DOI] [PubMed] [Google Scholar]
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Funk JA, Odejinmi S, Schnellmann RG. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther. 2010;333:593–601. doi: 10.1124/jpet.109.161992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. American journal of physiology. Renal physiology. 2012;302:F853–864. doi: 10.1152/ajprenal.00035.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WK, Wagener G, Zhu Y, Wang S, Lee HT. Urinary biomarkers in the early detection of acute kidney injury after cardiac surgery. Clin J Am Soc Nephrol. 2009;4:873–882. doi: 10.2215/CJN.04810908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Sueyasu K, Washida N, Tokuyama H, Tzukerman M, Skorecki K, Hayashi K, Itoh H. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem. 2010;285:13045–13056. doi: 10.1074/jbc.M109.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, Sato M, Horio Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15599–15604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. The Journal of biological chemistry. 1998;273:4135–4142. doi: 10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- Jin Q, Yan T, Ge X, Sun C, Shi X, Zhai Q. Cytoplasm-localized SIRT1 enhances apoptosis. Journal of cellular physiology. 2007;213:88–97. doi: 10.1002/jcp.21091. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Terada Y, Kuwana H, Tanaka H, Okado T, Kuwahara M, Tohda S, Sakano S, Sasaki S. Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int. 2008;73:1240–1250. doi: 10.1038/ki.2008.74. [DOI] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M, Krawczyk M, Irusta PM, Martin-Montalvo A, Hubbard BP, Zhang Y, Lehrmann E, White AA, Price NL, Swindell WR, Pearson KJ, Becker KG, Bohr VA, Gorospe M, Egan JM, Talan MI, Auwerx J, Westphal CH, Ellis JL, Ungvari Z, Vlasuk GP, Elliott PJ, Sinclair DA, de Cabo R. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitoris BA. Ischemia-induced loss of epithelial polarity: potential role of the actin cytoskeleton. Am J Physiol. 1991;260:F769–778. doi: 10.1152/ajprenal.1991.260.6.F769. [DOI] [PubMed] [Google Scholar]
- Molitoris BA, Chan LK, Shapiro JI, Conger JD, Falk SA. Loss of epithelial polarity: a novel hypothesis for reduced proximal tubule Na+ transport following ischemic injury. J Membr Biol. 1989;107:119–127. doi: 10.1007/BF01871717. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} The Journal of biological chemistry. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Funk JA, Jayavelu T, Green PT, Schnellmann RG. 5-hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J Pharmacol Exp Ther. 2010;332:632–639. doi: 10.1124/jpet.109.159947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. The Journal of biological chemistry. 2007;282:2355–2362. doi: 10.1074/jbc.M608009200. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. Isoflavones promote mitochondrial biogenesis. J Pharmacol Exp Ther. 2008;325:536–543. doi: 10.1124/jpet.107.134882. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS letters. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohas LM, St-Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7933–7938. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo-Mayer PW, Thulin G, Zhang L, Alves DS, Ardito T, Kashgarian M, Caplan MJ. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am J Physiol Renal Physiol. 2011;301:F1346–1357. doi: 10.1152/ajprenal.00420.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirin Y, Susztak K. Notch in the kidney: development and disease. The Journal of pathology. 2012;226:394–403. doi: 10.1002/path.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, Carney DP, Johnson RJ, Lavu S, Iffland A, Elliott PJ, Lambert PD, Elliston KO, Jirousek MR, Milne JC, Boss O. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst Biol. 2009;3:31. doi: 10.1186/1752-0509-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub M, Springate JE, Cutuli F. Targeting of renal proximal tubule Na,K-ATPase by salt-inducible kinase. Biochem Biophys Res Commun. 2010;393:339–344. doi: 10.1016/j.bbrc.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Morrison A, Mattison S, Qian S, Bryniarski M, Rankin B, Wang J, Thomas DP, Li J. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012 doi: 10.1096/fj.12-216473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, David S, Burstein D, Karumanchi SA, Stillman IE, Arany Z, Parikh SM. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. The Journal of clinical investigation. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Timmeren MM, van den Heuvel MC, Bailly V, Bakker SJ, van Goor H, Stegeman CA. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol. 2007;212:209–217. doi: 10.1002/path.2175. [DOI] [PubMed] [Google Scholar]
- Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. The Journal of clinical investigation. 1994;93:2175–2188. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C, Ding G, Liu B, Yang T, Zhang A. Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012;82:771–789. doi: 10.1038/ki.2012.188. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Lu B, Daubert RA, Chavin KD, Wang L, Schnellmann RG. Suramin promotes recovery from renal ischemia/reperfusion injury in mice. Kidney Int. 2009;75:304–311. doi: 10.1038/ki.2008.506. [DOI] [PubMed] [Google Scholar]