

Abstract
Enteroviruses are the most common human viral pathogens worldwide. This genus of small, non-enveloped, single stranded RNA viruses includes coxsackievirus, rhinovirus, echovirus, and poliovirus species. Infection with these viruses can induce mild symptoms that resemble the common cold, but can also be associated with more severe syndromes such as poliomyelitis, neurological diseases including aseptic meningitis and encephalitis, myocarditis, and the onset of type I diabetes. In humans, polarized epithelial cells lining the respiratory and/or digestive tracts represent the initial sites of infection by enteroviruses. Control of infection in the host is initiated through the engagement of a variety of pattern recognition receptors (PRRs). PRRs act as the sentinels of the innate immune system and serve to alert the host to the presence of a viral invader. This review assembles the available data annotating the role of PRRs in the response to enteroviral infection as well as the myriad ways by which enteroviruses both interrupt and manipulate PRR signaling to enhance their own replication, thereby inducing human disease.
Keywords: Enterovirus, type I IFN, toll-like receptor, RIG-I-like receptor
1. Introduction
1.1 Enteroviruses
Enteroviruses (EVs), which include coxsackievirus, rhinovirus, echovirus, and poliovirus species, are members of the picornavirus family. These small (~30nm), non-enveloped, single stranded RNA viruses consisting of a genome of ~7kB are the most common human viral pathogens worldwide [1, 2]. EVs, excluding rhinoviruses, are responsible for as many as 15 million symptomatic infections in the United States every year [3] and are commonly associated with neurological disease. As many as 10-15% of encephalitis cases in the U.S. and worldwide have been associated with non-poliovirus EV infections [4-7] and they are the leading causative agents of aseptic meningitis worldwide [8]. Although EV-induced CNS complications are more commonly associated with mortality in neonates and children, adult infections can also lead to severe complications (particularly when the adult has not been exposed to the EV serotype previously) [9]. Enterovirus 71 (EV71) has become an important public health concern in recent years, especially in Asia, as its incidence has increased in the region and the illness it causes is often associated with severe neurological complications and/or death [10]. Importantly, EVs, particularly coxsackievirus B (CVB), are also linked to the development of myocarditis with up to 50% of patients with myocarditis displaying evidence of an EV infection [11-13]. Finally, EV infections, specifically CVB4, have also been linked to the onset of type I diabetes [14-16]. In contrast, rhinoviruses are the causative agent of over 50% of human viral-induced acute respiratory tract infections [17], which are associated with nearly $40 billion in direct and indirect costs annually in the United States alone [18].
Studies detailing how these medically relevant viruses interact with the host immune system are described in this review, with a specific focus on how the innate immune system alerts the body to the presence of an enteroviral invader and how enteroviruses have evolved to attenuate this system in order to enhance their replication. In this review, we focus on the non-rhinovirus EVs.
1.2 Pattern Recognition Receptor Signaling
It has long been appreciated that the innate immune response is necessary for the induction of the subsequent adaptive immune response [19, 20]. Innate immunity to pathogens is largely mediated by pattern recognition receptors (PRRs), which recognize a variety of pathogen associated molecular patterns (PAMPs) that are highly conserved amongst classes of pathogens [21]. During a viral infection, PRRs induce an intracellular signaling cascade resulting in the alteration of the host cell’s transcription profile in response to recognition of their cognate PAMP. Two major classes of transcription factors are activated in response to this signaling: Interferon Regulatory Factors (IRFs) and NF-κB family members. These transcription factors act in concert to induce the expression of type I interferons (IFN)[22]. These auto- and paracrine signaling molecules serve to upregulate a cadre of genes, known as interferon stimulated genes (ISGs). The effects of type I IFNs and ISGs are legion; they are pro-inflammatory [23], enhance adaptive immunity [24], and are directly antiviral [25]. Additionally, NF-B activation induces a host of pro-inflammatory and pro-survival genes independently of type I IFN induction [26-29] and may be required for full induction of type I IFNs [27, 30].
Toll-like receptors (TLRs) 1-13 are transmembrane PRRs that recognize a diverse range of PAMPs. TLRs can be divided into two broad categories—those that are localized to the cell surface and those that are localized to the endosomal lumen. TLRs that are present on the cell surface are important in recognition of bacterial pathogens. In contrast, TLRs that are localized to the lumen of endosomes, TLRs 3, 7, 8, and 9, serve to recognize nucleic acids and are thus traditionally thought to be the most important in the promotion of an antiviral response. TLR3 recognizes dsRNA and the synthetic dsRNA structural homolog poly(I:C) [31]. TLR7 and TLR8 recognize ssRNA and imidazoquinoline compounds[32-35]. TLR9 recognizes unmethylated deoxycytidylate-phosphate-deoxyguanylate (CpG) DNA, found almost exclusively in bacteria [36, 37].
In addition to TLRs, cytoplasmic PRRs exist and are divided into two main groups—the NOD-like receptors (NLRs) and the RIG-I-like receptors (RLRs). There are three RLRs: RIG-I, MDA5, and LGP2. RIG-I recognizes cytoplasmic short dsRNA and 5′ppp-ssRNA[38-41]. MDA5 binds the internal duplex structure of cytoplasmic long dsRNA and cooperatively assembles into a filamentous oligomer composed of MDA5 dimers [41-47]. The role of LGP2 has not been thoroughly elucidated. Early studies suggested that it acted as a negative regulator of RIG-I and MDA5[48-50]. However, further studies revealed that LGP2 was essential for type I IFN response to picornavirus infections in mice and that LGP2 with active helicase activity is required for IFNβ production in response to various RNA viruses in dendritic cells (DCs) and mouse embryonic fibroblasts (MEFs)[51]. Further studies of LGP2 have yielded equally disparate results, as both overexpression of chicken LGP2 and knockdown of endogenous LGP2 in chicken cells resulted in reduced IFNβ production in response to avian influenza infection[52].
There are 22 human NLRs that can be further subdivided into five families: NLR families A, B, C, P, and X. These families are structurally related. All NLRs have three domains: an N-terminal domain involved in signaling, a nucleotide-binding NOD domain, and a C-terminal leucine rich region (LRR) important for ligand recognition (reviewed in [53, 54]). The NLR most traditionally associated with response to viral infection is NALP3, a member of the NLRP family. NALP3, also known as cryopyrin, is a member of the NALP3 inflammasome, which is responsible for the processing of the proinflammatory cytokine IL-1β to its mature form[55]. NALP3 has been shown to be a sensor for bacterial peptidoglycans[56], endogenous uric acid crystals (associated with gout)[57], bacterial RNA [58], and, importantly, imidazoquinolines and viral RNA [58, 59]. Recent data has shown that NOD2, a member of the NLRC family traditionally viewed as a sensor for bacterial muramyl dipeptide[60, 61], also serves to sense viral ssRNA[62]. Finally, there has been conflicting data published on the role of NLRX1 in the negative regulation of RLR antiviral signaling, with initial studies showing that the presence of NLRX1 dampens RLR signaling[63, 64], but subsequent studies showing no role for NLRX1 in RLR signaling[65, 66].
As summarized above, the activation of various PRRs by PAMPs produced by viral infection leads to an altered transcription profile of the infected cell. The induction of type I IFN signaling is important for the control of EV infections in vivo, as evidenced by enhanced EV-induced lethality in type I IFN receptor (IFN-R) null mice [67-69] and increased viral susceptibility in IFN -deficient mice [70]. In addition, purified IFN treatment of patients diagnosed with EV-induced myocarditis significantly improves cardiac function [71], underscoring the role of this cytokine in the control of human EV infections. Below we review what is known regarding the sensing of non-rhinovirus EVs and how these viruses target a variety of components within both the TLR and RLR pathways to promote their replication and/or spread.
2. Recognition of enteroviral infections
The literature shows that TLRs, RLRs, and NLRs, the three broad categories of PRRs described above, all play an important role in the sensing of EV infections. Below we summarize these studies based upon the subtype of PRRs responsible for this sensing.
2.1 Toll-Like Receptors
TLR3 has been shown to play an essential and non-redundant role in the response to EVs, and may be considered the TLR identified as most critical in the control of EV infections. TLR3-deficient mice exhibit significantly increased mortality in response to a dose of coxsackievirus B4 (CVB4) that is sublethal in TLR3-expressing mice [72]. In addition, mice deficient in TLR3 or TIR-domain-containing adaptor-inducing IFNβ (TRIF), a key adaptor in TLR3 signaling, are more susceptible to poliovirus (PV) infection, displaying increased mortality and viral load which were correlated with an inability to produce type I IFNs [73, 74]. TLR3 also plays a protective role in restricting CVB3 infection in the heart as TLR3-/- mice infected with CVB3 display increased mortality and myocarditis [75] due at least in part to an increase in IL-4 in TLR3-/- mice upon CVB3 infection and a subsequent shift from a protective Th1 response to a Th2 response in the hearts of these mice [76, 77]. TRIF-/- mice infected display increased viral replication in myocytes, decreased left ventricular functioning, and increased cardiac fibrosis [78]. Further supporting a role for TLR3 in EV innate immune signaling, human patients diagnosed with EV-induced myocarditis have increased frequencies of two single-nucleotide polymorphisms (SNPs) in TLR3 which result in variants that exhibit a reduced capacity to promote type I IFN and NF-κB signaling in vitro in response to poly(I:C) or CVB3 infection [79]. This suggests that a reduced ability to sense viral invasion through TLR3 results in an increased risk of developing virally induced cardiac inflammation.
In addition to TLR3, several other TLRs have been shown to be important in the sensing of EV infections. TLR4, which is localized to the cell surface, has also been shown to be important in the detection of EVs, although it is mainly studied in the context of bacterial pathogens. Infection with CVB4 is implicated in the development of type I diabetes, and the damage to the pancreatic beta cells is thought to be mediated by pro-inflammatory cytokines. It has been shown that TLR4 mediates the production of TNFα and IL-6 in pancreatic cells infected with CVB4, suggesting a role for TLR4 in recognizing not only bacterial LPS, but viral proteins as well [80]. Additionally, the level of TLR4 expression and the level of EV RNA present in endomyocardial tissue of patients with myocarditis have been shown to be positively correlated [81]. However, in contrast to the studies described above related to TLR3 signaling, much less is known regarding the consequences of TLR4 signaling on EV infection in vivo.
The ssRNA sensors TLR7 and TLR8 have also been shown to play some role in the induction of antiviral signaling in response to CVB3 infection, although their precise function remains largely unclear [82-84]. TLR7 has been shown to be required in plasmacytoid dendritic cells (pDCs), also known as interferon-producing cells because of their role in producing copious amounts of type I IFNs [85], for the production of IFNα and IL-12p40 in response to CVB3, although this role was dependent on CVB3 specific antibody-mediated opsonization of the virus [84]. It has been shown that knockdown of endogenous TLR8 in primary human cardiac cells critically abrogates the capacity of those cells to produce IL-6 in response to CVB3 infection [82]. This suggests that the damaging cardiac inflammation seen in EV-induced myocarditis may be mediated through sensing of viral RNA by TLR8. However, little is known regarding the role of these TLRs in the control of EV infections in vivo.
2.2 RIG-I-like Receptors
Our initial understanding of RLR mediated recognition of EVs was based on the role of RLRs in the detection of a related picornavirus, encephalomyocarditis virus (EMCV). As picornaviruses do not generate 5′-ppp RNA, but instead utilize RNA covalently linked at the 5′ end to the VPg protein [86, 87](Figure 1), they are not expected to be recognized by RIG-I. Indeed, the RLR mediated recognition of EMCV occurs primarily through MDA5 [88, 89]. Further, loss of the mitochondrial antiviral-signaling protein (MAVS), a downstream adaptor for RLRs, results in enhanced replication and deficient antiviral signaling in response to EMCV infection [90]. In addition, it has been shown that recognition of EMCV infection by the RLR pathway is partially reliant on the amplification of antiviral signaling mediated by RNase L as infection of RNase L-deficient mice with EMCV resulted in a decrease in serum IFNβ as compared to wild-type controls [92]. RNase L is an interferon-inducible antiviral endoribonuclease that has been shown to generate RNA ligands from self-RNA for MDA5 and RIG-I, enhancing antiviral signaling [91].
Figure 1. Schematic of the EV genome.
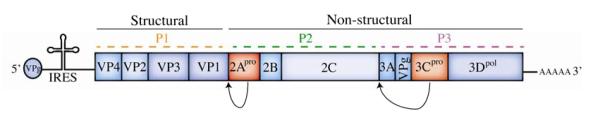
The positive sense single stranded RNA genome undergoes IRES dependent translation into a single polypeptide. This polypeptide is then processed into individual viral proteins by two viral proteases: 2Apro and 3Cpro (shown in red), as indicated by arrows. These viral proteases also act upon a wide range of host cell proteins.
Further research into the role of RLRs in the recognition of the picornavirus EMCV showed that mice deficient in LGP2 were unable to produce a Type I IFN response upon EMCV infection. However, these same LGP2 deficient mice were resistant to lethal doses of VSV, building further support for the role of LGP2 as both a positive and negative regulator of RLR signaling [93].
More recent work confirms the role of MDA5 in the recognition of picornaviruses and conclusively shows that MDA5 serves as the cytoplasmic sensor for EVs. In vitro, MDA5-/- MEFs but not RIG-/- MEFS were unable to produce type I IFN in response to transfection of CVB3 genomic RNA [94]. MDA5 mediated responses to CVB3 RNA, as well as that of multiple other EVs, have been shown to be largely dependent on direct interaction of MDA5 with the dsRNA replicative intermediate form [45, 94], a dsRNA generated during genome replication of EVs. The role of MDA5 also seems relevant in vitro, as in one study both MAVS and MDA5 deficient mice showed increased mortality and decreased systemic and tissue specific type I IFN upon CVB3 infection [95]. While a second study confirmed that MDA5 deficient mice were indeed more susceptible to CVB3 infection, as demonstrated by increased mortality, this study found that MDA5 seemed to be dispensable for production of systemic IFNα and tissue specific IFNβ in CVB3-infected mice [96]. These disparate results may be due to differences in the MDA5-/- mice used in the studies: Wang et al [95] used an MDA5-/- mouse on a SvJ background whereas the strain used by Huhn et al [96] was on a B6 background.
The role for MDA5 in the innate immune response to EV infections was further studied in the setting of PV infections. MDA5, but not RIG-I, was found to be essential in vitro for the production of type I IFNs [74]. However, MDA5 or MAVS deficient mice transgenically expressing the PV receptor did not display increased mortality [73, 74], defects in IFNα production, or enhanced viral replication [74] upon PV infections. This suggests that MDA5 may be capable of sensing EV infections in vitro or in specific cell types, but that the TLR3/TRIF pathway carries the main burden of EV recognition in vivo. EV71 infection has also been shown to be sensed by MDA5 in vitro, as loss of MDA5, but not RIG-I, was found to result in a loss of IRF3 activation[97] and type I IFN production in response to EV71 RNA [94, 97].
A link between the development of fulminant Type I diabetes and the sensing of EV infection via RLRs has also been suggested. Both α- and β-cells in the pancreas of fulminant type I diabetics with EV infection showed hyperexpression of RIG-I and MDA5 whereas non-fulminant diabetics without EV infections did not show this association [98]. However, the molecular basis for these results requires further study.
2.3 NOD-Like Receptors
Little is known regarding the role of NLRs in the sensing of EV infections. One study has shown that NLRX1 hinders the association of MDA-5 with MAVS upon EMCV infection without affecting the level of type I IFN production [64]. The relevance of this to viral infection is unclear, as is whether a similar function for NLRX1 is found during EV infections. Additionally, recent work has shown that NALP3 is activated during EMCV infection [99, 100]. This activation is proposed to be triggered by alterations in Ca2+ homeostasis induced by the EMCV viroporin 2B, and was also associated with EV71 and PV 2B proteins [100].
3. Enteroviruses: Evading detection
Viruses have evolved various strategies to counter or bypass innate immune defenses in order to promote their replication and/or spread. Viruses may accomplish this evasion in at least two possible ways. First, viruses may avoid detection by directly disabling the PRR mediated pathways described above. Alternatively, viruses may block the host response to the transcriptional changes that result from PRR engagement by directly targeting antiviral ISGs or rendering the host cells nonresponsive to type I IFNs. In reality, many viruses possess the ability to avoid immune control through both of these strategies and may possess further means of targeting antiviral signaling. In the following sections, the body of literature regarding the strategies utilized by EVs to alter PRR-mediated signaling will be summarized. Defining the mechanisms by which EVs manipulate host cell signaling pathways in order to avoid detection by the innate immune system is an excellent means by which to study the host innate immune system, as evolutionary pressure has ‘taught’ the virus what key signaling pathways it must dismantle or manipulate in order to survive. We as scientists can then, in turn, identify important host innate immune components that restrict viral infection by identifying the molecules and/or pathways targeted by the virus.
During infection, the EV viral genome is translated as a precursor polyprotein that requires proteolytic processing by the virally-encoded 2Apro and 3Cpro cysteine proteases (Figure 1). These proteases preferentially target conserved consensus cleavage sites located throughout the viral polyprotein (Tyr-Gly for 2Apro and Glu-Gly for 3Cpro). In addition, these proteases target a variety of host cell components that possess target cleavage sites (although there are additional determinants for site cleavage which might include accessibility of the conserved sites and/or cellular localization) [101]. There is a significant body of work detailing the ability of the EV proteases 2Apro or 3Cpro to cleave a number of factors involved in host-cell transcription and translation including eukaryotic initiation factor 4G (eIF4-G)[102], transcription factor IIIC (TFIIIC)[103], and the TATA-binding protein (TBP)[104] in addition to many others. Although this is obviously a very broad attack on the host cell, interfering with transcription and translation precludes production of type I IFN and ISGs, thus potently abrogating many downstream aspects of innate immune signaling. Further studies have pointed to a direct role for both 2Apro and 3Cpro in the potent attenuation of many aspects of antiviral innate immune signaling by EVs. These strategies are detailed in the following sections and summarized in Table 1 and Figure 2.
Table 1.
A summary of EV mediated evasion of PRR signaling.
| Enteroviral Protein | Role in Attenuating Innate Immune Signaling |
Reference |
|---|---|---|
| CVB3 3Cpro | Cleaves TRIF | [105] |
| CVB3 3Cpro | Cleaves MAVS | [105] |
| CVB3 3Cpro | Cleaves FAK | [109] |
| EV71 3Cpro | Cleaves TRIF | [106] |
| EV71 3Cpro | Structurally inhibits MAVS recruitment to RIG-I | [111] |
| EV71 3Cpro | Cleaves IRF7 | [112] |
| EV71 2Apro | Cleaves MAVS | [110] |
| EV71 induced caspases | Cleaves MDA5 | [97] |
| PV induced caspases | Cleaves MDA5 | [107] |
| PV induced caspases | Cleaves MAVS | [108] |
Figure 2. Interference of PRR-mediated signaling by EVs.

EVs have evolved multiple mechanisms to attenuate and/or modulate PRR signaling at a number of diverse stages. This results in a reduction of type I IFN production and/or NF-κB mediated transcription and allows the virus to evade detection by the innate immune system.
3.1 Evading TLR3 detection
As TLR3 has been identified as a key TLR for sensing EV infection (detailed in Section 2.1 above), it follows that EVs directly target this pathway in order to interrupt this arm of the innate immune system. Several EVs render the key TLR3 adaptor molecule TRIF non-functional. The CVB3 protease 3Cpro has been shown to cleave TRIF upon infection. These CVB3-generated TRIF cleavage fragments were unable to induce NF-B signaling or apoptosis, two roles of full-length TRIF [105]. In addition, 3Cpro encoded by EV71 also cleaves TRIF, resulting in an inhibition of TRIF-mediated IFNβ and NF-κB promoter activation [106]. Additional components of the TLR3 pathway are also directly targeted by CVB3 3Cpro (Harris and Coyne, unpublished data) as a means of suppressing this key pathway at multiple stages. Given the large relative contribution of this pathway in the control of EV infections, it is not surprising that these viruses specifically target this pathway at multiple, non-redundant points.
3.2 Evading RLR detection
PV infection has been shown to result in the cleavage of MDA5 [107]. Interestingly, this cleavage was not due to a virally-encoded protease, but instead was mediated by caspases that were activated in response to viral infection. Perplexingly, this cleavage event may enhance type I IFN signaling as induction of IFNβ was reduced in PV-infected cells treated with a caspase inhibitor to block MDA5 cleavage [107]. PV also targets the RLR adaptor MAVS for cleavage in a caspase-dependent manner [108], suggesting that PV has evolved mechanisms to utilize components of the host cell to directly target the RLR pathway.
MAVS is also targeted for cleavage by CVB3 3Cpro [105]. In this case, the 3Cpro-dependent cleavage of MAVS attenuated IFNβ signaling and led to the generation of cleavage fragments that were functionally deficient in NF-κB and type I IFN signaling when compared to full-length MAVS [105]. CVB3 3Cpro also targets the RLR signaling pathway through direct cleavage of Focal Adhesion Kinase (FAK) which is recruited to mitochondria upon viral infection and potentiates MAVS signaling by an as-yet-undefined mechanism [109].
EV71 also targets the RLR pathway at several points. The EV71 protease 2Apro cleaves MAVS during infection to abrogate downstream signaling [110]. EV71 3Cpro also targets the RLR pathway through a mechanism distinct from that of CVB3, functioning as a structural inhibitor of recruitment of MAVS to RIG-I. This results in a failure of interferon regulatory factor 3 (IRF3) to localize to the nucleus and consequently in a reduction in RIG-I mediated IFNβ expression [111]. The EV71 protease 3Cpro also cleaves IRF7 directly [112], thus suppressing IFN transcriptional induction. Further, in a manner similar to PV, EV71-induced caspase activation results in the degradation of MDA5 [97]. Finally, EMCV has also been shown to target the RLR pathway. EMCV infection results in cleavage of RIG-I [113, 114]. This cleavage is mediated by both the EMCV encoded 3Cpro and host cell caspases [114].
To our knowledge, no work has been published demonstrating the manipulation of the NLR signaling pathways by EVs.
4. Conclusion
Despite their small size, EVs are adept at suppressing the host innate immune system through a variety of highly evolved strategies. Both TLRs and RLRs have critical, well-established roles to play in the recognition of EV infections. Further work is required to determine what, if any, role NLRs might play in the recognition of EV infections. The study of the targeting of the innate immune system by EVs has the potential to provide many insights into novel components and pathways important in the control of antiviral innate immune signaling.
Acknowledgements
We wish to thank Dr. Saumendra Sarkar (University of Pittsburgh) for helpful discussions. We apologize to any colleagues whose work we may have neglected to cite due to space limitations. Our work on the interplay between EVs and the innate immune system is supported by the NIH R01-AI081759 (C.B.C.) and T32-AI-049820 (K.H.). In addition, C.B.C. is a recipient of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- [1].Centers for Disease Control Non-Polio Enterovirus Infections. 2011 [Google Scholar]
- [2].International Committee on Taxonomy of Viruses Virus Taxonomy: 2012 Release (current) 2012 [Google Scholar]
- [3].Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance--United States, 1970-2005. MMWR Surveill Summ. 2006;55:1–20. [PubMed] [Google Scholar]
- [4].Huang C, Morse D, Slater B, Anand M, Tobin E, Smith P, et al. Multiple-year experience in the diagnosis of viral central nervous system infections with a panel of polymerase chain reaction assays for detection of 11 viruses. Clin Infect Dis. 2004;39:630–5. doi: 10.1086/422650. [DOI] [PubMed] [Google Scholar]
- [5].Koskiniemi M, Rantalaiho T, Piiparinen H, von Bonsdorff CH, Farkkila M, Jarvinen A, et al. Infections of the central nervous system of suspected viral origin: a collaborative study from Finland. J Neurovirol. 2001;7:400–8. doi: 10.1080/135502801753170255. [DOI] [PubMed] [Google Scholar]
- [6].Xu Y, Zhaori G, Vene S, Shen K, Zhou Y, Magnius LO, et al. Viral etiology of acute childhood encephalitis in Beijing diagnosed by analysis of single samples. Pediatr Infect Dis J. 1996;15:1018–24. doi: 10.1097/00006454-199611000-00017. [DOI] [PubMed] [Google Scholar]
- [7].Fowlkes AL, Honarmand S, Glaser C, Yagi S, Schnurr D, Oberste MS, et al. Enterovirus-associated encephalitis in the California encephalitis project, 1998-2005. J Infect Dis. 2008;198:1685–91. doi: 10.1086/592988. [DOI] [PubMed] [Google Scholar]
- [8].Rotbart H. Meningitis and Encephalitis. In: Rotbart H, editor. Human Enterovirus Infections. American Society for Microbiology; Washington, DC: 1995. pp. 271–89. [Google Scholar]
- [9].Katz S, Gershon AA, Hotez PJ. Krugman’s infectious diseases of children. In: DeYoung L, editor. Enteroviruses. St Louis: Mosby-Year Book; 1998. pp. 81–97. [Google Scholar]
- [10].Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. Clinical features, diagnosis, and management of enterovirus 71. Lancet neurology. 2010;9:1097–105. doi: 10.1016/S1474-4422(10)70209-X. [DOI] [PubMed] [Google Scholar]
- [11].Andreoletti L, Leveque N, Boulagnon C, Brasselet C, Fornes P. Viral causes of human myocarditis. Arch Cardiovasc Dis. 2009;102:559–68. doi: 10.1016/j.acvd.2009.04.010. [DOI] [PubMed] [Google Scholar]
- [12].Tam PE. Coxsackievirus myocarditis: interplay between virus and host in the pathogenesis of heart disease. Viral Immunol. 2006;19:133–46. doi: 10.1089/vim.2006.19.133. [DOI] [PubMed] [Google Scholar]
- [13].Martino T, Liu P, Petric M, Sole M. Enteroviral myocarditis and dilated cardiomyopathy: A review of clinical and experimental studies. In: Rotbart H, editor. Human Enterovirus Infections. American Society for Microbiology; Washington, DC: 1995. pp. 291–351. [Google Scholar]
- [14].Jaidane H, Sauter P, Sane F, Goffard A, Gharbi J, Hober D. Enteroviruses and type 1 diabetes: towards a better understanding of the relationship. Rev Med Virol. 2010;20:265–80. doi: 10.1002/rmv.647. [DOI] [PubMed] [Google Scholar]
- [15].Rewers M, Atkinson M. The Possible Role of Enteroviruses in Diabetes Mellitus. In: Rotbart H, editor. Human Enterovirus Infections. American Society for Microbiology; Washington, DC: 1995. pp. 353–85. [Google Scholar]
- [16].Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35. doi: 10.1136/bmj.d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Monto AS, Ullman BM. Acute respiratory illness in an American community. The Tecumseh study. JAMA. 1974;227:164–9. [PubMed] [Google Scholar]
- [18].Fendrick AM, Monto AS, Nightengale B, Sarnes M. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Archives of internal medicine. 2003;163:487–94. doi: 10.1001/archinte.163.4.487. [DOI] [PubMed] [Google Scholar]
- [19].Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- [20].Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Current opinion in immunology. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- [21].Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- [22].Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell research. 2006;16:141–7. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- [23].de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. Journal of leukocyte biology. 2001;69:912–20. [PubMed] [Google Scholar]
- [24].Lopez CB, Hermesh T. Systemic responses during local viral infections: type I IFNs sound the alarm. Current opinion in immunology. 2011;23:495–9. doi: 10.1016/j.coi.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Taylor RT, Lubick KJ, Robertson SJ, Broughton JP, Bloom ME, Bresnahan WA, et al. TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell host & microbe. 2011;10:185–96. doi: 10.1016/j.chom.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- [27].Balachandran S, Beg AA. Defining emerging roles for NF-kappaB in antivirus responses: revisiting the interferon-beta enhanceosome paradigm. PLoS pathogens. 2011;7:e1002165. doi: 10.1371/journal.ppat.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li M, Shillinglaw W, Henzel WJ, Beg AA. The Rela(p65) subunit of NF-kappaB is essential for inhibiting double-stranded RNA-induced cytotoxicity. The Journal of biological chemistry. 2001;276:1185–94. doi: 10.1074/jbc.M006647200. [DOI] [PubMed] [Google Scholar]
- [29].Hoyos B, Ballard DW, Bohnlein E, Siekevitz M, Greene WC. Kappa B-specific DNA binding proteins: role in the regulation of human interleukin-2 gene expression. Science. 1989;244:457–60. doi: 10.1126/science.2497518. [DOI] [PubMed] [Google Scholar]
- [30].Thanos D, Maniatis T. The high mobility group protein HMG I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell. 1992;71:777–89. doi: 10.1016/0092-8674(92)90554-p. [DOI] [PubMed] [Google Scholar]
- [31].Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- [32].Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- [33].Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- [34].Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- [36].Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- [37].Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98:9237–42. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- [39].Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- [40].Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–7. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- [41].Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. The Journal of experimental medicine. 2008;205:1601–10. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, et al. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–9. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A. 2011;108:21010–5. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. The EMBO journal. 2012;31:1714–26. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Triantafilou K, Vakakis E, Kar S, Richer E, Evans GL, Triantafilou M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. Journal of cell science. 2012;125:4761–9. doi: 10.1242/jcs.103887. [DOI] [PubMed] [Google Scholar]
- [46].Berke IC, Yu X, Modis Y, Egelman EH. MDA5 assembles into a polar helical filament on dsRNA. Proc Natl Acad Sci U S A. 2012;109:18437–41. doi: 10.1073/pnas.1212186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–89. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- [48].Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–8. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- [49].Komuro A, Horvath CM. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006;80:12332–42. doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104:582–7. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. 2010;107:1512–7. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liniger M, Summerfield A, Zimmer G, McCullough KC, Ruggli N. Chicken cells sense influenza A virus infection through MDA5 and CARDIF-signaling involving LGP2. J Virol. 2012;86:705–17. doi: 10.1128/JVI.00742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–37. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Inohara Chamaillard, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- [55].Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- [56].Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–34. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- [57].Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- [58].Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–6. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- [59].Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. The Journal of biological chemistry. 2006;281:36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- [60].Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. The Journal of biological chemistry. 2003;278:8869–72. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- [61].Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. The Journal of biological chemistry. 2003;278:5509–12. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- [62].Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–80. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–7. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- [64].Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 2011;34:854–65. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, Tschopp J. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell death and differentiation. 2011;18:1387. doi: 10.1038/cdd.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate immunity. 2012 doi: 10.1177/1753425912467383. [DOI] [PubMed] [Google Scholar]
- [67].Wessely R, Klingel K, Knowlton KU, Kandolf R. Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation. 2001;103:756–61. doi: 10.1161/01.cir.103.5.756. [DOI] [PubMed] [Google Scholar]
- [68].Caine EA, Partidos CD, Santangelo JD, Osorio JE. Adaptation of enterovirus 71 to adult interferon deficient mice. PloS one. 2013;8:e59501. doi: 10.1371/journal.pone.0059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, et al. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol. 2005;79:4460–9. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation. 2004;110:3540–3. doi: 10.1161/01.CIR.0000136824.73458.20. [DOI] [PubMed] [Google Scholar]
- [71].Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, et al. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107:2793–8. doi: 10.1161/01.CIR.0000072766.67150.51. [DOI] [PubMed] [Google Scholar]
- [72].Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PloS one. 2009;4:e4127. doi: 10.1371/journal.pone.0004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Oshiumi H, Okamoto M, Fujii K, Kawanishi T, Matsumoto M, Koike S, et al. The TLR3/TICAM-1 Pathway Is Mandatory for Innate Immune Responses to Poliovirus Infection. J Immunol. 2011;187:5320–7. doi: 10.4049/jimmunol.1101503. [DOI] [PubMed] [Google Scholar]
- [74].Abe Y, Fujii K, Nagata N, Takeuchi O, Akira S, Oshiumi H, et al. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J Virol. 2012;86:185–94. doi: 10.1128/JVI.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci U S A. 2008;105:20446–51. doi: 10.1073/pnas.0810372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, et al. TLR3 deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirus B3 infection: role for IL-4. American journal of physiology Regulatory, integrative and comparative physiology. 2013;304:R267–77. doi: 10.1152/ajpregu.00516.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Abston ED, Coronado MJ, Bucek A, Bedja D, Shin J, Kim JB, et al. Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clinical & developmental immunology. 2012;2012:129486. doi: 10.1155/2012/129486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S, et al. TRIF is a critical survival factor in viral cardiomyopathy. J Immunol. 2011;186:2561–70. doi: 10.4049/jimmunol.1002029. [DOI] [PubMed] [Google Scholar]
- [79].Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. The Journal of biological chemistry. 2010;285:23208–23. doi: 10.1074/jbc.M109.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Triantafilou K, Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J Virol. 2004;78:11313–20. doi: 10.1128/JVI.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Satoh M, Nakamura M, Akatsu T, Shimoda Y, Segawa I, Hiramori K. Toll-like receptor 4 is expressed with enteroviral replication in myocardium from patients with dilated cardiomyopathy. Lab Invest. 2004;84:173–81. doi: 10.1038/labinvest.3700031. [DOI] [PubMed] [Google Scholar]
- [82].Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM, et al. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol. 2005;7:1117–26. doi: 10.1111/j.1462-5822.2005.00537.x. [DOI] [PubMed] [Google Scholar]
- [83].Coyne CB, Bozym R, Morosky SA, Hanna SL, Mukherjee A, Tudor M, et al. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell host & microbe. 2011;9:70–82. doi: 10.1016/j.chom.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang JP, Asher DR, Chan M, Kurt-Jones EA, Finberg RW. Cutting Edge: Antibody-mediated TLR7-dependent recognition of viral RNA. J Immunol. 2007;178:3363–7. doi: 10.4049/jimmunol.178.6.3363. [DOI] [PubMed] [Google Scholar]
- [85].Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–83. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U S A. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Flanegan JB, Petterson RF, Ambros V, Hewlett NJ, Baltimore D. Covalent linkage of a protein to a defined nucleotide sequence at the 5−-terminus of virion and replicative intermediate RNAs of poliovirus. Proc Natl Acad Sci U S A. 1977;74:961–5. doi: 10.1073/pnas.74.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- [89].Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–64. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. The Journal of experimental medicine. 2006;203:1795–803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chakrabarti A, Jha BK, Silverman RH. New insights into the role of RNase L in innate immunity. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2011;31:49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Malathi K, Dong B, Gale M, Jr., Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–9. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–55. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- [94].Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell reports. 2012;2:1187–96. doi: 10.1016/j.celrep.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol. 2010;84:254–60. doi: 10.1128/JVI.00631-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Huhn MH, McCartney SA, Lind K, Svedin E, Colonna M, Flodstrom-Tullberg M. Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after Coxsackievirus infection. Virology. 2010;401:42–8. doi: 10.1016/j.virol.2010.02.010. [DOI] [PubMed] [Google Scholar]
- [97].Kuo RL, Kao LT, Lin SJ, Wang RY, Shih SR. MDA5 Plays a Crucial Role in Enterovirus 71 RNA-Mediated IRF3 Activation. PloS one. 2013;8:e63431. doi: 10.1371/journal.pone.0063431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Aida K, Nishida Y, Tanaka S, Maruyama T, Shimada A, Awata T, et al. RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes. 2011;60:884–9. doi: 10.2337/db10-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol. 2011;85:4167–72. doi: 10.1128/JVI.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ito M, Yanagi Y, Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS pathogens. 2012;8:e1002857. doi: 10.1371/journal.ppat.1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Blom N, Hansen J, Blaas D, Brunak S. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein science : a publication of the Protein Society. 1996;5:2203–16. doi: 10.1002/pro.5560051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Krausslich HG, Nicklin MJ, Toyoda H, Etchison D, Wimmer E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J Virol. 1987;61:2711–8. doi: 10.1128/jvi.61.9.2711-2718.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Clark ME, Hammerle T, Wimmer E, Dasgupta A. Poliovirus proteinase 3C converts an active form of transcription factor IIIC to an inactive form: a mechanism for inhibition of host cell polymerase III transcription by poliovirus. The EMBO journal. 1991;10:2941–7. doi: 10.1002/j.1460-2075.1991.tb07844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kundu P, Raychaudhuri S, Tsai W, Dasgupta A. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease-mediated cleavage of the TATA-binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J Virol. 2005;79:9702–13. doi: 10.1128/JVI.79.15.9702-9713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS pathogens. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol. 2011;85:8811–8. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, et al. MDA-5 is cleaved in poliovirus-infected cells. J Virol. 2007;81:3677–84. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rebsamen M, Meylan E, Curran J, Tschopp J. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell death and differentiation. 2008;15:1804–11. doi: 10.1038/cdd.2008.119. [DOI] [PubMed] [Google Scholar]
- [109].Bozym RA, Delorme-Axford E, Harris K, Morosky S, Ikizler M, Dermody TS, et al. Focal adhesion kinase is a component of antiviral RIG-I-like receptor signaling. Cell host & microbe. 2012;11:153–66. doi: 10.1016/j.chom.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, et al. Enterovirus 71 Protease 2A(pro) Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS pathogens. 2013;9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, et al. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol. 2010;84:8051–61. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol. 2013;87:1690–8. doi: 10.1128/JVI.01855-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG-I is cleaved during picornavirus infection. Virology. 2009;391:171–6. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, et al. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology. 2009;393:311–8. doi: 10.1016/j.virol.2009.08.009. [DOI] [PubMed] [Google Scholar]