

Abstract
Sustained β-adrenergic receptor stimulation is associated with cardiomyopathy, an affect thought to result from cAMP-associated cardiac injury. Using a murine line with adenylyl cyclase 6 gene deletion (AC6KO), we tested the hypothesis that AC6 deletion, by limiting cAMP production, would attenuate cardiomyopathy in the setting of sustained β-adrenergic receptor stimulation. During 7d isoproterenol infusion, there was unexpected higher mortality in AC6KO mice compared to wild type control mice (p<0.0001). However, left ventricular function was similarly impaired in isoproterenol-infused control and AC6KO mice. There were no group differences in left ventricular hypertrophy, apoptosis, and fibrosis. Telemetric electrocardiography showed progressive prolongation of PR interval (p<0.0001), QRS duration (p<0.0005), and QTc (p<0.0001), as well as reduction in heart rate (p<0.0001), in AC6KO mice during isoproterenol infusion. These defective electrophysiological properties in isoproterenol-infused AC6KO mice were associated with decreased longitudinal ventricular conduction velocity (p<0.05) and reduced phosphorylation of connexin 43 at S368 in left ventricular samples (p=0.006). Taken together, these data demonstrate that limiting cAMP production does not prevent sustained β-adrenergic receptor stimulation-induced cardiomyopathy. Moreover, AC6 deletion impairs electrophysiological properties and increase mortality during sustained β-adrenergic receptor stimulation. Decreased connexin 43 phosphorylation and impaired ventricular conduction may be of mechanistic importance for the defective electrophysiological properties.
Keywords: LV function, ventricular conduction velocity, connexin 43, cAMP
1. INTRODUCTION
Heart failure is an inexorable disease associated with high morbidity and mortality, despite advanced pharmaceutical treatments and surgical interventions [1]. A hallmark of heart failure is increased circulating catecholamines and abnormal β-adrenergic receptor (βAR) signaling [2]. It is of considerable interest and importance to study the roles of each element of βAR signaling pathway in the development of cardiomyopathy and heart failure [3]. Indeed, chronic β-adrenergic receptor stimulation is associated with cardiomyopathy [4], which is generally thought to result from cAMP-associated cardiac injury. Cardiac-directed expression of β1AR [5, 6], β2ARs [7], and Gαs [8] all result in cardiac myocyte hypertrophy, cardiac fibrosis, and eventual cardiomyopathy. Expression of adenylyl cyclase 6 (AC6) does not have these deleterious effects shared with other members of the βAR-Gαs-AC signaling pathway [9, 10]. These studies suggest the theory that increasing cAMP is uniformly toxic to the heart may be overly simplistic; however, direct evidence is still lacking.
We have previously generated a murine line with AC6 deletion [11]. Left ventricular (LV) membranes from mice with AC6 deletion have showed a 60% reduction in cAMP generating capacity. We reasoned that if cAMP is central to the pathogenesis of cardiomyopathy and heart failure, mice with AC6 deletion might be less susceptible to cardiac dysfunction in the presence of elevated circulating catecholamines. We tested this hypothesis in AC6-deleted (AC6KO) mice in the setting of sustained βAR stimulation [4, 12].
2. METHODS
2.1. Animals
Four-month-old AC6KO mice in congenic C57BL/6N background were used [11]. Age and sex-matched wild type C57BL/6N mice served as controls (CON). Mouse genotyping was performed by PCR, and absence of AC6 protein expression was confirmed by Western blotting as previously described [11]. The Institutional Animal Care and Use Committee at the VA San Diego Healthcare System, in accordance with NIH and AAALAC guidelines, approved this study.
2.2. Iso Infusion
Osmotic minipumps (Alzet Model; DuRECT Corp., Cupertino, CA) filled with Iso (dissolved in saline supplemented with 0.1% ascorbic acid as anti-oxidant) were implanted subcutaneously in mice under light anesthesia with inhalation of isoflurane (5% for 1 min induction; 1% maintenance). Infusion rate of Iso was controlled at 60 mg/d/kg body weight as previously described [13, 14]. Infusion of vehicle (saline supplemented with 0.1% ascorbic acid as anti-oxidant) alone had no effects on mortality. Physical activity was monitored, and mortality was recorded daily for 4d. Telemetric electrocardiography was used to monitor electrophysiological abnormalities for 7d in another cohort of mice. To further determine the underlying mechanisms for the effects by AC6 deletion, other experiments were conducted on mice or on LV samples from mice received 4d continuous Iso infusion. For echocardiography study, the osmotic pumps were removed 24 h before experiment to avoid the confounding effects of Iso on measurements of LV dimensions and function. All physiological studies were performed without knowledge of group identity.
2.2. Electrocardiography
PhysioTel® ETA-F20 transmitters (Data Sciences International, St. Paul, MN) were implanted subcutaneously, and continuous electrocardiographic recording was initiated 7d after transmitter implantation. Two days later, osmotic pumps containing Iso were implanted.
2.4. Echocardiography
Anesthesia was induced with 5% isoflurane (at a flow rate of 1 L/min oxygen) and maintained with 1% isoflurane in oxygen. With mice in supine position, echocardiographic images were obtained using a 16L MHz linear probe and Sonos 5500® Echocardiograph system (Philips Medical Systems, Bothell, MA), as previously reported [11].
2.5. Optical Mapping
Hearts isolated from CON and AC6KO infusion with Iso for 4d were perfused with oxygenated modified Krebs-Henseleit solution (24.9 mM NaHCO3, 1.2 mM KH2PO4, 11.1 mM dextrose, 1.2 mM MgSO4, 4.7 mM KCl, 118.0 mM NaCl, and 2.5 mM CaCl2) at a constant pressure of 70 mmHg. Optical mapping was performed as previously described [15]. Briefly, a bolus of voltage sensitive fluorescent dye (Di-4-ANEPPS: 8 ml, 3.25 μM; Invitrogen, Carlsbad, CA) was injected into the perfusion line. The ventricular epicardium was illuminated by LED lamps (470 nm), and the fluorescence signal filtered by a long-pass >610 nm filter and recorded with a high speed CMOS camera (MiCAM Ultima L; Brainvision, Japan). To measure ventricular conduction velocity, the LV epicardium was imaged while the LV midwall was paced with a platinum unipolar electrode at a cycle length of 200 ms using a DS8000 digital stimulator (World Precision Instruments, Sarasota, FL). The ventricular epicardium from the anterior and posterior views were simultaneously imaged to measure global epicardial activation time during atrial pacing at a cycle length of 200 ms. Blebbistatin (10 μM; Sigma, St. Louis, MO) was included to the perfusate during imaging to attenuate motion artifact. Optical images were imported into Matlab (Mathworks, Natick, MA) and analyzed. Activation time at each pixel was identified at the maximum signal derivative during the action potential upstroke and conduction velocity was calculated from the spatial gradient of activation time.
2.6. Necropsy
After 4d continuous Iso infusion, the hearts were arrested in diastole and excised. Body weight and LV (with interventricular septum) weight were recorded. A short axis midwall LV ring was fixed in formalin and embedded in paraffin. The rest of LV samples were quickly frozen in liquid nitrogen and stored at −80°C. Liver weight and lung weight were also recorded.
2.7. LV Apoptosis
De-waxed LV sections (5 μm) were re-hydrated and used for terminal 2′-deoxyuridine 5′-triphosphate nick end-labeling (TUNEL) assay as previously described [16].
2.8. LV Fibrosis
LV sections (5 μm) were de-waxed, re-hydrated, and stained with picrosirius red for 1 h. Fractional collagen area was quantified using NIH image software Image J [17].
2.9. Quantitative RT-PCR
Total RNA was extracted from mouse LV samples using RNA STAT-60 (Tel-Test Inc., Friendswood, TX), purified with RNeasy Mini kit (Qiagen, Valencia, CA), and treated with RNase-free DNase (Qiagen) to eliminate genomic DNA contamination. Quantitative reverse-transcriptase PCR (RT-PCR) was conducted using primer pairs as previously described [17], under the following conditions: 5 min at 98°C, 40 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C. RNA equivalents were normalized to simultaneously determined glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels in each sample.
2.10. Western Blotting
LV samples were homogenized at 4°C in homogenization buffer (25 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 0.5 mM EGTA, 50 mM β-glycerophosphate, 10 mM NaF, 1 mM Na3VO4) in the presence of protease inhibitor cocktail (Roche, Indianapolis, IN). LV homogenates were subjected to SDS-PAGE and electrotransferred to polyvinylidene difluoride membranes. Membranes were incubated with the antibodies to connexin 43 (Cx43; Cat # C6219, Sigma, St. Louis, Missouri) or phospho-Cx43 (S368) (Cat # 3551, Cell Signaling Technology, Danvers, MA). Binding of the primary antibody was detected by an enhanced chemiluminescence method (ECL+, Amersham Biosciences, Piscataway, NJ) using horseradish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Equal loading of samples and even transfer efficiency were monitored by reprobing stripped blots with the antibody to GAPDH. Quantification of protein expression was performed using Gel-Pro® Analyzer (Media Cybernetics, Silver Spring, MD) [17].
2.11. Immunohistochemistry
LV sections (5 μm) were de-waxed and rehydrated. Antigen retrieval was carried out by boiling LV sections in 1% ZnSO4 for 10 min. LV sections were incubated overnight with phospho-Cx43 (S368) antibody (Cell Signaling Technology) in blocking buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20, 3% bovine serum albumin, 5% normal rabbit serum). Following extensive washing with TBST (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20), LV sections were incubated for 3 h with horseradish peroxidase-conjugated goat anti-rabbit IgG (Dako North America, Carpinteria, CA). LV sections were then incubated with Stable DAB (Invitrogen, Carlsbad, CA), counterstained with Meyer’s hematoxylin, dehydrated through a graded ethanol series, and mounted with Permont. Stained sections were examined using a Nikon E600 microscope, and images were acquired with a CoolSnap-Pro digital camera and Image Pro Plus software (Media Cybernetics, Silver Spring, MD).
2.12. Cyclic AMP Generation
cAMP production in LV homogenates was measured as described previously [18].
2.13. PKA and PKC Activities
LV homogenates was used to measure PKA and PKC activities as previously described [19].
2.14. Statistical Analysis
Results are presented as mean±SEM. Survival curves underwent Kaplan-Meier analysis and the log-rank test. Group differences for other data were analyzed using Student’s t-test (unpaired, two-tailed) or 2-way ANOVA. The null hypothesis was rejected when p<0.05.
3. RESULTS
3.1. Mortality
During 7d continuous Iso infusion, AC6KO mice had accrued a higher mortality compared to age and sex-matched wild type control mice (AC6KO: 64% mortality; CON: 7% mortality; p<0.0001) (Figure 1).
Figure 1. Kaplan-Meier analysis shows increased mortality in AC6KO mice, compared to CON mice, during 7d isoproterenol (Iso) infusion.
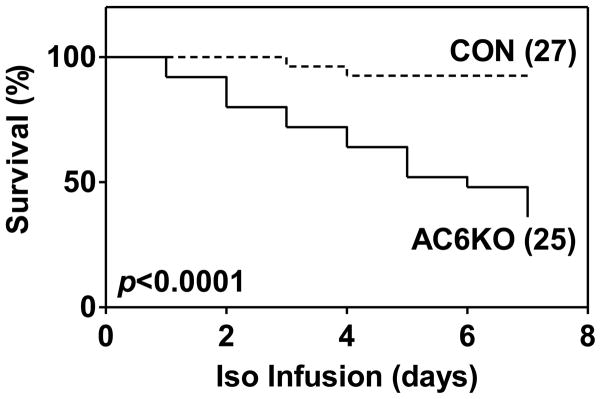
Numbers in parentheses indicate animal numbers. Probability value shown is from log rank test.
3.2. LV Function
To explore whether AC6 deletion increases mortality by adversely affect LV remodeling and function in Iso-infused hearts, we measured LV dimensions and function by echocardiography before and after 4d Iso infusion. In CON mice, Iso infusion reduced LV ejection fraction and increased LV systolic and diastolic diameters (Table 1), consistent with a deleterious effect of sustained βAR stimulation on LV function demonstrated previously. There were similar increase in LV diameters and reduction in LV function between CON and AC6KO after 4d Iso infusion (Table 1) and 14d Iso infusion (Supplementary Data, Table), indicating that other mechanisms, rather than LV remodeling and dysfunction, contributes to the increased mortality in AC6 mice.
Table 1.
Echocardiography
| Before Iso
|
4d Iso
|
p
|
|||||
|---|---|---|---|---|---|---|---|
| CON (8) | AC6KO (8) | CON (8) | AC6KO (8) | Interaction | AC6 effect | Iso effect | |
| HR (bpm) | 552±19 | 521±33 | 484±14 | 444±17 | ns | ns | <0.002 |
| EDD (mm) | 3.8±0.1 | 3.8±0.1 | 4.1±0.2 | 4.2±0.2 | ns | ns | <0.04 |
| ESD (mm) | 2.7±0.1 | 2.4±0.1 | 3.2±0.2 | 3.2±0.3 | ns | ns | <0.002 |
| PWTh (mm) | 0.7±0.02 | 0.7±0.02 | 0.6±0.02 | 0.6±0.02 | ns | ns | <0.001 |
| IVSTh (mm) | 0.7±0.02 | 0.7±0.01 | 0.7±0.02 | 0.7±0.02 | ns | ns | ns |
| LVEF (%) | 62±2 | 66±2 | 43±3 | 48±4 | ns | ns | <0.0001 |
| Vcfc (cir/s) | 23±1 | 25±1 | 14±1 | 15±2 | ns | ns | <0.0001 |
HR, heart rate; EDD, end-diastolic diameter; ESD, end-systolic diameter; PWTh, posterior wall thickness; IVSTh, interventricular septum thickness; LVEF, left ventricular ejection fraction; Vcfc, heart rate-corrected velocity of circumferential fiber shortening; ns, not significant. Data are mean±SEM. Probability values are from 2-way ANOVA for interaction, AC6 effect (absence of AC6), and Iso (isoproterenol) effect.
3.3. Electrocardiography
Using telemetric electrocardiography, we compared heart rate, PR interval, QRS duration, and QT interval corrected for heart rate (QTc) between CON and AC6KO mice during chronic Iso infusion (Figure 2). There were no group differences in these indices at baseline (before Iso infusion) (Figure 2C–E). However, after 4d Iso infusion, group differences emerged. For example, AC6 deletion was associated with progressive prolongation of the PR interval (Figure 2C), QRS duration (Figure 2D), and QTc (Figure 2E). Heart rate was also progressively decreased in AC6KO mice (Figure 2F).
Figure 2. AC6 deletion was associated with electrocardiographic abnormalities during chronic Iso infusion.

(A) Representative electrocardiograms showing abnormalities in AC6KO mouse hearts during Iso infusion. (B) Representative electrocardiograms showing prolongation of PR interval, QTc interval, and QRS duration in surviving AC6KO mice after 7d Iso infusion. Quantitative analysis shows prolongation of the PR interval (C), QTc interval (D), and QRS duration (E) in CON and AC6KO mice during Iso infusion. Heart rate, determined by telemetric electrocardiography, was also decreased (F) in AC6KO mice after Iso infusion. Numbers in parentheses indicate animal numbers, and error bars denote 1 SEM. Probability values shown are for AC6 deletion effect from 2-way ANOVA.
3.4. LV cAMP Generation and PKA Activity
After 4d Iso infusion, LV samples from both groups showed reductions in basal, Iso/GTPγS-stimulated, and NKH477-stimulated cAMP production, and also in PKA activity (Table 2). The relative reductions in PKA activity were similar in both groups (33–34%), but the absolute reduction was greater in AC6KO mice, due to their pre-Iso impairment in PKA activity.
Table 2.
LV cAMP Production, PKA Activity, and PKC Activity
| Before Iso
|
4d Iso
|
p
|
|||||
|---|---|---|---|---|---|---|---|
| CON (8) | AC6KO (8) | CON (8) | AC6KO (8) | Interaction | AC6 effect | Iso effect | |
| cAMP Production (fmol/min/μg) | |||||||
| • Basal | 37±3 | 30±5 | 35±3 | 25±4 | ns | <0.04 | ns |
| • Iso/GTPγS | 77±6 | 33±3 | 50±2 | 26±3 | <0.0001 | <0.0001 | <0.004 |
| • NKH477 | 299±12 | 62±3 | 278±34 | 59±7 | ns | <0.0001 | ns |
|
| |||||||
| PKA Activity (pmol/mg/min) | 971±54 | 513±9 | 652±9 | 341±18 | <0.02 | <0.0001 | <0.0001 |
|
| |||||||
| PKC Activity (pmol/mg/min) | |||||||
| • Cytosolic fraction* | 68±5 | 71±9 | 69±3 | 66±3 | ns | ns | ns |
| • Membrane fraction* | 74±8 | 76±10 | 70±7 | 79±6 | ns | ns | ns |
Values are mean±SEM. Probability values are from 2-way ANOVA for interaction, AC6 effect (absence of AC6), and Iso (isoproterenol) effect. NKH477, a water soluble forskolin analog; ns, not significant.
n=6.
3.5. Necropsy
Iso infusion for 4d increased LV weight in both CON and AC6KO mice (Table 3). However, no group differences in LV weight and LV/TL (tibial length) ratio were observed between CON and AC6KO mice before and after 4d Iso infusion (Table 3). In addition, we observed no group difference in liver weight and lung weight between CON and AC6KO mice before and after 4d Iso infusion.
Table 3.
Necropsy
| Before Iso
|
4d Iso
|
p
|
|||||
|---|---|---|---|---|---|---|---|
| CON (8) | AC6KO (8) | CON (8) | AC6KO (8) | Interaction | AC6 effect | Iso effect | |
| LV (mg) | 92.5±4.8 | 91.0±3.9 | 105.1±6.5 | 102.1±7.2 | ns | ns | <0.05 |
| BW (g) | 25.6±0.7 | 26.7±1.0 | 29.7±1.8 | 28.5±1.5 | ns | ns | <0.04 |
| LV/BW (mg/g) | 3.7±0.1 | 3.6±0.2 | 3.9±0.3 | 3.7±0.3 | ns | ns | ns |
| TL (mm) | 17.5±0.2 | 17.6±0.3 | 17.4±0.2 | 17.6±0.3 | ns | ns | ns |
| LV/TL (mg/mm) | 5.3±0.2 | 5.1±0.2 | 6.6±0.4 | 5.8±0.4 | ns | ns | <0.004 |
| Liver (g) | 1.2±0.1 | 1.2±0.1 | 1.3±0.1 | 1.2±0.2 | ns | ns | ns |
| Lung (mg) | 182.0±5.6 | 188.4±13.4 | 177.0±12.6 | 181.5±10.2 | ns | ns | ns |
LV, left ventricle; BW, body weight; LV/BW, left ventricle/body weight ratio; TL, tibial length; LV/TL, left ventricle weight/tibial length ratio; ns, not significant. Data are mean±SEM. Probability values are from 2-way ANOVA for interaction, AC6 effect (absence of AC6), and Iso (isoproterenol) effect.
3.6. LV Apoptosis
There were very few apoptotic cardiac myocytes (<0.002%) in LV samples from CON and AC6KO mice after 4d Iso infusion, as assessed by TUNEL assay, and the group difference was statistically insignificant. There was no group difference in Bcl2 protein content in LV samples after 4d Iso infusion [CON: 131±22 densitometric units (du), n=7; AC6KO: 101±22 du, n=6; p=0.37]. Similarly, there were no group differences in caspase 3/7 activity (CON: 335±20 U/mg, n=7; AC6KO: 294±12 U/mg, n=6; p=0.12).
3.7. LV Fetal Gene Expression
No group differences were detected in activation of fetal genes associated with LV hypertrophy (Table 4). Expression of BNP (brain natriuretic peptide) and FHL1 (four and a half LIM domain 1) also showed no group differences (Table 4).
Table 4.
Fetal Gene and Extracellular Matrix Protein Expression (RT-PCR)
| Gene - Fold Control - | Before Iso
|
4d Iso
|
p
|
||||
|---|---|---|---|---|---|---|---|
| CON (6) | AC6KO (6) | CON (6) | AC6KO (6) | Interaction | AC6 effect | Iso effect | |
| ANF | 1.00±0.14 | 0.92±0.12 | 2.10±0.22 | 1.89±0.50 | ns | ns | <0.002 |
| α-SK actin | 1.00±0.10 | 1.06±0.19 | 1.32±0.23 | 1.56±0.65 | ns | ns | ns |
| β-MHC | 1.00±0.12 | 0.57±0.22 | 0.57±0.06 | 0.64±0.14 | ns | ns | ns |
| BNP | 1.00±0.07 | 1.34±0.20 | 4.25±0.45 | 2.83±0.78 | ns | ns | <0.0001 |
| FHL1 | 1.00±0.05 | 0.85±0.09 | 2.34±0.12 | 2.48±0.25 | ns | ns | <0.0001 |
| Col Iα1 | 1.00±0.07 | 0.79±0.09 | 5.15±0.93 | 2.90±1.32 | ns | ns | <0.001 |
| Col IIIα1 | 1.00±0.05 | 0.85±0.18 | 7.16±1.26 | 4.42±2.70 | ns | ns | <0.004 |
| Periostin | 1.00±0.08 | 0.83±0.09 | 14.71±3.11 | 9.80±6.57 | ns | ns | <0.005 |
ANF, atrial natriuretic factor; α-SK actin, α-smooth muscle actin; β-MHC, β-myosin heavy chain; BNP, brain natriuretic peptide; FHL1, four and a half LIM domain 1; col Iα1, collagen Iα1; col IIIα1, collagen IIIα1, ns, not significant. Data are mean±SEM. Probability values are from 2-way ANOVA for interaction, AC6 effect (absence of AC6), and Iso (isoproterenol) effect.
3.8. LV Fibrosis
There was no group difference in LV fractional collagen area, assessed by picrosirius red staining, after 4d Iso infusion (CON: 2.5±0.3%, AC6KO: 2.6±0.4%, p=0.92, n=8). The LV mRNA contents of the extracellular matrix proteins collagen Iα1, collagen IIIα1, and periostin also showed no group differences (Table 4).
3.9. Optical Mapping
Hearts from AC6KO mice infused with Iso for 4d showed decreased LV epicardial longitudinal conduction velocity during ventricular pacing (200 ms cycle length) (Fig. 3A, B). There was no group difference in transverse ventricular conduction velocity between CON and AC6KO mouse hearts after 4d Iso infusion (Fig. 3A, C). Group differences were not seen in ventricular epicardial activation time (95-5%): (CON: 4.4±1.5 ms, n=5; AC6KO: 2.8±0.1 ms, n=5; p=0.2) during atrial pacing (200 ms cycle length).
Figure 3. AC6 deletion was associated with decreased ventricular conduction after 4d Iso infusion.

(A) Representative activation maps of left ventricular epicardium of Iso-infused CON and AC6KO mice during midwall epicardial pacing. Optical mapping revealed slower conduction in the direction of maximum conduction velocity (arrow) in AC6KO hearts compared to control hearts. Isochrones are at 1 ms intervals. (B) AC6 deletion was associated with decreased longitudinal conduction velocity (CVmax) after 4d Iso infusion. (C) AC6 deletion was not associated with decreased transverse conduction velocity (CVmin) after 4d Iso infusion (p=0.40). Probability values are from Student’s t-test (unpaired, 2-tailed). Error bars denote 1 SEM; numbers in bars indicate group size.
3.10. Cx43 Phosphorylation
LV samples from AC6KO mice infused with Iso for 4d showed decreased phosphorylation of Cx43 at S368 (Figure 4A, B). Total LV Cx43 protein and mRNA contents showed no group differences (Figure 4A, C). Immunohistochemistry also showed decreased protein content of phospho-Cx43 (S368) between cardiac myocytes in LV samples from AC6KO mice after 4d Iso infusion (Figure 4D). Atrial Cx45 expression was reduced by Iso infusion similarly in both groups (before Iso: CON: 1.00±0.08 fold, AC6KO: 1.07±0.09 fold; after 4d Iso: CON: 0.75±0.04 fold, AC6KO: 0.66±0.03 fold; n=6), but atrial Cx30.2 expression was unchanged in atrial samples from CON and AC6KO mice (before Iso: CON: 1.00±0.05 fold, AC6KO: 1.13±0.15 fold; after 4d Iso: CON: 0.97±0.06 fold, AC6KO: 1.08±0.04 fold; n=6).
Figure 4. AC6 deletion was associated with decreased Cx43 phosphorylation after 4d Iso infusion.

(A) Representative Western blots showing reduced phospho-Cx43(S368) content and normal total Cx43 content in LV samples from CON and AC6KO mice before and after 4d Iso infusion. (B) Quantification of data from Western blotting showing decreased phospho-Cx43(S368) content in LV samples from Iso-infused AC6KO mice (Iso effect: p<0.0001; AC6 effect: p<0.02). (C) There was similar total Cx43 protein content in LV samples from CON and AC6KO mice before and after 4d Iso infusion (Iso effect: p=0.96, AC6 effect: p=0.53). (D) Immunohistochemical staining showing decreased phospho-Cx43(S368) staining in LV samples from AC6KO mice. Probability values are from 2-way ANOVA. Error bars denote 1 SEM; group size: 8. du, densitometric unit.
To determine whether differences in kinase activities were associated with differences in Cx43 phosphorylation, we assessed PKC and PKA activities in LV samples from CON and AC6KO mice before and after 4d continuous Iso infusion. No group differences were found in cytosolic or membrane LV PKC activity either before or after 4d continuous Iso infusion (Table 2). In contrast, AC6 deletion was associated with a 47% reduction in LV PKA activity before Iso infusion (Table 2). Following 4d of Iso infusion, both groups showed similar relative reductions in LV PKA activity, but the absolute PKA activity was 48% lower in AC6KO mouse hearts (Table 2).
4. DISCUSSION
The most important findings of this study were: (1) limiting cAMP production does not prevent sustained β-adrenergic receptor stimulation-induced cardiomyopathy; (2) AC6 deletion, in the setting of sustained βAR stimulation, is associated with marked electrocardiographic abnormalities, including prolongation of PR interval, QRS duration, and QTc interval. These electrocardiographic abnormalities are associated with dire physiological consequences including impaired ventricular conduction velocity and high mortality. It appears that the difference in mortality was due to something other than differences in LV dysfunction.
Previous studies indicate that cardiac myocyte-specific expression of AC6 is associated with beneficial electrophysiological effects [9, 20, 21], including facilitation of AV conduction [20], normalization of prolonged action potential duration [21], and reduced mortality in cardiomyopathy [9]. However, since AC6 expression was also associated with increased LV function, it was not clear whether the beneficial effects by AC6 expression were a consequence of increased contractile function or an electrophysiological effect per se. Data from the current study suggest that normal AC6 expression may be required to maintain normal AV conduction and ventricular conduction velocity during stress. That AC6 deletion was also associated with QTc prolongation indicates that AC6 may be required for normal repolarization, although such speculations would require patch clamp studies to confirm.
It is important to note, however, that none of the electrocardiographic abnormalities was present in the AC6KO mouse hearts prior to sustained Iso stimulation. These abnormalities began at 48 h after initiation of continuous Iso infusion, and progressively worsened for the duration of the infusion. These was a 64% mortality rate during 7d Iso infusion in AC6KO mice., in which agonal rhythm uniformly was high grade AV block. Therefore, we must seek a link between βAR-stimulation and abnormal electrophysiological consequences in the AC6KO mice.
4.1. Cx43
Cx43 is a major component of ventricular gap junctions that promotes electrical coupling between cardiac myocytes [22, 23]. Although reduced Cx43 protein content was reported to slow LV conduction velocity [24], phosphorylation of Cx43 also plays a critical role in gap junction formation and remodeling [25]. Indeed, decreased Cx43 phosphorylation at S368 has a causal effect on arrhythmias and sudden death [26, 27]. Decreased Cx43 phosphorylation at S368 is also seen in failing LV in association with impaired LV conduction velocity [28]. Therefore, decreased LV Cx43 phosphorylation at S368 in AC6KO mice during chronic βAR stimulation could be a contributing factor for the impaired LV conduction velocity and increased mortality in our experimental setting. It is noteworthy that Cx43 phosphorylation at S368 was unaffected by sustained Iso stimulation in wild type CON mice. It appears that AC6 deletion renders Cx43 more susceptible to Iso-induced change of phosphorylation states, indicating that the interplay of chronic βAR stimulation and AC6 deletion is the basis for the difference. A logical question is whether AC6 deletion influences kinases and phosphatases that govern Cx43 phosphorylation.
The primary kinase regulating Cx43 phosphorylation is debated. Computational models predict the S368 site of Cx43 to be a consensus PKA phosphorylation site [29]; other studies indicate that S368 is a consensus PKC phosphorylation site. Radiolabeling of Cx43 after cAMP stimulation in vitro has identified S364 as the primary PKA phosphorylation site even though Cx43 affinity for PKA is relatively low [30]. In the present study, we found no group differences in LV PKC activity either before or after Iso infusion. AC6 deletion alone (pre-Iso) was associated with a 47% reduction in PKA activity, but with a 2-fold increase in Cx43 phosphorylation at S368. After Iso, both groups displayed similar reductions (33–34%) in LV PKA activity, but Cx43 phosphorylation was unchanged in control while markedly diminished in AC6KO mouse hearts. These observations indicate that the metric linking PKA activity with proportional alterations in Cx43 phosphorylation at S368 is overly simplistic and must be rejected.
A single point mutation of S364 to non-phosphorylatable proline decreases Cx43-mediated cell-cell communication, and is associated with cardiac malformation.[31] In contrast, microinjection of active PKA increases cell-cell communication regulated by native Cx43 but not by S364P mutant [31]. The unavailability of a commercially available specific phospho-Cx43 (S364) antibody prevented us from assessing phosphorylation level at this PKA consensus site and determining its direct effects on gap junction channels in hearts from Iso-infused mice. However, phosphorylation of Cx43 at S364 by PKA potentiates Cx43 phosphorylation at S368 by PKC [30], providing possible link between decreased PKA activity and decreased Cx43 phosphorylation at S368 in hearts from Iso-infused AC6KO mice. Alternatively, AC6 may act as a scaffold by recruiting other molecules to regulate connexin 43 function after chronic Iso stimulation. In summary, although it seems plausible that Iso-related reduced Cx43 phosphorylation in AC6KO mice is of mechanistic importance in reduced LV conduction velocity, its precise molecular underpinnings will require further studies.
4.2. Abnormal AV Conduction
The previously described facilitation of AV conduction by cardiac myocyte-specific AC6 expression [21], the progressive PR prolongation during Iso infusion in AC6KO mice, together with the uniformity of high grade AV block as the agonal rhythm, makes AV node dysfunction seem the most plausible mechanism for mortality. However, the small size of the mouse heart is widely believed to preclude re-entrant tachycardia and therefore the absence of ventricular tachycardia must be viewed with this caveat. Moreover, many of the underpinnings that would facilitate ventricular arrhythmia are present after 4d Iso infusion in AC6KO mouse hearts. For example, we observed progressive QTc prolongation and reduced LV conduction velocity, abnormalities known to presage ventricular tachycardia [32, 33]. Finally, it is worth noting that high grade AV block was a late occurrence and was preceded not only by PR prolongation but also by QTc prolongation. Therefore, abnormalities in both AV and LV conduction presaged increased mortality in Iso-infused AC6KO mice.
If Cx43 phosphorylation was of mechanistic importance in QTc prolongation and impaired LV conduction velocity, were similar abnormalities seen in AV node connexin expression or phosphorylation? The dominant connexins in AV node are Cx45 and Cx30.2, and no antibodies are available to assess their phosphorylation. Levels of mRNA for Cx45 and Cx30.2 in the right atrium, atrial septum and AV node, however, were not altered by AC6 deletion before or after 4d Iso infusion. Therefore the mechanism for impaired AV conduction in Iso-infused AC6KO mouse hearts remains obscure.
4.3. Iso Cardiomyopathy
The degrees of LV hypertrophy, dilation and dysfunction were identical in AC6KO and wild type CON mice after 4d Iso infusion. There also were no group differences in LV apoptosis and fibrosis. AC6 deletion, despite impaired LV cAMP generating capacity and reduced LV PKA activity, did not attenuate Iso-induced LV hypertrophy, dilation, or dysfunction. This was somewhat surprising, although marked abnormalities in Ca2+ handling in AC6KO mouse hearts [11] augur poorly for adaptive responses to stress in general in this line. Our data indicate that high levels of cAMP generation are not a sine qua non for Iso-induced LV dysfunction. Further study on AC6-deleted mice infused with Iso at a more moderate dose may provide insight on the interaction of AC6 deletion and chronic Iso-induced cardiomyopathy.
LV fibrosis with its attendant accumulation of extracellular matrix proteins and myofibroblasts is known to impair LV conduction and provoke arrhythmias [34, 35], and is anticipated to occur, accompanied by increased apoptosis, following sustained Iso infusion [36]. However, our 7d infusion was not sufficiently long to cause increased fibrosis or increased apoptosis in LV of CON or AC6KO mice. Although there were Iso-related inductions in extracellular matrix protein expression, the inductions were similar between CON and AC6KO groups. These observations compel one to seek other causes for the striking electrocardiographic abnormalities and mortality seen in AC6KO mice.
4.4. Conclusions
Limiting cAMP production does not prevent sustained β-adrenergic receptor stimulation-induced cardiomyopathy. Moreover, AC6 deletion was associated with increased mortality in mice during chronic Iso infusion. Marked reduction in Cx43 phosphorylation, specific for the Iso-infused AC6KO mouse hearts, was associated with QTc prolongation, and impaired LV conduction velocity, both harbingers of increased mortality in clinical settings. These data suggest that reduced Cx43 phosphorylation, a sequalea of AC6 deletion and βAR stimulation, is mechanistically important in increased mortality.
Supplementary Material
Highlights.
Limiting cAMP production does not prevent Iso-induced cardiomyopathy.
Unexpected higher mortality in AC6KO mice during 7d Iso infusion.
Progressive prolongation of PR, QRS, and QTc in AC6KO mice during Iso infusion.
Decreased longitudinal ventricular conduction in AC6KO mice during Iso infusion.
Acknowledgments
We thank for Jay Parikh and Sherry Zhuang for technical assistance. This work was supported by a Grant-in-Aid from the American Heart Association (TT), NIH grants P01 HL66941, HL088426, and HL081741 (HKH), and a VA Merit Review Award 1I01BX000783 (HKH).
Footnotes
DISCLOSURES
Dr. Hammond is founder, consultant, and equity holder in Renova Therapeutics, which had no role in funding, design, or interpretation of data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–11. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 3.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–28. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 4.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111:131–50. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- 5.Bisognano JD, Weinberger HD, Bohlmeyer TJ, Pende A, Raynolds MV, Sastravaha A, et al. Myocardial-directed overexpression of the human beta(1)-adrenergic receptor in transgenic mice. J Mol Cell Cardiol. 2000;32:817–30. doi: 10.1006/jmcc.2000.1123. [DOI] [PubMed] [Google Scholar]
- 6.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–64. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, et al. Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101:1707–14. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 8.Iwase M, Uechi M, Vatner DE, Asai K, Shannon RP, Kudej RK, et al. Cardiomyopathy induced by cardiac Gs alpha overexpression. Am J Physiol. 1997;272:H585–9. doi: 10.1152/ajpheart.1997.272.1.H585. [DOI] [PubMed] [Google Scholar]
- 9.Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, et al. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105:1989–94. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- 10.Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, et al. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099–102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 11.Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, et al. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008;117:61–9. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 12.El-Armouche A, Wittkopper K, Degenhardt F, Weinberger F, Didie M, Melnychenko I, et al. Phosphatase inhibitor-1-deficient mice are protected from catecholamine-induced arrhythmias and myocardial hypertrophy. Cardiovasc Res. 2008;80:396–406. doi: 10.1093/cvr/cvn208. [DOI] [PubMed] [Google Scholar]
- 13.De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, et al. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2001;98:3322–7. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama H, Bodi I, Correll RN, Chen X, Lorenz J, Houser SR, et al. alpha1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J Clin Invest. 2009;119:3787–96. doi: 10.1172/JCI39724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim BK, Xiong D, Dorner A, Youn TJ, Yung A, Liu TI, et al. Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J Clin Invest. 2008;118:2758–70. doi: 10.1172/JCI34777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–96. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- 17.Tang T, Lai NC, Hammond HK, Roth DM, Yang Y, Guo T, et al. Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J Am Coll Cardiol. 2010;55:1476–86. doi: 10.1016/j.jacc.2009.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, et al. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation. 2004;110:330–6. doi: 10.1161/01.CIR.0000136033.21777.4D. [DOI] [PubMed] [Google Scholar]
- 19.Tang T, Gao MH, Roth DM, Guo T, Hammond HK. Adenylyl cyclase type VI corrects cardiac sarcoplasmic reticulum calcium uptake defects in cardiomyopathy. Am J Physiol Heart Circ Physiol. 2004;287:H1906–12. doi: 10.1152/ajpheart.00356.2004. [DOI] [PubMed] [Google Scholar]
- 20.Sastry A, Arnold E, Gurji H, Iwasa A, Bui H, Hassankhani A, et al. Cardiac-directed expression of adenylyl cyclase VI facilitates atrioventricular nodal conduction. J Am Coll Cardiol. 2006;48:559–65. doi: 10.1016/j.jacc.2006.01.082. [DOI] [PubMed] [Google Scholar]
- 21.Timofeyev V, He Y, Tuteja D, Zhang Q, Roth DM, Hammond HK, et al. Cardiac-directed expression of adenylyl cyclase reverses electrical remodeling in cardiomyopathy. J Mol Cell Cardiol. 2006;41:170–81. doi: 10.1016/j.yjmcc.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–72. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, et al. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99:1991–8. doi: 10.1172/JCI119367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen HH, Baty CJ, Maeda T, Brooks S, Baker LC, Ueyama T, et al. Transcription enhancer factor-1-related factor-transgenic mice develop cardiac conduction defects associated with altered connexin phosphorylation. Circulation. 2004;110:2980–7. doi: 10.1161/01.CIR.0000146902.84099.26. [DOI] [PubMed] [Google Scholar]
- 26.Remo BF, Qu J, Volpicelli FM, Giovannone S, Shin D, Lader J, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res. 2011;108:1459–66. doi: 10.1161/CIRCRESAHA.111.244046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glukhov AV, Fedorov VV, Kalish PW, Ravikumar VK, Lou Q, Janks D, et al. Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy. Circulation. 2012;125:1835–47. doi: 10.1161/CIRCULATIONAHA.111.047274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller EE, Momen A, Masse S, Zhou YQ, Liu J, Backx PH, et al. Electrical remodelling precedes heart failure in an endothelin-1-induced model of cardiomyopathy. Cardiovasc Res. 2011;89:623–33. doi: 10.1093/cvr/cvq351. [DOI] [PubMed] [Google Scholar]
- 29.Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & Cellular Proteomics. 2008;7:1598–608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah MM, Martinez AM, Fletcher WH. The connexin43 gap junction protein is phosphorylated by protein kinase A and protein kinase C: in vivo and in vitro studies. Mol Cell Biochem. 2002;238:57–68. doi: 10.1023/a:1019902920693. [DOI] [PubMed] [Google Scholar]
- 31.Britz-Cunningham SH, Shah MM, Zuppan CW, Fletcher WH. Mutations of the Connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N Engl J Med. 1995;332:1323–9. doi: 10.1056/NEJM199505183322002. [DOI] [PubMed] [Google Scholar]
- 32.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–9. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–29. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- 34.Kawara T, Derksen R, de Groot JR, Coronel R, Tasseron S, Linnenbank AC, et al. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation. 2001;104:3069–75. doi: 10.1161/hc5001.100833. [DOI] [PubMed] [Google Scholar]
- 35.Rohr S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhythm Electrophysiol. 2012;5:442–52. doi: 10.1161/CIRCEP.110.957647. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Weber KT. Animal models of cardiac fibrosis. Methods Mol Med. 2005;117:273–90. doi: 10.1385/1-59259-940-0:273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.