

Abstract
Alpha/beta-hydrolase domain containing 6 (ABHD6) is a transmembrane serine hydrolase that hydrolyzes the endogenous cannabinoid 2-arachidonoylglycerol (2-AG) to regulate certain forms of cannabinoid receptor-dependent signaling in the nervous system. The full spectrum of ABHD6 metabolic activities and functions is currently unknown and would benefit from selective, in vivo-active inhibitors. Here, we report the development and characterization of an advanced series of irreversible (2-substituted)-piperidyl-1,2,3-triazole urea inhibitors of ABHD6, including compounds KT182 and KT203, which show exceptional potency and selectivity in cells (< 5 nM) and, at equivalent doses in mice (1 mg kg-1), served as systemic and peripherally-restricted ABHD6 inhibitors, respectively. We also describe an orally-bioavailable ABHD6 inhibitor KT185 that displays excellent selectivity against other brain and liver serine hydrolases in vivo. We thus describe several chemical probes for biological studies of ABHD6, including brain-penetrant and peripherally-restricted inhibitors that should prove of value for interrogating ABHD6 function in animal models.
Introduction
The alpha/beta-hydrolase domain containing 6 (ABHD6) gene encodes a ∼35 kDa protein containing an N-terminal transmembrane region followed by a catalytic domain that includes the canonical GXSXG active-site motif of serine hydrolases (SHs). ABHD6 is a unique and highly conserved enzyme in mammals (94% sequence identity between human and mouse orthologues, ∼25% identity to the nearest homologous enzyme - SERHL) and is expressed prominently in brain, liver, kidney, and brown adipose tissue based on global gene expression analysis (http://biogps.gnf.org/) and activity-based protein profiling (ABPP).1 As a member of the SH class, ABHD6 is predicted to hydrolyze ester, amide, or thioester bonds in substrates that could include small-molecules, lipids, or peptides, although the full range of substrates regulated by this enzyme in vivo is currently unknown.2
Chemical proteomic studies led to the discovery that ABHD6 can hydrolyze the endogenous cannabinoid (endocannabinoid), 2-arachidonoylglycerol (2-AG) in vitro3 and, in support of this metabolic function, inactivation of ABHD6 by first-generation carbamate inhibitors4 alters cannabinoid receptor-dependent synaptic signaling in mouse cortical slices.3, 5 Recent studies have also shown that ABHD6 carbamate inhibitors produce anti-inflammatory and neuroprotective effects in a mouse model of traumatic brain injury.6 Whether the neuroprotective effects of these inhibitors are mediated by elevations in 2-AG or a novel ABHD6-regulated metabolite, however, remains unknown. While first-generation carbamate compounds exhibit good inhibitory activity against ABHD6 in vitro (IC50 value of ∼70-85 nM),4, 7 next-generation inhibitors with improved potency, selectivity, and in vivo activity would facilitate the functional analysis of ABHD6 in mammalian biology and disease.
In recent studies focused on developing inhibitors of the endocannabinoid biosynthetic enzyme DAGLβ,8, 9 we identified a (2-phenyl)-piperidyl-1,2,3-triazole urea [(2-phenyl)-Pip-1,2,3-TU] inhibitor of ABHD6 termed compound 1 (KT195),8 which is predicted to irreversibly inhibit ABHD6 by carbamoylation of the enzyme's serine nucleophile.8 Here, we describe the further optimization of (2-substituted)-Pip-1,2,3-TU inhibitors of ABHD610 and show that the addition of polar substituents onto the biphenyl-triazole group can fine-tune the potency, selectivity, and in vivo activity of compounds, resulting in development of the highly potent (IC50 values ∼ 1 nM) and selective ABHD6 inhibitors, 9 (KT182) and 20 (KT203), that show systemic and peripherally restricted activity, respectively, as well as the first orally-active ABHD6-selective inhibitor, 11 (KT185). These findings highlight the versatility of 1,2,3-TUs as inhibitors of ABHD6, which combine simplified synthetic routes with the ability to achieve excellent potency and selectivity and controlled access to the central nervous system (CNS) for developing peripherally-restricted chemical probes.
Results
A clickable probe to evaluate the proteome-wide selectivity of compound 1
Previous studies using both gel- and MS-based competitive ABPP8 showed that compound 1 (Table 1) exhibits excellent potency (in vitro IC50 of ∼10 nM) and selectivity for ABHD6 across the SH family, but did not address potential for cross-reactivity with other proteins in the proteome. To assess the broader, proteome-wide selectivity of compound 1, we synthesized an alkynylated analog 2 (Figure 1A), such that the alkyne group would serve as a latent affinity handle suitable for conjugation to reporter tags by copper-catalyzed azide alkyne cycloaddition11 (CuAAC or click chemistry). We confirmed that compound 2 maintained good inhibitory activity against ABHD6 as measured by gel-based competitive ABPP in mouse neuroblastoma Neuro2A cell and mouse brain proteomes (Figure 1B, C). Next, we treated Neuro2A cells with varying concentrations of compound 2 for 1 hr. Cells were then lysed and the membrane proteomes conjugated by click chemistry with an azide-Rh tag,12 separated by SDS-PAGE, and probe-labeled proteins visualized by in-gel fluorescence scanning (Figure 1D). This analysis revealed a single major protein target of ∼ 35 kDa, matching the molecular mass of ABHD6, that could be detected at concentrations of compound 2 as low as 10 nM (Figure 1D). At higher concentrations (80-600 nM) of 2, some limited cross-reactivity was observed, mainly with a 60 kDa protein that likely represents fatty acid amide hydrolase (FAAH), a known, lower affinity off-target of compound 1 (Table 1). We confirmed that compound 2 is cross-reactive with FAAH in the mouse brain proteome at concentrations of 0.4 – 10 μM as judged by competitive ABPP (Figure 1C). Considering that compound 1 completely inactivates ABHD6 (with negligible cross-reactivity with FAAH) at concentrations of 25 nM in living cells,8 our data argue that 1 exhibits excellent proteome-wide selectivity at concentrations required to inhibit ABHD6 in situ. When combined with complementary studies of clickable probes for 2-substituted Pip-1,2,3-TU inhibitors of DAGL enzymes, which also showed good proteome-wide selectivity at concentrations of 80 nM or below,9 our results suggest further that the (2-substituted)-Pip-1,2,3-TU chemotype is generally suitable for creating selective SH inhibitors, especially if the in situ potencies of these agents can be optimized to the low (< 100 nM) range.
Table 1.
Structure-activity relationship of lead ABHD6 inhibitors.
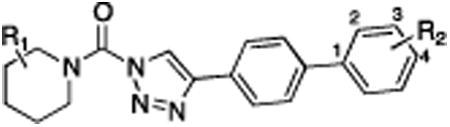
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | R1= | R2 = | DAGLβ % Inhibition (100 nM) | DAGLβ % Inhibition (10 μM) | ABHD6 % Inhibition (10 nM) | Off-targets (10 μM) |
| 1 (KT195) | 2-Ph | 4-OMe | 0 | 0 | 30 | FAAH |
| 3 | 2-Bn | 2-OMe | 66 | 100 | 89 | MGLL |
| 4 | 2-Bn | 3-CH2OH | 37 | 91 | 91 | none |
| 5 | 2-Bn | 4-piperidineamide | 15 | 58 | 79 | none |
| 6 | 2-Ph | 2-OMe | 14 | 70 | 82 | FAAH, MGLL |
Figure 1.
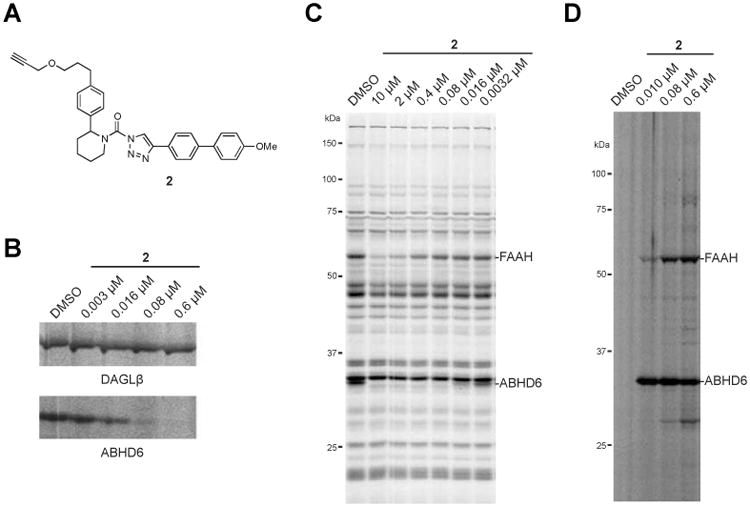
Structure and activity of compound 2, a clickable analogue of 1. (A) Chemical structure of compound 2. (B) In vitro potency of compound 2 against DAGLβ and ABHD6 in Neuro2A membrane proteome as measured by gel-based competitive ABPP using the tailored activity-based probe HT-01. Neuro2A proteome (1 mg/mL) was incubated with the indicated concentrations of 2 (30 min, 37 °C) followed by labeling with 1 μM HT-01 (30 min, 37 °C), and DAGLβ and ABHD6 activity visualized by SDS-PAGE and in-gel fluorescence scanning. (C) Selectivity of compound 2 against mouse brain membrane SH enzymes as measured by gel-based competitive ABPP using the broad-spectrum, SH-directed probe FP-Rh. (D) Click chemistry-ABPP of Neuro2A cells treated in situ with compound 2. Neuro2A cells were treated with the indicated concentrations of compound 2 (1 hr, 37 °C), lysed, and compound 2-labeled proteins visualized in the membrane proteome by click chemistry reaction with azide-Rh followed by SDS-PAGE and in-gel fluorescence scanning. Fluorescent gels are shown in gray scale. Assignment of serine hydrolase enzyme activities in competitive ABPP gels are based on gel migration patterns consistent with past studies.8, 9, 13
Optimization of (2-substituted)-Pip-1,2,3-TUs as ABHD6 inhibitors
In our previous studies, we used compound 1 primarily as a control probe for evaluating the activity of structurally related DAGLβ inhibitors.8 Consequently, the structure-activity relationship for (2-substituted)-Pip-1,2,3-TU-ABHD6 interactions was not explored. Here, we set out to address this question by testing the activity of structurally diverse analogues of 1 to identify ABHD6 inhibitors with improved in vitro potency and in vivo activity. We first compared the activity of several compounds that contained polar groups on the biphenyl triazole group (Table 1 and Figure 2). As reported previously, 2-benzyl compounds, such as 3 (KT172),8 4 (KT123),9 and 5 (KT125),9 exhibited high-potency for ABHD6, but also cross-reacted with DAGLβ (Figure 2A, B and Table 1). Inclusion of polar groups at the 3 or 4 positions of the distal phenyl ring on the biphenyl triazole leaving group improved selectivity against DAGLβ (Figure 2A, B and Table 1), as well as eliminating monoacylglycerol lipase (MGLL) as an off-target at higher concentrations (Figure 2C and Table 1). Substitution of the 2-benzyl for a 2-phenyl group (compounds 1 and 6) nearly eliminated DAGLβ cross-reactivity with minimal loss in potency towards ABHD6 (Figure 2A, B and Table 1). However, this alteration also introduced FAAH as an additional off-target at higher inhibitor concentrations (10 μM).
Figure 2.
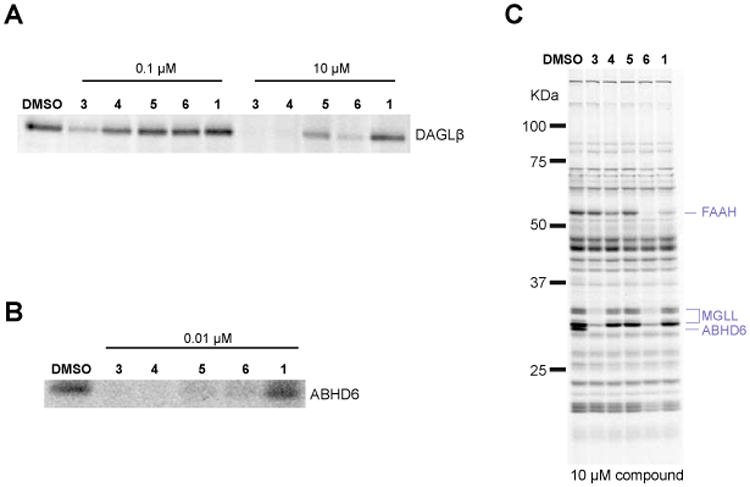
Potency and selectivity of lead ABHD6 inhibitors. Potency of (2-substituted)-Pip-1,2,3-TUs against (A) recombinant DAGLβ expressed by transient transfect in HEK293T cells (DAGLβ-HEK293T lysates), and (B) endogenous ABHD6 in Neuro2A membrane proteomes as measured by gel-based competitive ABPP using the HT-01 probe. (C) Selectivity of inhibitors against mouse brain membrane SH enzymes as measured by gel-based competitive ABPP using the FP-Rh probe. For the gel-based ABPP assays, proteomes were incubated with compound (0.01, 0.1 or 10 μM) for 30 min at 37 °C followed by reaction with fluorescent ABPP probes (HT-01 or FP-Rh, 1 μM, 30 min, 37 °C). Fluorescent gel images are shown in gray scale. Serine hydrolase activities in gels were assigned as described in Figure 1.
We next aimed to improve the potency and selectivity of (2-phenyl)-Pip-1,2,3-TUs by further exploring various polar substitutions (hydroxymethyl, 8-10; secondary amide, 11 and 12; or primary amide, compound 13) on the biphenyl triazole group (Scheme 1). In brief, the 4-bromo-triazole ring was synthesized using the copper-catalyzed azide-alkyne cycloaddition reaction14 and coupled to 2-phenyl piperidine to generate compound 7 as previously described.8, 15 Compounds 8-13 were synthesized using 7 as the key intermediate for subsequent Suzuki coupling reactions (Scheme 1). We compared the in vitro potency of compounds against endogenous ABHD6 in mouse brain or Neuro2A membrane proteomes by gel-based competitive ABPP utilizing the broad-spectrum and tailored SH activity-based probes, fluorophosphonaterhodamine (FP-Rh) and HT-01, respectively, as described previously.8
Scheme 1. Synthesis of (2-phenyl)-piperidine-1,2,3-TUs.

All of the synthesized derivatives (8-13), when tested at 1 μM, selectively inhibited ABHD6 among mouse brain SHs, but showed variable levels of off-target activity at 10 μM (Figure 3A, B and Table 2). We consistently observed increased inhibitory activity against ABHD6 for compounds with modifications at the 2- (8) and 3-positions (9 and 11) compared with their respective 4-substituted derivatives (10, 12, and 13). From this series of compounds, we selected compounds 9 and 11 as suitable for further investigation. Compound 9 was chosen based on its improved selectivity over 8 (i.e., attenuated activity against LYPLA1 and LYPLA2; Figure 3B) and its smaller molecular weight in comparison with 12 (MW < 500). Compound 11 was selected based on its enhanced selectivity against FAAH and other SHs when tested at high concentrations in the mouse brain (10 μM, Figure 3B).
Figure 3.
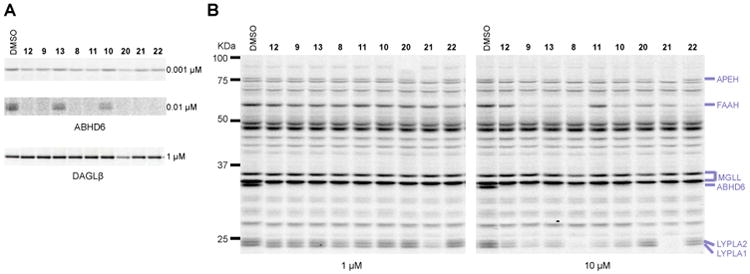
Potency and selectivity of advanced ABHD6 inhibitors. (A) Potency of (2-substituted)-Pip-1,2,3-TUs against ABHD6 in Neuro2A cell membranes or DAGLβ-HEK293T lysates by gel-based competitive ABPP using the HT-01 probe. (B) Selectivity of (2-substituted)-Pip-1,2,3-TUs against mouse brain SHs as measure by gel-based competitive ABPP using the FP-Rh probe. Gel-based ABPP assays were performed as described in Figure 1. Fluorescent gel images are shown in gray scale. Serine hydrolase activities in gels were assigned as described in Figure 1.
Table 2. Activity of (2-phenyl)-piperidine-1,2,3-TUs.
| Compound | ABHD6 % Inhibition (10 nM) | ABHD6 % Inhibition (1 nM) | DAGLβ Inhibition (1 μM) | Off-targets (1 μM) | Off-targets (10 μM) |
|---|---|---|---|---|---|
| 8 | 98 | 38 | 0 | none | FAAH, LYPLA1, LYPLA2 |
| 9 | 84 | 29 | 0 | none | FAAH, LYPLA1, LYPLA2 |
| 10 | 47 | 18 | 0 | none | FAAH, LYPLA1, LYPLA2 |
| 11 | 100 | 51 | 0 | none | LYPLA1, LYPLA2 |
| 12 | 94 | 35 | 0 | none | LYPLA1, LYPLA2 |
| 13 | 27 | 17 | 0 | none | FAAH, LYPLA1, LYPLA2 |
| 20 | 100 | 62 | 50 | none | FAAH, DAGLβ |
| 21 | 100 | 36 | 6 | APEH, LYPLA1, LYPLA2 | FAAH, APEH, LYPLA1, LYPLA2 |
| 22 | 94 | 33 | 4 | none | FAAH |
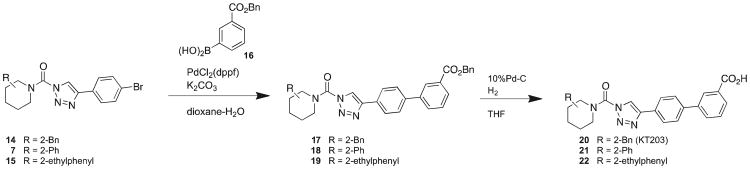
We also synthesized a series of carboxylic acid derivatives by coupling amines with a 4-bromo-triazole group to form intermediates 7, 14, and 15, which were then converted into corresponding 3-benzylcarboxyphenyl derivatives by Suzuki coupling reactions (17 – 19) (Scheme 2). Subsequent treatment of benzylcarboxyphenyl intermediates with 10% Pd-C under hydrogen atmosphere generated the carboxylic acid derivatives (20 – 22). We found that substitution of a hydroxymethyl for a carboxylate group at the 3-position of the distal phenyl ring of the triazole-biphenyl group preserved potency for ABHD6 (compare profiles of 9 and 21 in Figure 2A and Table 2), but increased off-target interactions in the mouse brain proteome, where 21 (at 1 μM) also inhibited APEH, LYPLA1, and LYPLA2 (Figure 3B and Table 2). Substitution of the 2-phenyl (21) for a 2-benzyl (20) but not a 2-phenethyl (22) group improved both potency and overall selectivity with no observable SH off-targets at 1 μM other than a modest 50% inhibition of DAGLβ (Figure 3A, B and Table 2). Given its potency, selectivity, and increased polarity, compound 20 was designated as a third ABHD6 inhibitor worthy of more detailed analysis.
Scheme 2. Synthesis of carboxylate-modified (2-substituted)-Pip-1,2,3-TU ABHD6 inhibitors.
(2-substituted)-Pip-1,2,3-TUs are potent and selective ABHD6 inhibitors in situ
We next measured in vitro potencies for compounds 9, 11, and 20 and found that all three compounds potently inhibited ABHD6 as measured by gel-based competitive ABPP (IC50 values between 0.8 - 1.7 nM, Figure 4A, B) and 2-AG hydrolysis assays (IC50 values between 3.9 – 15.1 nM, Figure 4C). In situ potencies were then measured by treating Neuro2A cells with varying concentrations of compounds for 4 hr, after which cells were lysed and proteomes subjected to gel-based ABPP as previously described.8 We found that all three compounds inhibited ABHD6 in situ with IC50 values in the subnanomolar range (0.2 - 0.3 nM, Figure 4D, E). For comparison, compounds 9, 11, and 20 were ∼10-fold and ∼5-fold more potent in vitro and in situ, respectively, compared to compound 1.8
Figure 4.
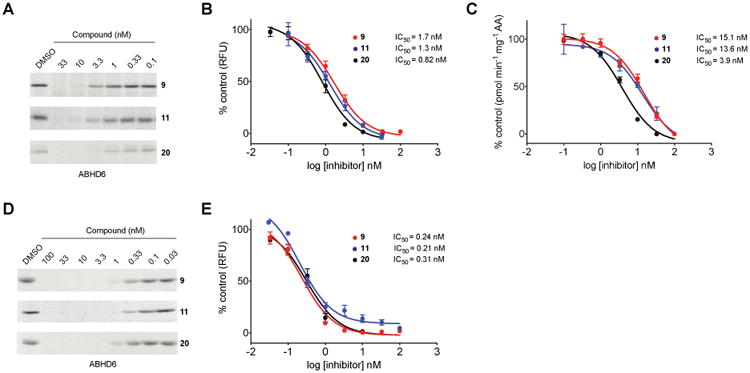
In vitro (A-C) and in situ (D, E) IC50 values for compounds 9, 11, and 20 against ABHD6 as determined by gel-based competitive ABPP with the activity-based probe, HT-01 (A, B, D, E) and 2-AG substrate hydrolysis assays (C). For in vitro gel-based ABPP studies (A, B), Neuro2A membrane proteomes were preincubated with varying concentrations of compounds for 30 min at 37 °C, treated with HT-01 (1 μM, 30 min, 37 °C), quenched in SDS-PAGE loading buffer, and analyzed by in-gel fluorescence scanning. For 2-AG hydrolysis assays (C), inhibitors were tested against ABHD6 recombinantly expressed in HEK293T cells as described in the Experimental Section. For in situ studies (D, E), Neuro2A cells were treated with varying amounts of compounds for 4 hr, lysed, and subjected to competitive ABPP as described above. Integrated band intensities of probe-labeled proteins were calculated using ImageJ and used to generate concentration-dependent inhibition curves for both in vitro (B) and in situ (E) studies.
To more comprehensively profile the selectivity of compounds 9, 11, and 20 in cells, we utilized a quantitative mass-spectrometry (MS)-based proteomic method termed ABPP-SILAC.8, 15, 16 In brief, Neuro2A cells cultured in “light” (12C614N2-lysine and 12C614N2-arginine) or “heavy” medium (13C615N2-lysine and 13C615N2-arginine) were treated with DMSO or inhibitors (3 nM, 4 hr), respectively, lysed, proteomes separated into soluble and membrane fractions, and SHs enriched with FP-biotin (10 μM, 2 hr). Heavy (inhibitor-treated) and light (DMSO-treated) fractions were then mixed 1:1, enriched with avidin, digested on-bead with trypsin, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an LTQ-Orbitrap instrument. Light and heavy signals were quantified from parent ion peaks (MS1) and the corresponding proteins identified from product ion profiles (MS2) using the SEQUEST search algorithm. The depicted bar graphs represent the average ratios of quantified heavy/light tryptic peptides for >50 SHs identified in the Neuro2A proteome (Figure 5). We found that both compound 9 and 20 blocked > 90% of ABHD6 activity, while 11 blocked > 80% of ABHD6 activity in Neuro2A cells, and, importantly, none of the compounds showed substantial cross-reactivity (< 50%) against the ∼50 other SHs detected in this cell line (Figure 5). Collectively, these results show that compounds 9, 11, and 20 potently and selectively inhibit ABHD6 in living cells.
Figure 5.

In situ activity and selectivity of ABHD6 inhibitors in Neuro2A cells. ABPP-SILAC analysis of SH activities from Neuro2A cells treated in situ with 3 nM of compound 9 (A), 11 (B), or 20 (C) for 4 hr. Error bars represent mean ± s.e.m. of heavy/light ratios for the multiple quantified peptides observed for each enzyme (minimum of 3 unique peptides per enzyme) in both soluble and membrane fractions.
Compounds 9 and 20 are potent and selective ABHD6 inhibitors in vivo
Mice were treated intraperitoneally (i.p., 18:1:1 solution of saline/ethanol/PEG40 (ethoxylated castor oil), 10 μL g-1) with compound 9 or 20 at various doses (0.1 – 1 mg kg-1) for 4 hr, sacrificed, and brain and liver tissue collected for analysis by gel-based competitive ABPP using HT-01 and FP-Rh probes. Both compound 9 and 20 produced near-complete blockade of ABHD6 in the liver at the highest dose tested (1 mg kg-1, HT-01 gels, Figure 6A). At lower doses, the compounds maintained ∼80% inhibition of ABHD6 in the liver with 20 showing slightly enhanced potency compared to 9 at the lowest dose tested (0.1 mg kg-1, Figure 6A). Notably, both compound 9 and 20 showed impressive selectivity in the mouse liver even at higher doses (1 mg kg-1), exhibiting little cross-reactivity against the numerous carboxylesterase enzymes (CESs) that are common off-targets of mechanism-based SH inhibitors in rodents.1, 8, 17 Compound 9 also completely inactivated ABHD6 in the mouse brain at 1 mg kg-1 (Figure 6B). In contrast, compound 20 showed negligible inhibition of brain ABHD6 at all doses tested (Figure 6B). Both compounds showed good selectivity against other brain and liver SHs as judged by gel-based ABPP (FP-Rh gels, Figure 6), which revealed only a single off-target, carboxylesterase-1 (CES1), a plasma esterase that exhibits broad reactivity with various classes of SH inhibitors1 and was inhibited in our study by 9, but not 20. These data confirm that compounds 9 and 20 are potent and selective ABHD6 inhibitors in vivo, showing systemic- and peripherally-restricted activity, respectively. Considering that compounds 9 and 20 displayed similar potencies against liver ABHD6 in vivo, we believe that the reduced activity of 20 against brain ABHD6 likely reflects diminished CNS penetrance possibly due to it unique carboxylic acid substituent.
Figure 6.
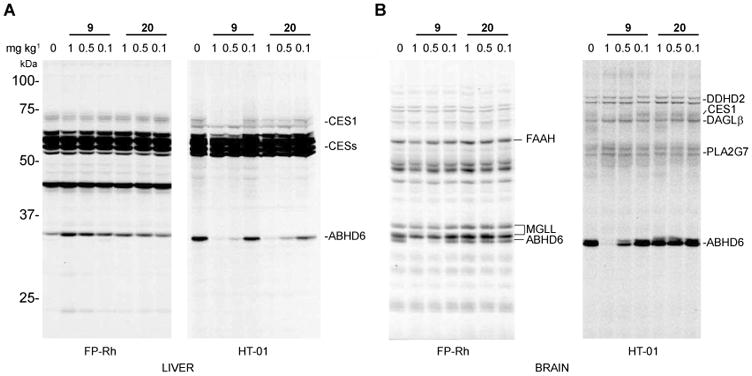
Comparative analysis of the potency and selectivity of compounds 9 and 20 in vivo in mouse liver (A) and brain (B). Mice were administered vehicle or test compounds (1, 0.5, or 0.1 mg kg-1, i.p.), and, after 4 hr, sacrificed and their brain and liver tissues harvested, the membrane fractions isolated, and subjected to gel-based competitive ABPP with FP-Rh or HT-01 (1 μM, 30 min, 37 °C). Compound 9 produced near-complete inactivation of ABHD6 at 1 mg kg-1 in both liver (A, HT-01 gel) and brain (B, FP-Rh or HT-01 gels), while 20 showed peripherally restricted activity with near-complete inactivation of ABHD6 in liver, but not brain at at 1 mg kg-1. For gel-based ABPP experiments, proteomes were labeled with FP-Rh or HT-01 (1 μM probe) for 30 min at 37 °C. Note that ABHD6 was too low in abundance in liver for robust detection by the FP-Rh probe under the described assay conditions. Serine hydrolase activities in gels were assigned as described in Figure 1.
Compound 11 is an orally-bioavailable and selective ABHD6 inhibitor in vivo
Our in vivo studies with 11 revealed that this compound was systemically active, but exhibited reduced potency in mice compared to 9, requiring ∼40 mg/kg (i.p.) to completely block ABHD6 in the brain (Figure 7A). In the liver we observed near-complete blockade of ABHD6 at doses as low as 5-10 mg/kg (i.p., Supplementary Figure 2) suggesting that this compound shows reduced central activity akin to 20. However, at all doses tested, compound 11 maintained excellent selectivity for ABHD6 over other brain SHs, including FAAH (Figure 7B), against which this compound showed superior selectivity compared with compounds 9 and 20 at high concentrations in vitro (Figure 3B). We also found that compound 11 was orally active in mice (p.o., PEG300 vehicle, 4 μL g-1, 4 hr), producing complete blockade of brain ABHD6 at 40 mg/kg as measured by gel-based competitive ABPP using HT-01 and FP-Rh (Figure 7B) with the only detectable off-target being CES1. These results contrasted with the activity of compounds 9 and 20, which showed cross-reactivity with FAAH in the brains of mice treated by oral gavage with doses as low as 10 mg kg-1 (Supplementary Figure 1). We also observe near-complete blockade of ABHD6 at doses of 10 – 40 mg/kg and minimal cross-reactivity with CESs (other than CES1) in liver of compound 11-treated mice (Supplementary Figure 2). These data confirm that compound 11 is an orally-bioavailable and selective ABHD6 inhibitor in vivo, showing minimal cross-reactivity with other SHs in the mouse brain and liver across a broad dose-range.
Figure 7.

Potency and selectivity of compound 11 in the mouse brain in vivo. (A) Mice were administered vehicle or compound 11 (1-40 mg kg-1, i.p.), and, after 4 hr, sacrificed and brain tissue harvested, the membrane fractions subjected to gel-based competitive ABPP with FP-Rh or HT-01 (1 μM, 30 min, 37 °C). Compound 11 completely inactivated ABHD6 at 40 mg kg-1 with partial blockade at lower doses (>70 % inhibition at 20 mg kg-1) and showed excellent selectivity against other brain SHs, including negligible cross-reactivity with FAAH across the entire tested dose-range. (B) Compound 11 administered to mice by oral gavage (p.o.) produced near-complete and selective inactivation of brain ABHD6 at a dose of 40 mg kg-1. For gel-based ABPP experiments, proteomes were labeled with FP-Rh or HT-01 (1 μM probe) for 30 min at 37 °C. Serine hydrolase activities in gels were assigned as described in Figure 1.
Conclusion
Despite emerging roles for ABHD6 in endocannabinoid metabolism, neural signaling, and neurodegeneration,3, 5, 6 the full spectrum of endogenous substrates regulated by this enzyme remains poorly characterized due in part to the lack of optimized inhibitors for functional studies in vivo. Here, we have addressed this problem by developing several new chemical probes including brain-penetrant and peripherally-restricted inhibitors of ABHD6.
In previous studies, we described compound 1 as a lead (2-phenyl)-Pip-1,2,3-TU inhibitor of ABHD6 and its use as a control probe in our characterization of structurally-related DAGLβ inhibitors.8 Here, we aimed to improve the in vitro and in vivo activity of 2-substituted Pip-1,2,3-TU inhibitors by performing a more comprehensive exploration of the structure-activity relationship underlying their interactions with ABHD6. We discovered that the potency and selectivity of lead compounds could be affected by the addition of polar substituents to the triazole group. This modification, combined with replacing the 2-benzyl with a 2-phenyl moiety on the piperidine group eliminated most of the off-targets for (2-substituted)-Pip-1,2,3-TUs, including DAGLβ. Our ABPP-guided medicinal chemistry efforts culminated in the development of the optimized ABHD6 inhibitors, 9, 11, and 20 that all showed improved potency (∼10-fold enhancement, Figure 4A and C) and selectivity in vitro compared with compound 1.8
We proceeded to demonstrate that compounds 9, 11, and 20 were potent and selective ABHD6 inhibitors in cells (IC50 values of 0.2 – 0.3 nM, Figure 4C and D), displaying negligible cross-reactivity against the other 50+ SHs detected in Neuro2A cells as evaluated by ABPP-SILAC8, 15, 16 (Figure 5). While compounds 9 and 20 shared similar properties with respect to potency and selectivity in cells, they displayed different activity profiles in vivo. Treatment of mice with compound 9 inactivated ABHD6 in both the brain and liver (Figure 6). In contrast, compound 20 produced negligible inhibition of ABHD6 in the brain, but near-complete blockade of ABHD6 in the liver (Figure 6). Both compounds displayed good selectivity in vivo as judged by gel-based competitive ABPP with the FP-Rh and HT-01 probes. Our studies thus indicate that compounds 9 and 20 can be used as paired probes to distinguish the effects of peripheral and central inactivation of ABHD6 in vivo. We also found that compound 11 selectively inactivated ABHD6 in mice when administered intraperitoneally or by oral gavage. Taken together, our data demonstrates that (2-substituted)-piperidyl-1,2,3-triazole ureas can be fine-tuned to create selective ABHD6 inhibitors with good in vivo activity. These inhibitors should serve as valuable chemical probes for characterizing ABHD6 function in cell and animal models.
Experimental Section
Materials Used for Biological Experiments
Pharmacological studies were conducted in C57Bl/6 mice. The fluorophosphonate and HT-01 activity-based probes were synthesized as previously described.8, 18, 19 HEK293T and Neuro2A cells were purchased from ATCC. All lipids used in substrate assays were purchased from Cayman Chemicals or Avanti Lipids.
Synthetic Methods
Commercially-available chemicals and reagents were purchased from the following vendors: Sigma-Aldrich, Acros, Fisher, Fluka, Maybridge, Combi-Blocks, BioBlocks, and Matrix Scientific and used without further purification unless noted otherwise. Dry solvents were obtained by passing commercially available pre-dried, oxygen-free formulations through activated alumina columns. Reactions were performed under a nitrogen atmosphere using oven-baked glassware unless otherwise noted. Flash chromatography was performed using 230-400 mesh silica gel. Reactions were monitored by analytical thin-layer chromatography (TLC) on precoated, glass backed silica gel 60 F254 plates. Reactions were purified either by pTLC, also on silica gel 60 F254 plates or by flash chromatography on 40-60 MYM mesh silica gel as specified. 1H-NMR and spectra were recorded in CDCl3 on a Varian Mercury-300 spectrometer, a Varian Inova-400 or a Bruker DRX-600 spectrometer, and were referenced to trimethylsilane (TMS). Chemical shifts were reported in ppm relative to TMS and J values were reported in Hz. High resolution mass spectrometry (HRMS) experiments were performed at The Scripps Research Institute Mass Spectrometry Core on an Agilent mass spectrometer using electrospray ionization-time of flight (ESI-TOF). All final compounds were determined to be >95% pure by HPLC analysis (Agilent 1100 LC/MS).
(4-(3′-(Hydroxymethyl)-[1,1′-biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)(2-phenylpiperidin-1-yl)methanone (9)
A solution of KT1798 (7) (0.70 g, 1.7 mmol) in dioxane (30 mL) and H2O (3 mL) was treated with 3-hydroxymethylphenyl boronic acid (0.39 g, 2.6 mmol, 1.5 equiv), K2CO3 (0.70 g, 5.1 mmol, 3.0 equiv) and PdCl2(dppf) (62 mg, 0.085 mmol, 0.2 equiv), and the reaction mixture was stirred for 2 hr at 80 °C under N2. The mixture was poured into H2O and extracted with ethyl acetate. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated under reduced pressure. Chromatography (150 g, ethyl acetate:hexane=1:1) afforded 9 (0.55 g, 1.26 mmol, 74%). The final compound was purified as the racemate, which was suitable for biological studies.9
1H NMR (CDCl3, 300 MHz) δ 8.44 (s, 1H), 7.96 (d, 2H, J = 8.3 Hz), 7.70 (d, 2H, J = 8.3 Hz), 7.65 (s, 1H), 7.58 (m, 1H), 7.48-7.25 (m, 7H), 5.93 (br, 1H), 4.78 (br, 2H), 4.38 (brd, 1H, J = 13.5 Hz), 3.19 (m, 1H), 2.53 (brd, 1H, J = 14.1 Hz), 2.16 (m, 1H), 1.90-1.65 (m, 4H). 13C NMR (CDCl3, 151 MHz) δ 176.96, 149.78, 146.88, 141.86, 141.52, 141.10, 138.20, 129.45, 129.31, 128.95, 128.01, 127.53, 126.92, 126.65, 126.48, 125.94, 121.34, 65.68, 28.13, 26.19, 19.66. HRMS calculated for C27H27N4O2 [M+H]+ 439.2128, found 439.2116.
(2-Phenylpiperidin-1-yl)(4-(3′-(piperidine-1-carbonyl)-[1,1′-biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)methanone (11)
A solution of 7 (10 mg, 0.024 mmol) in dioxane (1 mL) and H2O (0.1 mL) was treated with (3-(piperidine-1-carbonyl)phenyl)boronic acid (8 mg, 0.036 mmol, 1.5 equiv), K2CO3 (10 mg, 0.072 mmol, 3.0 equiv) and PdCl2(dppf) (4 mg, 0.0049 mmol, 0.2 equiv), and the reaction mixture was stirred for 2 hr at 80 °C under N2. The mixture was poured into H2O and extracted with ethyl acetate. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated under reduced pressure. The crude oil was purified by pTLC (ethyl acetate:hexane = 1:1) to give 11 (4 mg, 0.008 mmol, 32%).
1H NMR (CDCl3, 300 MHz) δ 8.45 (s, 1H), 7.96 (d, 2H, J = 8.3 Hz), 7.72-7.64 (m, 4H), 7.53-7.25 (m, 7H), 5.93 (br, 1H), 4.38 (brd, 1H, J = 12.7 Hz), 3.74 (br, 2H), 3.40 (br, 2H), 3.19 (m, 1H), 2.54 (brd, 1H, J = 14.3 Hz), 2.16 (m, 1H), 1.90-1.50 (m, 10H). 13C NMR (CDCl3, 151 MHz) δ 176.97, 170.44, 149.76, 146.78, 141.04, 140.95, 138.19, 137.52, 129.24, 128.24, 128.01, 127.53, 126.92, 126.72, 126.18, 125.72, 121.41, 49.20, 43.50, 30.04, 28.18, 26.98, 26.18, 25.97, 24.94, 19.66. HRMS calculated for C32H34N5O2 [M+H]+ 520.2707, found 520.2708.
Benzyl 4′-(1-(2-benzylpiperidine-1-carbonyl)-1H-1,2,3-triazol-4-yl)-[1,1′-biphenyl]-3-carboxylate (17)
A solution of 14 (1.2 g, 2.9 mmol) in dioxane (40 mL) and H2O (4 mL) was treated with 3-carboxybenzylphenyl bronic acid (1.1 g, 4.4 mmol, 1.5 equiv), K2CO3 (1.2 g, 8.7 mmol, 3.0 equiv) and PdCl2(dppf) (0.11 g, 0.15 mmol, 0.05 equiv), and the reaction mixture was stirred for 2 hr at 80 °C under N2. The mixture was poured into H2O and extracted with ethyl acetate. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated under reduced pressure. Chromatography (150 g, ethyl acetate:hexane=1:3) afforded 17 (1.6 g, quantitative).
1H NMR (CDCl3, 300 MHz) δ 8.36 (s, 1H), 8.08 (d, 1H, J = 7.5 Hz), 7.89 (br, 2H), 7.84 (d, 1H, J = 7.3 Hz), 7.71 (d, 2H, J = 8.4 Hz), 7.56-7.33 (m, 6H), 7.30-6.90 (m, 5H), 5.42 (s, 2H), 4.86 (br, 1H), 4.37 (d, 1H, J = 13.3 Hz), 3.48-2.69 (m, 3H), 2.05-1.65 (m, 6H). HRMS calculated for C35H33N4O3 [M+H]+ 557.2547, found 557.2552.
4′-(1-(2-Benzylpiperidine-1-carbonyl)-1H-1,2,3-triazol-4-yl)-[1,1′-biphenyl]-3-carboxylic acid (20)
A solution of 17 (1.6 g, 2.9 mmol) in THF (30 mL) was treated with 10% Pd-C (0.30 g) and the mixture was stirred overnight at room temperature under N2. The mixture was passed through Celite and the filtrate was concentrated under reduced pressure. Crystallization from ethyl acetate and hexane afforded 20 (KT203) (1.2 g, 2.6 mmol, 89%).
1H NMR (CDCl3, 300 MHz) δ 8.41 (s, 1H), 8.12 (d, 1H, J = 7.8 Hz), 7.95-7.84 (m, 3H), 7.73 (d, 2H, J = 8.3 Hz), 7.59 (t, 1H, J = 7.8 Hz), 7.50-6.95 (m, 5H), 5.30 (br, 1H), 4.37 (brd, 1H, J = 13.8 Hz), 3.48-2.60 (m, 3H), 2.05-1.65 (m, 6H). 13C NMR (CDCl3, 151 MHz) δ 176.95, 171.95, 149.63, 146.22, 141.22, 140.27, 138.29, 132.54, 130.30, 129.54, 129.51, 129.45, 129.07, 129.03, 127.99, 126.94, 126.63, 121.03, 57.75, 41.27, 36.98, 29.23, 25.64, 19.21. HRMS calculated for C28H27N4O3 [M+H]+ 467.2078, found 467.2077.
2-AG substrate hydrolysis assay
The activity of ABHD6 was determined using recombinant mouse ABHD6 protein overexpressed in HEK293T cells using a previously described 2-AG LC-MS hydrolysis assay with some minor modifications.3 HEK293T-ABHD6 membrane lysates were diluted to 0.2 mg/mL in assay buffer (PBS + 0.05% Triton X-100, 50 μL reaction volume). Lysates were treated with DMSO or compound for 30 min at 37 °C. The substrate was prepared by sonicating 2-arachidonoylglycerol (2-AG) in PBS + 0.05% Triton X-100. The substrate was added to the sample reaction (50 μL, 100 μM final concentration of 2-AG) and incubated for 30 min at 37 °C. The reaction was quenched by adding 300 μL of 2:1 v/v CHCl3:MeOH, doped with 1 nmol of pentadecanoic acid standard, vortexed and then centrifuged (1,400 × g, 3 min) to separate the phases. The organic phase was subjected to LC-MS analysis and arachidonic acid release was quantified as previously described.3
Preparation of cell line proteomes
HEK293T and Neuro2A cells were grown in DMEM supplemented with 10% fetal bovine serum at 37 °C with 5% CO2. For in vitro experiments, cells were grown to 80-90% confluency, washed twice with cold PBS (pH 7.5) and scraped. Cell pellets were then isolated by centrifugation at 1,400 × g, for 3 min at 4 °C. The pellets were resuspended in 500 μL of cold PBS (pH 7.5), sonicated, and centrifuged (100,000 × g, 45 min) to generate soluble and membrane fractions. Mouse DAGLβ was transiently overexpressed in HEK293T cells as previously described.8 Total protein concentration of membrane and soluble fractions was determined using a protein assay kit (Bio-Rad). Samples were stored at -80 °C until use.
Preparation of mouse tissue proteomes
Brains and livers from C57Bl/6 mice were Dounce-homogenized in PBS, pH 7.5, followed by a low-speed spin (1,400 × g, 5 min) to remove debris. The supernatant was then centrifuged at 100,000 × g, for 45 min to generate the cytosolic fraction in the supernatant and the membrane fraction as a pellet. The pellet was washed and resuspended in PBS buffer by sonication. The total protein concentration in each fraction was determined using a protein assay kit (Bio-Rad). Samples were stored at -80 °C until further use.
Gel-based competitive ABPP
Gel-based competitive ABPP experiments were performed as previously described.1, 8, 20 Proteomes (1 mg/mL) were pretreated with compounds at indicated concentrations (30 min, 37 °C) followed by labeling with either FP-rhodamine or HT-01 (1 μM final concentration) in a 50 μL total reaction volume. After 30 min or 1 hr at 37 °C, the FP-rhodamine- or HT-01-labeled reactions, respectively, were quenched with SDS-PAGE loading buffer. After separation by SDS-PAGE (10% acrylamide), samples were visualized by in-gel fluorescence scanning using a flatbed fluorescent scanner (Hitachi FMBio IIe). For measurement of recombinant DAGLβ activity, proteomes were diluted to 0.3 mg/mL in assay buffer (50 mM HEPES, pH 7.2, 100 mM NaCl, 5 mM CaCl2, 0.1% v/v TX-100, 10% v/v DMSO) and subjected to ABPP analysis as described above.
Abpp-Silac
Neuro2A cells were grown for 10 passages in either light or heavy SILAC DMEM medium supplemented with 10% (v/v) dialyzed FCS and 2 mM L-glutamine. Light medium was supplemented with 100 μg/mL L-arginine and 100 μg/mL L-lysine while heavy medium was supplemented with 100 μg/mL [13C615N4]-L-Arginine and 100 μg/mL [13C615N2]-L-Lysine. Heavy cells (in 10 mL medium) were treated with test compound and light cells were treated with DMSO for 4 hr at 37 °C. Cells were washed with PBS (2×), harvested, and lysed by sonication in DPBS. Membrane and soluble proteomes were isolated as described above and adjusted to a final concentration of 2.0 mg/mL and labeled with 10 μM FP-biotin (500 μL total reaction volume) for 2 hr at 25 °C. After incubation, light and heavy proteomes were mixed in 1:1 ratio, and excess FP-biotin removed by CHCl3/MeOH extraction. To the light and heavy mixed proteomes (1 mL volume), we added 2 mL MeOH, 0.5 mL CHCl3, 1.5 mL H2O, vortexed, and centrifuged at 1,400 × g for 3 min. The top (aqueous) and bottom (organic) layers were removed and 600 μL of MeOH was added to the protein interface, which was subsequently transferred to a microfuge tube. Next, 150 μL of CHCl3 and 600 μL of H2O was added, vortexed, and centrifuged as described above. The top and bottom layers were removed, the protein interface sonicated in 600 μL of MeOH, and then centrifuged at 14,000 rpm for 5 min to pellet protein. The MeOH was removed and pellet resuspended in 6 M urea/25 mM ammonium bicarbonate. Samples were then reduced with 10 mM DTT for 15 min (65 °C) and alkylated with 10 mM iodoacetamide for 30 min at 25 °C in the dark. Afterwards, samples were treated with SDS (2% final, 5 min) and biotinylated proteins enriched with avidin beads (50 μL beads; conditions: 1 hr, 25 °C, 0.5% SDS in PBS). The beads were washed three times with 1% SDS in PBS followed by three washes with PBS. The urea concentration was reduced to 2 M with 2x volume DPBS. On-bead digestions were performed for 12 hr at 37 °C with sequence-grade modified trypsin (Promega; 2 μg) in the presence of 2 mM CaCl2. Peptide samples were acidified to a final concentration of 5% (v/v) formic acid and stored at -80 °C prior to analysis. LC-MS/MS analysis of ABPP-SILAC samples were performed as previously described.8, 15, 16
In vivo studies with compounds 9, 11, and 20
Mice were injected with compounds 9, 11, or 20 i.p. in 18:1:1 (v/v/v) solution of saline/ethanol/PEG40 (ethoxylated castor oil, 10 μl g-1). For p.o. studies with 11, mice were administered compound by oral gavage in PEG300 (4 μl g-1). For dose-response studies, mice were treated with varying doses of compounds for 4 hr, anesthetized with isoflurane, and euthanized by cervical dislocation. Animal experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Determination of IC50 values
For in vitro analyses, proteomes were pretreated with inhibitor for 30 min at 37 °C at the indicated concentrations (n = 3) followed by labeling with HT-01 or FP-rhodamine (1 μM probe) for 30 min at 37 °C. For in situ measurements, Neuro2A cells were treated with varying concentrations of compound (n = 3) in serum-free media (4 hr, 37 °C with 5% CO2), lysed, and proteomes prepared for ABPP analysis as described above. After quenching, SDS-PAGE, and in-gel visualization, the percentage of enzyme activity remaining was determined by measuring the integrated optical intensity of the bands using ImageJ software. Nonlinear regression analysis was used to determine the IC50 values from a dose-response curve generated using GraphPad Prism.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grants DA017259 (B.F.C.) and DA033760 (B.F.C.), a Hewitt Foundation Postdoctoral Fellowship (K.L.H.), the Skaggs Institute for Chemical Biology, and Dainippon Sumitomo Pharma (K.T.)
Abbreviations Used
- ABPP
activity-based protein profiling
- SH
serine hydrolase
- 2-AG
2-arachidonoylglycerol
- Pip-1,2,3-TU
piperidyl-1,2,3-triazole urea
- CuAAC
copper-catalyzed azide alkyne cycloaddition
- FP-Rh
fluorophosphonate-rhodamine
- SILAC
stable isotope labeling by amino acids in cell culture
Footnotes
Supporting Information: Supplemental methods, synthetic procedures and characterization of 1,2,3-triazole ureas, 1H NMR spectra, HRMS data, and supplemental figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare the following competing financial interest(s): Dr. Cravatt is a founder and adviser to Abide Therapeutics a biotechnology company interested in developing serine hydrolase inhibitors as drugs to treat human disease.
References
- 1.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blankman JL, Simon GM, Cravatt BF. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 5.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tchantchou F, Zhang Y. Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury. J Neurotrauma. 2013;30:565–579. doi: 10.1089/neu.2012.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navia-Paldanius D, Savinainen JR, Laitinen JT. Biochemical and pharmacological characterization of human alpha/beta-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12) J Lipid Res. 2012;53:2413–2424. doi: 10.1194/jlr.M030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol. 2012;8:999–1007. doi: 10.1038/nchembio.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu KL, Tsuboi K, Whitby LR, Speers AE, Pugh H, Inloes J, Cravatt BF. Development and optimization of piperidyl-1,2,3-triazole ureas as selective chemical probes of endocannabinoid biosynthesis. J Med Chem. 2013 doi: 10.1021/jm400898x. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu KL, Tsuboi K, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P, Rosen H. Probe Reports from the NIH Molecular Libraries Program [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2012. Optimization and characterization of triazole urea inhibitors for abhydrolase domain containing protein 6 (ABHD6) updated Mar 14 2013. [PubMed] [Google Scholar]
- 11.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Speers AE, Adam GC, Cravatt BF. Activity-Based Protein Profiling in Vivo Using a Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 13.Nagano JM, Hsu KL, Whitby LR, Niphakis MJ, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder P, Srinivasan P, Gonzalez TD, Rosen H, Bahnson BJ, Cravatt BF. Selective inhibitors and tailored activity probes for lipoprotein-associated phospholipase A(2) Bioorg Med Chem Lett. 2013;23:839–843. doi: 10.1016/j.bmcl.2012.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalisiak J, Sharpless KB, Fokin VV. Efficient Synthesis of 2-Substituted-1,2,3-triazoles. Org Lett. 2008;10:3171–3174. doi: 10.1021/ol8006748. [DOI] [PubMed] [Google Scholar]
- 15.Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Click-generated triazole ureas as ultrapotent in vivo–active serine hydrolase inhibitors. Nat Chem Biol. 2011;7:469–478. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc Natl Acad Sci U S A. 2011;108:6811–6816. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 20.Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.