

Abstract
Rhodopsin, the membrane protein responsible for dim-light vision, until recently was the only G protein-coupled receptor (GPCR) with a known crystal structure. As a result, there is enormous interest in studying its structure, dynamics, and function. Here we report the results of three all-atom molecular dynamics simulations, each at least 1.5 microseconds, which predict that substantial changes in internal hydration play a functional role in rhodopsin activation. We confirm that the increased hydration is specific to the Meta-I intermediate with 1H magic angle spinning NMR. The internal waters interact with several conserved residues, suggesting that changes in internal hydration may be important during the activation of other GPCRs. The results serve to illustrate the synergism of long timescale molecular dynamics simulations and NMR in enhancing our understanding of GPCR function.
Introduction
The G protein-coupled receptors (GPCRs) are arguably the single most important family of membrane proteins known. They are the largest superfamily in the human genome, and are of enormous biological 1; 2; 3 and pharmaceutical importance 3. However, until the structure of the β2 adrenergic receptor was published earlier this year,4 only the dim-light receptor rhodopsin was known to atomic-resolution 5; 6; 7; 8; 9; 10; 11. As a result, a great deal of effort has been expended to explore rhodopsin’s activation 1.
The basic mechanism by which rhodopsin is activated is well understood: dark-state rhodopsin is stabilized by the presence of an 11-cis retinal moiety bound via a protonated Schiff base linkage to Lys-296 in the protein interior. Upon absorption of a photon, retinal isomerizes to the all-trans form, and the protein relaxes via a series of intermediate states to Metarhodopsin-I (MI), an inactive state which exists in equilibrium with the active Metarhodopsin-II (MII) state. A number of intermediate states have been identified largely via vibrational and NMR spectroscopy experiments, often at low temperature, although X-ray structures for Lumirhodopsin (formed within the first few hundred nanoseconds) and an MII-like state were recently published 12; 13, as well as a low-resolution electron-cryomicroscopy structure of MI 9.
However, at the atomic level, many details of the activation process remain unclear. In particular, two contrasting models have emerged describing the role of two highly conserved glutamates (Glu-181 and Glu-113) in activation (see Figure 1). Both models assert that the protonated Schiff base is associated with Glu-113 in the dark state, and that this interaction is diminished upon formation of MI. However, the first mechanism, articulated by Yan et al. 14, postulates that Glu-181 is protonated in the dark state, and that during the activation process the proton is transferred to Glu-113, resulting in the Schiff base switching counterions. By contrast, Lüdeke et al. 15 suggest that both residues are charged throughout activation, and that MI formation is accompanied by the formation of a complex counterion. We will refer to these two competing models as the “counterion switch” and “complex counterion”.
Figure 1.

Proton transfer mechanisms in the formation of MI. We test two separate models for proton transfer in the retinal binding pocket after photoactivation. The complex counterion model 14 begins with Glu-181 protonated and Glu-113 deprotonated; after retinal isomerizes, a proton is transferred from Glu-181 to Glu-113, leading to the counterion switch in MI. In the complex counterion model15), Glu-181 and Glu-113 are both charged until MII formation.
In this work we present three microsecond-scale all-atom molecular dynamics simulations of rhodopsin in lipid bilayers. Specifically, we ran simulations of 1.5 and 2 μs to model the complex counterion and counterion switch mechanisms, applying an additional potential to the cis double bond of retinal to induce isomerization to the all-trans form, emulated the effects of light absorption. We also ran an additional 1.5 μs simulation of dark state rhodopsin, to serve as a control. These simulations are based on careful development of methodologies for modeling the role of polyunsaturated lipids 16; 17 and cholesterol 18 in rhodopsin function 19. Although other groups have performed molecular dynamics simulations of activated rhodopsin 20; 21; 22; 23, the present trajectories are at least an order of magnitude longer, and are thus able to explore more of the protein’s relaxation after retinal isomerization, yielding new insights into the protein’s path toward activation. Indeed, in both simulations the retinal moiety’s behavior is consistent with the MI formation mechanism predicted by the underlying activation model; in the counterion switch trajectory, the protonated Schiff based cleanly switches from Glu-113 to Glu-181, while a complex counterion is formed in the other. Comparison of the simulations to 2H NMR experiments suggests that the latter mechanism is more likely to be correct 24. Thus, we believe the complex counterion simulation reaches a reasonable approximation of the MI state, even though experimental measurements suggest that MI formation should take significantly longer25; this discrepancy may indicate flaws in the long timescale kinetics of molecular dynamics force fields, which have yet to be extensively validated. For this reason, we prefer to focus on structural comparisons.
The simulations also make a startling new prediction: in both activated trajectories, rhodopsin activation is accompanied by a significant increase in internal hydration. We confirm this surprising result experimentally, using magic angle spinning (MAS) 1H NMR experiments on bovine rod outer segment disks to demonstrate that hydration is selectively enhanced in the MI state. This observation, combined with the unprecedented atom-level description of the photointermediates, demonstrates the power of molecular dynamics to generate new insights into GPCR activation.
Results and Discussion
The intermediate states in rhodopsin’s activation were defined using optical methods, with the result that detailed structural information is somewhat limited. Although the situation has improved recently, with the release of a crystal structure for Lumirhodopsin (Lumi) 12 and a low-resolution MI structure 9, this still suggests an opportunity for long timescale molecular dynamics to make significant contributions. In order to do so credibly, especially given the unprecedented scale of the calculations, the simulations must first be shown to be consistent with experimental results. For example, Borhan et al. 26 used crosslinking experiments with modified retinals to conclude that retinal’s ionone ring approaches Ala-169 during Lumi formation and remains there in MI and MII. We see a similar migration of the ionone ring in both activated simulations, but not in the simulation of the dark state, where it remains in contact with Trp 265. Further, we can track MI formation by examining the interaction of the protonated Schiff base with its counterion. As shown by Figure 2, the mechanism differs significantly in the two trajectories. Figure 2a shows the time course for the separations between the two glutamates and the Schiff base in the counterion switch simulation, where the proton was explicitly moved from Glu-113 to Glu-181; the proton exchange (manually applied 500 ns after retinal isomerization) immediately induces a small reduction of the distance to Glu-181. Roughly 500 ns later, the tight association between Glu-113 and the Schiff base breaks, and within 50 ns a new salt bridge forms with Glu-181. The complex counterion simulation produces a very different result, as shown in Figure 2c: the salt bridge between Glu-113 and the Schiff base breaks almost immediately after isomerization, and after a ~500 ns migration period, the Schiff base is shared by the two charged glutamate side chains, although Glu-181 is on average roughly 1 Å closer. In contrast to the other simulation, no discrete switching event occurs. Although we only have one trajectory for each mechanism, that fact that both behave exactly as predicted by the underlying MI formation model strongly suggests that both activated simulations reach an approximation of MI. This is encouraging, but does not help distinguish which of the two models is correct. In order to draw this distinction, we used the orientation of the retinal in the latter part of each simulation to compute theoretical 2H NMR spectra, which we then compared to experiment 24. The counterion switch simulation produces theoretical spectra very different from those measured experimentally, while spectra computed from the complex counterion trajectory match the experiment very well. It is on the basis of this structural comparison with experiment, and not the time scale of the simulation, that we conclude that the complex counterion trajectory is sampling MI-like conformations.
Figure 2.

The counterion switch and internal hydration. Parts A and C track the time evolution of the distances between the ionone ring and Glu-113 and -181, for the counterion switch and complex counterion models respectively. Parts B and D show the number of water molecules inside the protein cavity for the counterion switch and complex counterion simulations, respectively. Part D also contains the equivalent time series for a control simulation of dark state rhodopsin. The number of internal waters at t=0 differs in the two simulations because the number of waters fluctuated during dark state equilibration.
Several internal waters were resolved in the X-ray crystal structures of dark state rhodopsin7. These waters are thought to play an important structural role7, stabilizing several charged or polar side chains which otherwise lack hydrogen bonding partners in the dark state27. However, Figures 2b and 2d show the most surprising result in the simulations: in both activated simulations, retinal isomerization is followed by a dramatic increase of internal hydration. Although the effect is more dramatic in the simulation of the complex counterion (Figure 2d), perhaps because of the presence of the extra charged side chain, the counterion switch trajectory also shows a significant increase in the number of waters inside the protein compared to the dark state. The dramatic increase in the number of internal waters seems to be directly related to the Schiff base-counterion interaction: in the counterion switch simulation, the number of internal waters increases rapidly beginning almost immediately after formation of the salt bridge between the Schiff base and Glu-181, while in the complex counterion trajectory the internal hydration process begins concurrently with the breaking of the Glu-113-Schiff base salt bridge, shortly after retinal isomerization. Figure 2d also shows the equivalent time series for dark state rhodopsin with both glutamates charged, a control for the complex counterion simulation; although internal hydration increases slightly over the first 300 ns or so, it then remains roughly constant over an additional microsecond of dynamics; unlike the activated trajectories, in this simulation the waters do not significantly interact with the retinal. The initial increase is not surprising, given that crystallography will identify only the most ordered water molecules in the system. Interestingly, water entry does not significantly perturb the overall structure of the protein; the RMS deviation of the backbone from the starting point remains under 3 Å, mostly due to the high flexibility of several loops and the C-terminal tail and consistent with experimental evidence that the protein core does not undergo rigid-body transformations upon entering MI 9.
Figure 3, a snapshot from the complex counterion simulation roughly 1 μs after activation, clearly shows that the hydration of the protein core is qualitatively different from that found in the crystal structures. The retinal moiety has become mostly hydrated, which is not the case in either the crystal structures or the control simulation. This water seems to play a functional role; it appears to drive Trp-265 away from the retinal ionone ring, allowing the ring to shift to its MI-like orientation. Given the importance of Trp-265 in stabilizing the dark state and its high degree of conservation across the class A family of GPCRs 28, these results suggests that the role played by water here may not be unique to rhodopsin. Along the same lines, much of the water entered the protein core via the highly conserved NPxxY and D(E)RY motifs on the cytoplasmic face, again suggesting that this mechanism could be relevant to other GPCRs.
Figure 3.

Internal water molecules solvate retinal during MI formation. A snapshot from the 1 μs point of the complex counterion simulation is shown, depicting the large number of waters inside the protein cavity and their interaction with the retinal. Trp-265 (green) has moved away from the retinal (yellow) head group. The image was created using PYMOL50.
The dramatic increase of internal hydration upon MI formation is surprising, but not totally without precedent in the literature. For example, several groups have noted changes in solvent accessibility associated with protein activation7; 29; 30; 31; 32; 33; 34, and in particular Mitchell and Litman demonstrated that there are differences in hydration between the MI and MII states35; 36 However, the notion that thirty or more waters enter the protein core as the protein transforms from the dark state to MI is novel, and to our knowledge has not been previously seen experimentally.
For this reason, we used 1H magic angle spinning (MAS) NMR, using rhodopsin in intact rod outer segment disks to track changes in rhodopsin hydration upon activation. Specifically, we measured magnetization transfer from water to the lipid chains via the protein, as summarized by the middle panel of Figure 437; 38. Saturating the water magnetization in the presence of rhodopsin leads to attenuation of NMR peaks of lipid hydrocarbon chains. This is conveniently detected at 2.8 ppm, the resonance frequency for the methylene protons of carbons 6, 9, 12, 15, and18 on the polyunsaturated chain. A comparison of the relevant portions of the spectra is shown in the bottom panel of Figure 4 (see Figure S1 for an example difference spectrum). The effect, which is absent when a lipid matrix without protein is used, is enhanced upon rhodopsin activation. Specifically, Figure 5 shows the time course of the relative attenuation of the 2.8 ppm resonance, which we interpret as indicative of the internal hydration of the protein. At pH 8, where [MI]/[MII] ≈ 0.71, the relative attenuation increases immediately after exposure to light. By contrast, the effect is absent at pH 7, where the population of MI is very small. This suggests that the increased hydration is specific to the MI state. The enhanced attenuation decays with a time constant of 8.9±2.4 minutes, consistent with the 8.2 minute decay time for MIII formation measured by spectrophotometry39. Although this experiment does not directly determine where on the protein hydration changes, magnetization transfer is most efficient when the interactions are both short-range (< 5 Å) and long-lived (1–10 ns), conditions best satisfied by hydration of the protein core. Such hydration is unlikely to occur in the loops, which are flexible, located far from the membrane’s hydrophobic core, and do not undergo significant conformational change during MI formation. By contrast, the simulations indicate that internal water exchanges with bulk on the 10–50 ns time scale, which is simultaneously slow enough for efficient magnetization transfer from water to the protons of rhodopsin, and fast enough that many generations of water are exchanged during the 0.5 s saturation pulse.
Figure 4.

1H NMR magnetization transfer experiment. The top portion shows the pulse sequence used in the experiment. The middle portion illustrates the route of magnetization transfer, from the water through the protein to the lipid. The lower panel shows the 1H MAS NMR spectrum of rod outer segment disk membranes before (solid line) and immediately after photoactivation of rhodopsin (dashed line) as described below. Immediately after photoactivation a significant increase (up to 11%) in the saturation of lipid proton resonances was observed. Reproducibility of signal intensities was 1%. The effect was recorded as a function of time by recording intensity of the well-resolved =CH-CH2-CH= resonance (2.8 ppm) of polyunsaturated hydrocarbon chains (see insert, and Fig. 4). Attenuation increased also for other resonances, in particular those with contributions to intensity from rhodopsin protons. See Figure S1 in supplemental information for an example difference spectrum.
Figure 5.
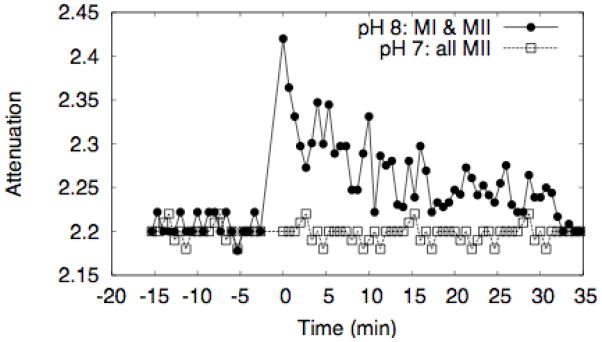
Time dependence of magnetization transfer from water to DHA via rhodopsin. The data shows the attenuation of the polyunsaturated resonance at 2.8 ppm. The open squares were measured at pH 7, where the MI/MII equilibrium strongly favors MII, while the filled circles were measured at pH 8 and T=20° C, where [MI]/[MII] ≈ 0.71. Photoactivation occurs at t=0.
The attenuation effect exists in dark state rhodopsin, consistent with the presence of water in the crystal structures, and with the control simulation presented here, which indicates that there are always roughly 10–20 waters inside the protein cavity, which exchange with the bulk on the timescale of tens of nanoseconds. However, the 2.8 ppm signal is not attenuated if lipid bilayers with a composition similar to ROS disk membranes but without rhodopsin are used in the experiment. Similarly, varying the pH without bleaching the rhodopsin did not affect magnetization transfer either, confirming that the effect is not due to the pH dependence of generic proton exchange between water and hydroxyl and amino groups on rhodopsin, but rather is due to pH effects on the conformational equilibrium of rhodopsin. Saturating the protein signal directly leads to attenuation of the hydrocarbon chain resonances, in particular those associated with polyunsaturated lipids but no differences were detected between dark adapted rhodopsin, MI and MII, indicating that the signal is not due to changes in lipid-protein packing in MI or MII38. Taken together, these controls confirm that the pH dependence demonstrated in Figure 5 is due to systematic changes in hydration during MI formation. Moreover, the reduction in hydration during the MI to MII transition measured in the experiment is consistent with the osmotic stress experiments of Mitchell and Litman36.
Recent experimental work has suggested that MI forms on a time scale significantly longer than that of our simulations25. Thus, it is possible that our simulation has not reached the full MI state, but is merely moving along the path toward MI. In this view, the simulation’s two main predictions – the retinal orientation consistent with 2H NMR and the increased hydration seen in the 1H NMR experiments – would have to be relatively early events in MI formation, and not the final determinants of the absorption spectrum; given the lack of information about the atomic details of MI formation, this hypothesis is not unreasonable. Alternatively, the experiments measure the net result of an ensemble of transition times, with the result that, as in single molecule experiments, a single trajectory’s timescale may not appear representative. Finally, it should be noted that force field development has historically focused on reproducing biomolecular structure and energetics, while the scarcity of microsecond-scale all-atom simulations means that their long timescale kinetic properties have not been tested extensively. For these reasons, we did not attempt to define the protein’s state kinetically, and instead focused on the structural validation of the simulation via comparison to the NMR results. This comparison suggests that the changes in retinal’s structure and environment seen in the complex counterion simulation are representative of MI40.
Conclusions
Despite enormous experimental and theoretical efforts, many details of rhodopsin activation remain poorly understood. In the present work, we have used molecular dynamics on an unprecedented scale41, combined with NMR experiments, to reveal a new feature of rhodopsin’s photocycle, a dramatic increase in the internal hydration upon moving from the dark state to MI. It appears likely that this water will play a functional role in the rest of the photocycle, and further experiments and simulations will be needed to clarify the implications of this finding for our understanding of the activation of rhodopsin and other GPCRs.
Methods
System Construction and Simulation Details
Both simulations consisted of a single rhodopsin molecule embedded in a bilayer containing a 2:2:1 mixture of SDPC:SDPE:cholesterol. Specifically, there were 49 SDPC molecules, 50 SDPE molecules, and 24 cholesterols, along with 7,400 water molecules, 14 sodium ions, and 16 chloride ions, for 43,222 atoms total. The “complex counterion” simulation was begun starting from the end of our previous 118 ns dark adapted simulation 18. The “counterion switch” simulation was constructed using the highest resolution crystal structure (PDB Code 1U19) using the protocol detailed in Grossfield et al. 16, and equilibrated for roughly 70 ns in the dark state. During this time, the number of the water molecules found inside the protein fluctuated significantly, although there was no systematic change. The control simulation of the dark state was constructed in a manner identical to the “counterion switch” simulation, except with both Glu-181 and Glu-113 charged; the first 100 ns of this simulation was part of the work published in Grossfield et al. 16.
In the activated simulations, isomerization of the 11-cis retinal to all-trans was achieved by applying a modified torsion term with a single minimum at the trans position and a barrier height of 60 kcal/mol to the 11–12 bond of retinal. Isomerization occurred within the first half picosecond. The modified torsion was not used for the remainder of the calculation.
After 500 ns post-flash dynamics in the counterion switch simulation, a proton was explicitly transferred from Glu-181 to Glu-113, in accord with the activation model of Yan, et al. 14 The manual procedure was required because in a classical molecular dynamics simulation, bonds are represented as harmonic springs, which prevents proton dissociation. Both glutamate sidechains were in contact with water at the time of transfer.
Electrostatic interactions were evaluated using the particle-particle-particle-mesh Ewald summation technique, using a 1283 grid for the fast Fourier transform, a charge-interpolation distance of 4 mesh points, and the Ewald α value set to 0.35 Å−1. Repulsion-dispersion interactions were smoothly truncated at 10 Å, and the CHARMM all-atom force field was used throughout, with waters represented using the TIP3P water model42; 43; 44. All bonds involving hydrogen were constrained to their equilibrium length using the RATTLE45 algorithm, with the tolerance set to 10−10, enabling us to run the dynamics with a 2 fs timestep using the velocity Verlet integrator 46. The simulations were performed in the NVE ensemble, in a rectilinear periodic box (55 × 77 × 102.65 Å for the complex counterion simulation, 56.5 × 79.2 × 95.5434 Å for the counterion switch and control simulations). The starting temperature for each simulation was 310 K, and drifted less than 1K over the course of the simulations. Production dynamics was performed on the Blue Gene/W supercomputer 47; 48 located at the T.J. Watson Research Center, typically on 4096 nodes, using Blue Matter, a simulation package developed at IBM specifically to take advantage of the Blue Gene architecture 48.
Internal waters were identified by constructing a rectangular 24 × 27 × 30 Å box centered on the protein, and counting the number of waters inside the box over the course of the trajectories. These dimensions were chosen because they adequately covered the dimensions of the protein interior, and although the absolute number of waters varied slightly with different definitions of the box, the overall results are robust.
1H MAS NMR Details
Intact disk-membranes were isolated from rod outer segments (ROS) of bovine retinas as described by Litman 49. Samples were kept at 4°C, and all manipulations were performed in the dark or under dim red light. Disk membranes were transferred into buffer containing 10mM Tris-HCl, 60 mM KCl, 30 mM NaCl, 2mM MgCl2, 50 μM DTPA, 1.5 mM DTT, 1.5 mg/mL aprotinin, pH = 8.0 or 7.0.
The 1H MAS NMR spectra were acquired at a resonance frequency of 500.13 MHz on a Bruker DMX500 spectrometer equipped with a triple resonance 1H/13C/2H, 4 mm MAS probehead (Bruker Biospin, Inc., Billerica, MA). Experiments were conducted at a MAS spinning frequency of 10 kHz, using the pulse sequence shown in Figure 4, with ten (n=10) Gaussian-shaped saturation pulses of length l=50 ms at a rf field strength of 0.12 kHz, and a 1H π/2 pulse length of 4 μs. The saturation frequency was set to the water resonance at 4.8 ppm. Typically four scans with a recycle delay of 10 s were acquired. Samples in the spinning MAS rotor were illuminated for one minute with light from a miniature light bulb inserted into the MAS stator.
Supplementary Material
Acknowledgments
We thank the IBM Watson Research Center and the Computational Biology Center for use of the BGW Blue Gene supercomputing facility. We extend a special thanks to the Blue Matter development team (B. Fitch, R. Germain, A. Rayshubskiy, T. J. C. Ward, M. Eleftheriou, F. Suits, Y. Zhestkov, R. Zhou, J. Pitera, and W. Swope). K.G. and O.S. were supported by the Intramural Research Program of NIAAA, NIH. S.E.F. thanks the NSF and NIH for support through Awards MCB-0543124 and PHS 2 PN2 EY016570B, respectively.
References
- 1.Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Prot Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 2.Lefkowitz RJ. Historical review: a brief history and personal restropective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;3:928–44. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Rasmussen SGF, Choi H-J, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Crystal structure of the human beta-2 adrenergic G-protein-coupled receptor. Nature. 2007 doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–38. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 6.Okada T. X-ray crystallographic studies for ligand-protein interaction changes in rhodopsin. Biochem Soc Trans. 2004;32:738–41. doi: 10.1042/BST0320738. [DOI] [PubMed] [Google Scholar]
- 7.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci U S A. 2002;99:5982–7. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 9.Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. Embo J. 2004;23:3609–20. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs) Biochemistry. 2001;40:7761–72. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel R, Ruprecht J, Villa C, Mielke T, Schertler GF, Siebert F. Rhodopsin photoproducts in 2D crystals. J Mol Biol. 2004;338:597–609. doi: 10.1016/j.jmb.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Nakamichi H, Okada T. Local peptide movement in the photoreaction intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103:12729–12734. doi: 10.1073/pnas.0601765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan EC, Kazmi MA, Ganim Z, Hou JM, Pan D, Chang BS, Sakmar TP, Mathies RA. Retinal counterion switch in the photoactivation of the G protein-coupled receptor rhodopsin. Proc Natl Acad Sci U S A. 2003;100:9262–7. doi: 10.1073/pnas.1531970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ludeke S, Beck M, Yan EC, Sakmar TP, Siebert F, Vogel R. The role of Glu181 in the photoactivation of rhodopsin. J Mol Biol. 2005;353:345–56. doi: 10.1016/j.jmb.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 16.Grossfield A, Feller SE, Pitman M. A role for direct interactions in the modulation of rhodopsin by omega-3 polyunsaturated lipids. Proc Natl Acad Sci U S A. 2006;103:4888–4893. doi: 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossfield A, Feller SE, Pitman MC. Contribution of omega-3 Fatty acids to the thermodynamics of membrane protein solvation. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:8907–9. doi: 10.1021/jp060405r. [DOI] [PubMed] [Google Scholar]
- 18.Pitman MC, Grossfield A, Suits F, Feller SE. Role of cholesterol and polyunsaturated chains in lipid-protein interactions: Molecular dynamics simulation of rhodopsin in a realistic membrane environment. J Am Chem Soc. 2005;127:4576–4577. doi: 10.1021/ja042715y. [DOI] [PubMed] [Google Scholar]
- 19.Grossfield A, Feller SE, Pitman M. Convergence of molecular dynamics simulations of membrane proteins. Proteins: Struct Funct Gen. 2006;67:31–40. doi: 10.1002/prot.21308. [DOI] [PubMed] [Google Scholar]
- 20.Lemaitre V, Yeagle P, Watts A. Molecular dynamics simulations of retinal in rhodopsin: from the dark-adapted state towards lumirhodopsin. Biochemistry. 2005;44:15082–15082. doi: 10.1021/bi0506019. [DOI] [PubMed] [Google Scholar]
- 21.Rohrig UF, Guidoni L, Laio A, Frank I, Rosthlsberger U. A molecular spring for vision. J Am Chem Soc. 2004;126:15328–15329. doi: 10.1021/ja048265r. [DOI] [PubMed] [Google Scholar]
- 22.Saam J, Tajkhorshid E, Hayashi S, Shulten K. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys J. 2002;83:3097–3112. doi: 10.1016/S0006-3495(02)75314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crozier PS, Stevens MJ, Woolf TB. How a small change in retinal leads to G-protein activation: initial events suggested by molecular dynamics calculations. Proteins: Struct Funct Gen. 2006;66:559–574. doi: 10.1002/prot.21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Mayorga K, Pitman MC, Grossfield A, Feller SE, Brown MF. Retinal counterion switch mechanism in vision evaluated by molecular simulations. J Am Chem Soc. 2006;128:16502–16503. doi: 10.1021/ja0671971. [DOI] [PubMed] [Google Scholar]
- 25.Epps J, Lewis JW, Szundi I, Kliger DS. Lumi I -> Lumi II: The last detergent independent process in rhodopsin photoexcitation. Photochem Photobio. 2006;82:1436–1441. doi: 10.1562/2006-02-01-RA-792. [DOI] [PubMed] [Google Scholar]
- 26.Borhan B, Souto ML, Imai H, Shichida Y, Nakanishi K. Movement of retinal along the visual transduction path. Science. 2000;288:2209–12. doi: 10.1126/science.288.5474.2209. [DOI] [PubMed] [Google Scholar]
- 27.Patel AB, Crocker E, Reeves PJ, Getmanova EV, Eilers M, Khorana HG, Smith SO. Changes in interhelical hydrogen bonding upon rhodopsin activation. J Mol Biol. 2005;347:803–12. doi: 10.1016/j.jmb.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 28.Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, Smith SO. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J Mol Biol. 2006;357:163–72. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 29.Altenbach C, Klein-Seetharaman J, Hwa J, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 59–75 connecting helices I and II in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38:7945–9. doi: 10.1021/bi990014l. [DOI] [PubMed] [Google Scholar]
- 30.Cai K, Klein-Seetharaman J, Farrens D, Zhang C, Altenbach C, Hubbell WL, Khorana HG. Single-cysteine substitution mutants at amino acid positions 306–321 in rhodopsin, the sequence between the cytoplasmic end of helix VII and the palmitoylation sites: sulfhydryl reactivity and transducin activation reveal a tertiary structure. Biochemistry. 1999;38:7925–30. doi: 10.1021/bi9900119. [DOI] [PubMed] [Google Scholar]
- 31.Klein-Seetharaman J, Hwa J, Cai K, Altenbach C, Hubbell WL, Khorana HG. Single-cysteine substitution mutants at amino acid positions 55–75, the sequence connecting the cytoplasmic ends of helices I and II in rhodopsin: reactivity of the sulfhydryl groups and their derivatives identifies a tertiary structure that changes upon light-activation. Biochemistry. 1999;38:7938–44. doi: 10.1021/bi990013t. [DOI] [PubMed] [Google Scholar]
- 32.Allan AE, Cooper A. Hydration of retinal and the nature of metarhodopsin II. FEBS Lett. 1980;119:238–40. doi: 10.1016/0014-5793(80)80261-4. [DOI] [PubMed] [Google Scholar]
- 33.Rath P, DeGrip WJ, Rothschild KJ. Photoactivation of rhodopsin causes an increased hydrogen-deuterium exchange of buried peptide groups. Biophys J. 1998;74:192–8. doi: 10.1016/S0006-3495(98)77779-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rafferty CN, Shichi H. The involvement of water at the retinal binding site in rhodopsin and early light-induced intramolecular proton transfer. Photochem Photobiol. 1981;33:229–34. doi: 10.1111/j.1751-1097.1981.tb05329.x. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell DC, Litman BJ. Effect of protein hydration on receptor conformation: decreased levels of bound water promote metarhodopsin II formation. Biochemistry. 1999;38:7617–23. doi: 10.1021/bi990634m. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell DC, Litman BJ. Effect of ethanol and osmotic stress on receptor conformation. Reduced water activity amplifies the effect of ethanol on metarhodopsin II formation. J Biol Chem. 2000;275:5355–60. doi: 10.1074/jbc.275.8.5355. [DOI] [PubMed] [Google Scholar]
- 37.Soubias O, Gawrisch K. Probing specific lipid-protein interaction by saturation transfer difference NMR spectroscopy. J Am Chem Soc. 2005;127:13110–1. doi: 10.1021/ja0538942. [DOI] [PubMed] [Google Scholar]
- 38.Soubias O, Teague WE, Gawrisch K. Evidence for specificity in lipid-rhodopsin interactions. J Biol Chem. 2006 doi: 10.1074/jbc.M603059200. [DOI] [PubMed] [Google Scholar]
- 39.Kibelbek J, Mitchell DC, Beach JM, Litman BJ. Functional equivalence of metarhodopsin II and the Gt-activating form of photolyzed bovine rhodopsin. Biochemistry. 1991;30:6761–8. doi: 10.1021/bi00241a019. [DOI] [PubMed] [Google Scholar]
- 40.Martínez-Mayorga K, Pitman MC, Grossfield A, Feller SE, Brown MF. Retinal counterion switch mechanism in vision evaluated by molecular simulations. J Am Chem Soc. 2006;128:16502–16503. doi: 10.1021/ja0671971. [DOI] [PubMed] [Google Scholar]
- 41.Fitch BG, Germain RS, Mendell M, Pitera J, Pitman M, Rayshubskiy A, Sham Y, Suits F, Swope WC, Ward TJC, Zhestkov Y, Zhou R. Blue Matter, an application framework for molecular simulation on Blue Gene. J Para Distrib Comp. 2003;63:759–773. [Google Scholar]
- 42.Feller SE, MacKerell AD., Jr An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem B. 2000;104:7510–7515. [Google Scholar]
- 43.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher IWE, Roux B, Schlenkrich M, Smith J, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 44.Pitman MC, Suits F, Mackerell AD, Jr, Feller SE. Molecular-level organization of saturated and polyunsaturated fatty acids in a phosphatidylcholine bilayer containing cholesterol. Biochemistry. 2004;43:15318–28. doi: 10.1021/bi048231w. [DOI] [PubMed] [Google Scholar]
- 45.Andersen HC. Rattle: A “Velocity” Version of the Shake Algorithm for Molecular Dynamics Calculations. Journal of Computational Physics. 1983;52:24–34. [Google Scholar]
- 46.Swope WC, Andersen HC, Berens PH, Wilson KR. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: application to small water clusters. J Chem Phys. 1982;76:636–649. [Google Scholar]
- 47.Gara A, Blumrich MA, Chen D, Chiu GL-T, Coteus P, Giampapa ME, Haring RA, Heidelberger P, Hoenicke D, Kopcsay GV, Liebsch TA, Ohmacht M, Steinmacher-Burow BD, Takken T, Vranas P. Overview of the Blue Gene/L system architecture. IBM Journal of Res and Dev. 2005:49. [Google Scholar]
- 48.Fitch BG, Rayshubskiy A, Eleftheriou M, Ward TJC, Giampapa M, Zhestkov Y, Pitman MC, Suits F, Grossfield A, Pitera J, Swope WC, Zhou R, Germain RS, Feller SE IBM. Blue Matter: Strong Scaling of Molecular Dynamics on Blue Gene/L. 2005. [Google Scholar]
- 49.Litman BJ. Purification of rhodopsin by concanavalin A affinity chromatography. Methods Enzymol. 1982;81:150–3. doi: 10.1016/s0076-6879(82)81025-2. [DOI] [PubMed] [Google Scholar]
- 50.DeLano WL. In: The PyMOL Molecular Graphics System. Scientific D, editor. Palo Alto, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.