

Abstract
Objective
The aim was to identify the clinical and laboratory predictors of clinical improvement in a cohort of myositis patients treated with rituximab.
Methods
We analyzed data for 195 myositis patients [75 adult polymyositis/72 adult dermatomyositis/48 juvenile dermatomyositis (JDM)] in the Rituximab in Myositis trial. Clinical improvement was defined as 20% improvement in at least 3 of 6 core set measures (CSM) of disease activity: physician and patient/parent global disease activity, manual muscle testing, physical function, muscle enzymes, and extramuscular disease activity. We analyzed the association of the following baseline variables with improvement: myositis clinical subgroup, demographics, myositis damage, clinical and laboratory parameters, CSM, rituximab treatment, and myositis autoantibodies (anti-synthetase, -Mi-2, -SRP, -TIF1-γ, -MJ, other and no autoantibodies). All measures were univariately assessed for association with improvement using time-to-event analyses. A multivariable time-dependent proportional hazards model was used to evaluate the association of individual predictive factors with improvement.
Results
In the final multivariable model, the presence of an anti-synthetase [primarily anti-Jo-1 (HR 3.08, p<0.01)], anti-Mi-2 (HR 2.5, p<0.01), or other autoantibody (HR 1.4, p=0.14) predicted a shorter time to improvement compared to the autoantibody negative subset. Lower physician global damage (HR 2.32, p< 0.01) and JDM (vs. adult myositis, HR 2.45, p<0.01) also predicted improvement. Unlike the autoantibody subset, the predictive effect of physician global damage and JDM diminished by week 20. Rituximab treatment did not affect these associations.
Conclusion
The presence of an anti-synthetase and anti-Mi-2 autoantibodies, JDM subset and lower disease damage strongly predicted clinical improvement in refractory myositis patients.
Keywords: predictor, clinical improvement, myositis, autoantibody
The idiopathic inflammatory myopathies (IIM) are a group of acquired, heterogeneous, systemic connective tissue diseases (CTD) that include polymyositis (PM), adult dermatomyositis (DM), childhood myositis (predominantly juvenile DM), myositis associated with cancer or another connective tissue disease, and inclusion body myositis (IBM) (1, 2). Over the last few decades, survival has improved in IIM, with patients experiencing less cumulative damage and better health related quality of life. Despite an improvement in survival, our knowledge about clinical and serological predictors of clinical improvement in IIM is limited by the lack of well-designed, long–term epidemiological studies and clinical trials. IIM patients have heterogeneous features from a mild rash to life threatening muscle weakness or lung involvement. Their course can be self-limited or may require long-term glucocorticoids and multiple immunosuppressive medications. The response to immunosuppressive drugs is quite variable and current data do not allow the accurate prediction of clinical improvement, which poses a significant challenge to treating physicians, as well as investigators. The varying clinical features of myositis are closely linked to myositis autoantibodies, some of which may contribute to the pathogenesis of IIM (3). Although these autoantibodies provide useful prognostic information on patient outcomes (4–6), this relationship has not been established in prospective cohorts with uniform treatment. Previous evidence suggested that patients possessing anti-Mi-2 autoantibodies had a better prognosis, while patients with anti-SRP fared worse and those with anti-synthetase autoantibodies had intermediate outcomes (6, 7). In addition, there is a paucity of literature regarding predictors of clinical improvement by IIM disease subgroups. IBM is associated with poor treatment responses but studies differentiating responses between PM, DM and JDM are lacking (6). Treatment delay, muscle damage and longer disease duration have also been shown to be associated with poor prognosis (7–10). However, published studies are limited by small sample sizes, retrospective design and a limited assessment of prognostic factors.
The availability of targeted therapies and validated outcome measures (11–13) spearheaded the recently completed Rituximab in Myositis (RIM) trial that was designed to evaluate the safety and efficacy of B cell depletion in adult and pediatric myositis patients (14). Rituximab has been studied in a wide variety of autoimmune diseases, as B cells play a critical role in the initiation and propagation of the immune response and are specifically implicated in the pathogenesis of myositis (15). As biologic agents are increasingly utilized in autoimmune diseases, it is important to elucidate the factors that predict a favorable outcome so that clinical trials can be designed with stratification of patients with a good and poor likelihood of improvement. The aim of this study was to identify the clinical and laboratory predictors of clinical improvement in a trial of refractory myositis subjects treated with B cell depletion. This is the first comprehensive study in myositis to evaluate factors associated with clinical improvement in a large prospective cohort.
Patients and Methods
A total of 200 patients (76 adult PM/76 adult DM/48 JDM) with refractory myositis (14), were treated with rituximab as part of a multicenter clinical trial (RIM) using a randomized placebo phase design (RPPD) (16). However, only 195 were enrolled for at least 2 weeks and available for analysis of achieving the definition of improvement (DOI). Refractory myositis was defined by the intolerance to or an inadequate response to glucocorticoids and at least one other immunosuppressive agent. Patients were randomized to either a ‘rituximab early’ (drug at weeks 0/1, placebo at weeks 8/9) or ‘rituximab late’ arm (placebo at week 0/1, drug at week 8/9), such that all patients received active drug at some point in the study
The DOI was the International Myositis Assessment and Clinical Studies (IMACS) Group preliminary validated response (11) of a ≥ 20% improvement in 3 of any 6 core set measures (CSM) (17), with no more than 2 CSM worsening by ≥ 25% [which could not include manual muscle testing (MMT)]. The primary endpoint was the time to achieve the DOI at two consecutive time-points. The 6 CSM (17) for this trial were: (1) Patient (or parent) global disease activity using a 10 cm visual analog scale (VAS); (2) MD global disease activity also using a 10 cm VAS; (3) Health Assessment Questionnaire (HAQ) or Childhood HAQ (CHAQ); (4) serum muscle enzyme [most abnormal of the creatine kinase (CK), aldolase, lactate dehydrogenase (LDH), alanine aminotransferase (ALT) or aspartate aminotransferase (AST)] (5) Global extra-muscular disease activity (based on the investigator’s composite assessment of disease activity on the constitutional, cutaneous, skeletal, gastrointestinal, pulmonary and cardiac scales of the Myositis Disease Activity Assessment Tool (MDAAT) (13); and (6) MMT, assessed using a validated measure, the MMT-8 (18). The myositis CSM that were assessed in determining the DOI were collected at 14 visits over a 44-week trial period.
Baseline predictor (independent) variables
Two of the authors (RA and CVO) selected a priori the baseline clinical, laboratory and serologic variables that were evaluated for their predictive potential of clinical improvement. Variable selection was based on clinical experience and a literature review of previous studies (19–21). The variables selected for analysis are listed in Table 1.
Table 1.
Baseline predictor variables analyzed for univariate analysis.
| Group | Variables | Comments |
|---|---|---|
| Demographic features | Age at trial entry, age at diagnosis, gender, ethnicity, disease duration | |
| Myositis clinical subgroup | Polymyositis, dermatomyositis, juvenile dermatomyositis | |
| Laboratory parameters | Total IgM and IgG immunoglobulin levels, hemoglobin, leukocyte count, platelet count, serum creatinine | |
| Autoantibody (autoAb)* | Anti-synthetase, anti-Mi-2, other autoAb, no autoAb | |
| Baseline myositis damage variables | Muscle, gastrointestinal (GI), pulmonary and physician global damage | Measured by Myositis Damage Index (MDI), a validated tool for assessing damage in muscle and extra-muscular organ systems on 10 cm VAS scale (42). Physician global damage was a 10 cm VAS measure of overall global damage as per the physician. |
| Baseline myositis disease activity variables | Skeletal (i.e. inflammatory arthritis), GI, pulmonary and muscle disease activity | Measured by the Myositis Disease Activity Assessment Tool (MDAAT), a validated measure of physician rated myositis and extra-muscular disease activity on 10 cm VAS scale (43) |
| Myositis core set activity measures | Physician and patient (or parent) global activity measured by using a 10 cm visual analog scale (VAS) Muscle enzyme designated as times the upper limit of normal of the most abnormal muscle enzyme [creatine kinase (CK), aldolase, lactate dehydrogenase (LDH), alanine aminotransferase (ALT) or aspartate aminotransferase (AST)] Global extramuscular disease activity was measured on the MDAAT tool (10 cm VAS composite assessment of disease activity on the constitutional, cutaneous, skeletal, gastrointestinal, pulmonary and cardiac scales). Muscle strength assessed using a validated measure, the MMT-8 (18). |
|
| Baseline medication | Early vs. late rituximab arm, number of total failed immunosuppressive agents at trial entry, and baseline glucocorticoid dose (in prednisone equivalents) | |
| Categorical baseline MDAAT variables | Dysphagia, arthritis, mechanics hands, active ILD | Active ILD defined as dyspnea or cough or parenchymal abnormalities on chest radiography or computed tomography or pulmonary function tests showing a ≥ 10% change in FVC or DLCO (with physician attributing the change to active reversible ILD) |
| Categorical baseline MDI variables | Calcinosis, muscle atrophy, radiographic pulmonary fibrosis, abnormal DLCO or FEV1 | |
| Other | Raynaud phenomenon, clinical trial site |
Defined in Methods section
Autoantibodies (autoAb) were detected using protein and RNA immunoprecipitation (IP) (14) and were placed into 4 groups: (1) Myositis autoAbs including (a) anti-aminoacyl tRNA synthetases (anti-Syn): anti-Jo-1,-PL-7, -PL-12, -KS, -OJ and -EJ, (b) anti-Mi-2, (c) anti-SRP (signal recognition particle), (d) anti-TIF1-γ (transcription intermediary factor 1-gamma), (e) anti-MJ; (2) Other known autoAbs (anti-PM-Scl, -U1RNP, -SSA/SSB, -Ku, -SAE, -U1/U2, -centromere) seen in myositis and/or other CTDs; (3) Undefined autoAb (i.e. unable to be definitively identified by IP); and (4) subjects with no detectable autoAbs. Since the Kaplan-Meier curves for Groups (1c–e), (2) and (3) above were overlapping and not statistically different from each other, these groups were consolidated and analyzed as one category termed ’other autoAb’. Thus, 4 autoAb subsets emerged for the final statistical analysis: ‘anti-Syn’, ‘anti-Mi-2’, ‘other autoAb’ and ‘no autoAb’.
Statistical Analysis
The baseline for this study was defined as week 0 of the RIM trial regardless of whether a subject was in the early or late treatment arm. As in the RIM trial, the primary outcome was the time to achieve the DOI assessed in time to event analyses. All baseline variables were univariately assessed for association with time to DOI. All univariate variables that had a potential for association with time to DOI were then considered in a multivariable model.
For univariate analyses the association of the individual variables with time to DOI was assessed using nonparametric comparisons of Kaplan-Meier curves (DOI-free survival curves). Multi-category variables were grouped according to the quartiles of the observed values and evaluated using tests for trend. Nominal variables were assessed using Wilcoxon homogeneity tests or Log-rank tests in case of substantial difference between the results. The tests were performed using PROC LIFETEST SAS v.9.3 (SAS Institute Inc., Cary, NC).
Results of the univariate analysis were illustrated with a hazard ratio (“hazard” of achieving the DOI) for factors dichotomized at the median. Hazard ratios for nominal variables were computed with respect to the selected reference category. If the dichotomized variable had a similar strength of association as its four-category representation (results not shown), the binary form of the variable was considered in subsequent model building. In addition, all variables were analyzed separately in each arm of the trial to verify absence of a masking effect of the treatment.
For the multivariable model, univariate factors with a p-value ≤ 0.1 were combined using a Cox proportional hazard model (PROC PHREG, SAS v.9.3). Variables in which a univariate association with DOI was time-dependent were evaluated as time-dependent variables. Within the model, individual factors were tested at the 0.05 significance level. For each factor included in the final model the hazard ratios were evaluated at several time points. 95% confidence intervals were adjusted for multiplicity using Scheffe’s approach (PROC PHREG, SAS. v.9.3).
In the secondary analyses to assess the influence of treatment on predictive factors, we analyzed the effect of treatment in the final multivariable model using treatment arm in both time-dependent and fixed variable approaches.
Results
A total of 195 of the 200 RIM trial subjects (75 PM/72 DM/48 JDM) were analyzed (93 early arm/102 late arm) for baseline clinical, laboratory, autoantibody and disease variables as predictors of clinical improvement in univariate analyses. As previously reported (14), there was no difference in clinical outcomes between the rituximab early and late treatment groups. Most patients were Caucasian (70%) and female (73%), with a mean disease duration exceeding 5 years. The RIM trial population was clearly refractory to therapy yet had active myositis features. That is, subjects had already failed a mean of 3.1 immunosuppressive agents, but their mean baseline physician MDAAT global disease activity and muscle activity VAS scores were 5.0 cm and 4.8 cm respectively. The average prednisone dose at entry was 21 mg/day.
Eighty percent of the cohort (157/195) possessed at least one autoAb by immunoprecipitation (22): this included 30 patients (15%) anti-Syn [28 anti-Jo-1, 1 anti-OJ, 1 anti-PL-7], 26 (13%) anti-Mi-2, 25 (13%) anti-SRP, 23 (12%) anti-TIF1-γ and 22 (11%) anti-MJ subjects. Twenty-four subjects had other autoAbs: e.g. anti-SSA/B, U1RNP, U1/U2, SAE, Ku, centromere, 9 had autoAbs that were present but not clearly defined and 39 subjects had no identifiable autoAbs. The autoAb breakdown of DM patients included 16 (21%) anti-Syn, 20 (26%) anti-Mi-2, 30 (39%) other autoAbs and 11 (14%) without autoAbs. PM patients included 15 (20%) anti-Syn, 1 (1%) Mi-2, 39 (51%) other autoAbs and 21 (28%) without autoAbs. The JDM subgroup of patients included only 1 (2%) anti-Syn, 5 (11%) anti-Mi-2, 34 (72%) other autoAbs and 7 (15%) without autoAbs.
Univariate analyses of baseline patient characteristics that predicted time to improvement
The results of the univariate analyses of 38 predictor variables are summarized in Table 2. Twelve variables were identified as primary candidates for inclusion in the multivariable model (p ≤ 0.1). Due to a similar DOI-free survival (i.e., time to DOI) for adult PM and DM RIM trial subjects, and a difference in the JDM subset, the myositis clinical subgroups were represented using a dichotomous factor differentiating adult (PM and DM) versus juvenile myositis. JDM demonstrated a trend toward better clinical improvement as compared to adult subgroups (p=0.06). Of these 12 primary candidates for the model, four had a significant (p<0.05) univariate association with improvement: AutoAb groups (p=0.001), muscle damage (p=0.009), physician global damage (p=0.003), and muscle atrophy (p=0.016). Male gender, shorter disease duration, diminished lung function, higher leukocyte count, higher baseline muscle enzyme levels, higher extra-muscular disease activity and higher HAQ/CHAQ disability index univariately demonstrated a trend for association with improvement, but were not significant after accounting for other factors in the multivariable analysis. No additional factors were identified by considering each treatment arm separately.
Table 2.
Baseline values of univariate variables and their association with time to improvement.
| Variables (category assessed) | Median (25th–75th percentiles) or # and (%) of patients | Hazard Ratios of DOI | p value* |
|---|---|---|---|
| Demographics: | |||
| Age at diagnosis (≤40) | 40 (19–50) | 1.12 | 0.11 |
| Age at trial entry (≤46) | 46 (27–55) | 1.06 | 0.16 |
| Disease duration (≤3.3) | 3.3 (1.56–6.76) | 1.34 | 0.06 |
| Sex (Male) | 53/195 (27.2%) | 1.27 | 0.10 |
| Race (Caucasian) | 138/195 70.5% | 1.28 | 0.22 |
| Disease subsets: | |||
| Adult vs. Juvenile (Juvenile) | 48/195 (24.6%) | 1.15 | 0.06 |
| Core set measures: | |||
| Manual Muscle Testing (≤74.6) | 74.6 (65–80) | 1.11 | 0.52 |
| MD Global Activity VAS (>51) | 51 (38–62) | 1.08 | 0.50 |
| Patient/Parent Global activity VAS (>70) | 70 (51–82.6) | 1.02 | 0.66 |
| HAQ/CHAQ Disability (>1.5) | 1.5 (1–2.13) | 1.09 | 0.08 |
| Muscle enzyme x ULN (>2.31) | 2.31 (1.17–7.35) | 1.30 | 0.10 |
| Extramuscular VAS (>25) | 25 (13.68–45.0) | 1.26 | 0.07 |
| Autoantibody groups: | <0.01+ | ||
| Anti-Synthetase (28 Jo-1) | 30/195 (15.4%) | 2.83 | <0.01 |
| Anti-Mi-2 | 26/195 (13.3%) | 2.48 | <0.01 |
| Other autoAb (see text) | 101/195 (51.8%) | 1.39 | 0.14 |
| No autoAb | 38/195 (19.5%) | 1.0 (reference) | |
| Medication: | |||
| Mean prednisone dose(>20) | 20 (10–25) | 1.07 | 0.40 |
| Number of failed immunosuppressive agents (≤3) | 3 (2–4) | 1.08 | 0.35 |
| Disease Activity VAS from Myositis Damage Index (MDI) | |||
| Muscle disease activity (>49.5) | 49.5 (30–63) | 1.27 | 0.85 |
| Skeletal disease activity (>0) | 0 (0–13) | 1.03 | 0.92 |
| GI disease activity (>0) | 0 (0–9) | 1.17 | 0.80 |
| Pulmonary disease activity (>3) | 3 (0–19) | 1.13 | 0.30 |
| Disease damage VAS | |||
| Muscle damage (≤23) | 23 (4–53) | 1.26 | 0.01 |
| GI damage (=0) | 0 (0–5.38) | 1.03 | 0.27 |
| Pulmonary damage (=0) | 0 (0–9) | 1.23 | 0.64 |
| Physician global damage (≤23) | 23 (10–45) | 1.30 | <0.01 |
| Other clinical variables | |||
| Mechanics hands (present) | 35/195 (17.9%) | 1.20 | 0.70 |
| Dysphagia (absent) | 152/195 (77.9%) | 1.10 | 0.44 |
| Arthritis (present) | 16 (8.2%) | 1.38 | 0.22 |
| Active ILD (present) | 34/169 (20.1%) | 1.12 | 0.99 |
| Calcinosis (present) | 32/194 (16.5%) | 1.33 | 0.26 |
| Raynauds (present) | 37/195 (19%) | 1.14 | 0.57 |
| Muscle atrophy (absent) | 70/193 (36.3%) | 1.45 | 0.02 |
| Pulmonary fibrosis (present) | 33/177 (18.6%) | 1.38 | 0.11 |
| Diminished lung function (present) | 30/195 (15.4%) | 1.51 | 0.06 |
| Laboratory variables: | |||
| Hemoglobin (≤39.9) | 39.9 (37.3–42.8) | 1.13 | 0.97 |
| Leukocyte (WBC) count (>8.3) | 8.3 (6.6–11) | 1.44 | 0.06 |
| Platelet count (>296) | 296 (248–363) | 1 | 0.59 |
| Total IgG (<1130) | 1130 (890–1470) | 1.02 | 0.67 |
| Total IgM (<112) | 112 (73–175) | 1.04 | 0.82 |
| Serum creatinine (>0.6) | 0.6 (0.4–0.7) | 1.12 | 0.57 |
comparison of strata using Wilcoxon test (proc lifetest, (SAS v.9.3). Log-rank test was used if result was significantly different from Wilcoxon.
p value for difference among four autoantibody groups
The presence of a myositis autoAb was most strongly associated with improvement and had a relatively constant effect on the time to DOI throughout the trial. Specifically, the presence of anti-Syn (primarily anti-Jo-1) and anti-Mi-2 were strongly related to achieving improvement (2–3 fold higher chances for improvement than for the ‘no autoAb subset, p<0.002), while the ‘no autoAb’ group was associated with the worst time to improvement (Figure 1). Patients with ‘other autoAbs’ were more likely to improve (hazard ratio 1.4) although were not statistically different from the ‘no autoAb’ group (p=0.14).
Figure 1.

Kaplan-Meier curves for probability of meeting the definition of improvement (DOI) according to myositis autoantibody (autoAb) subset. Patients were classified into 4 subsets: those with antisynthetase autoantibodies (including anti–Jo-1), those with anti–Mi-2, those with other autoantibodies, and those with no detectable autoantibodies.
Lower physician global damage and the clinical subgroup of JDM (as compared to adult PM and DM) were strong univariate predictors for time to improvement, but these effects decreased with time (Figures 2 and 3). There was no significant difference in time to improvement between the adult PM and adult DM clinical subgroups.
Figure 2.
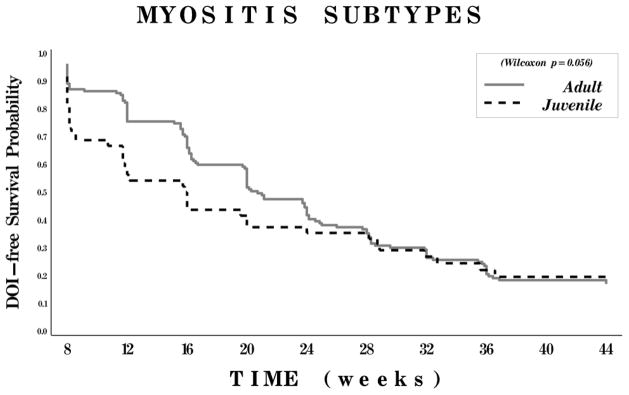
Kaplan-Meier curves for probability of meeting the definition of improvement (DOI) according to myositis clinical subgroups. Patients were classified as having either adult myositis (polymyositis or dermatomyositis) or juvenile
Figure 3.

Kaplan-Meier curves for probability of meeting the definition of improvement (DOI) according to degree of myositis disease damage. Patients were classified as having low damage (score of ≤23 a 100-mm visual analog scale [VAS]) or high damage (score of >23 on a 100-mm VAS).
Multivariable analysis of baseline patient characteristics that predicted time to improvement
The final multivariable model included the following three significant factors associated with clinical improvement: 1) autoAbs (anti-Syn, -Mi-2, other autoAb and no autoAb), 2) physician global damage (high: > 23; low ≤ 23 on 100 mm VAS scale dichotomized at median), and 3) myositis subtype (adult/juvenile). The hazard ratios for these three factors at week 8 and 20 are summarized in Table 3. Similar to the univariate assessment, after controlling for other factors in the multivariable model, patients with anti-Syn (primarily anti-Jo-1) and anti-Mi-2 showed a 2 to 3 fold higher chance of improvement as compared to the ‘no autoAb’ group. Lower physician global damage and JDM were associated with improvement in the final model. The time varying nature of the effects of physician global damage and myositis clinical subgroup were analyzed using interactions with time (as illustrated in Table 3). Over the first 8 weeks of the RIM trial, the time to improvement differed based on physician global damage scores and adult PM-DM versus JDM subgroups. However, after week 20 of the trial, there were no significant differences in the improvement between these groups. In contrast, the presence of autoAb was associated with substantial differences in improvement throughout the entire duration of the RIM trial.
Table 3.
Final multivariable model for predicting improvement
| Predictor Variable in Model | Hazard Ratio of DOI | 95% C.I. | p- value |
|---|---|---|---|
| Autoantibodies | |||
| No autoAb | 1.0 (reference) | ||
| Anti-Syn | 3.08 | 1.80–5.28 | <0.01 |
| Anti-Mi-2 | 2.5 | 1.42–4.41 | <0.01 |
| Other autoAb | 1.40 | 0.90–2.17 | 0.14 |
| Physician global damage (low vs. high) | 2.32 (week 8) 1.03 (week 20) |
1.09–4.90* 0.66–1.60* |
0.02* for week 8, 0.99* for week 20 |
| Myositis clinical subgroup (JDM vs. adult PM/DM) | 2.45 (week 8) 1.01 (week 20) |
1.16–5.15* 0.59–1.73* |
0.01* for week 8, 0.41* for week 20 |
Adjusted for multiplicity using Scheffe’s method (proc phreg, SAS, v.9.3)
The time-dependent effect of treatment (i.e. the 8-week lag for the “late” arm) was not associated with improvement and the results of multivariable analysis were not affected after controlling for treatment arm.
Discussion
Eighty three percent of RIM trial subjects improved (met DOI) while 30% of refractory myositis patients historically improve (23, 24). The current study indicates that autoAbs, especially the anti-synthetases (mainly anti-Jo-1) and anti-Mi-2, were the strongest predictors of clinical improvement in a cohort of rituximab-treated myositis patients, whereas the lack of definable autoAbs predicted no improvement. All other autoAb subgroups including anti-SRP had similar responses to each other, with little predictive capacity. We found no differences between anti-Jo-1 and anti-Mi-2 as predictors of improvement, although both were better than anti-SRP and other autoAbs. Previous reports indicated that anti-Jo-1 positive patients required more immunosuppressive medication as compared to patients with anti-Mi-2, although both responded better than anti-SRP positive patients (6, 7, 25). This is somewhat consistent with our results which showed the superior rate and time to improvement of anti-SRP and anti-Mi-2 positive subjects. We also found that JDM patients and subjects with a lower degree of myositis damage at baseline are more likely to have a favorable clinical improvement in a cohort of rituximab-treated myositis patients. In the RIM trial there was no difference in response rates between the early and late treatment arms. In this analysis we have not detected a treatment effect due to early and late treatment after accounting for significant predictors of improvement.
The importance of myositis autoAbs to identify phenotypically distinct subsets of myositis patients is well recognized, and our results expand their role as predictive factors for clinical improvement in myositis patients. Our finding that anti-Jo-1 predicts clinical improvement is even more intriguing when considered in the context of previous studies demonstrating that anti-Jo-1 autoAb levels may serve as a biomarker of myositis disease activity (26–28). Combined with additional RIM trial data showing that anti-Jo-1 autoAb levels correlate with myositis disease activity (29), the findings reported herein that Jo-1 positive patients have a better outcome suggests that immune responses related to anti-Jo-1 autoAbs may be pathogenic. In this regard, human tyrosyl-tRNA synthetase, a rare autoantigen in myositis, has chemoattractant and leukocyte activating properties after proteolytic cleavage (30, 31), while histidyl-tRNA synthetase (the target of anti-Jo-1) and asparaginyl-tRNA synthetase (the target of anti-KS) activate chemokine receptors on T lymphocytes and immature dendritic cells (32). Thus, autoAbs directed against these ubiquitous human aminoacyl-tRNA synthetases or the antigen themselves may contribute in some undetermined fashion to the perpetuation of pathogenic immune responses in muscle or other tissue (32).
Similarly, our results demonstrated that subjects with anti-Mi-2 also had better clinical improvement. Previous studies have shown anti-Mi-2 to be associated with more favorable outcomes (33, 34). In contrast, patients with anti-SRP generally have necrotizing, poorly responsive PM (6, 35), which is similar to our current finding of intermediate improvement (worse than anti-Jo-1 and anti-Mi-2) in the RIM trial. In a separate report analyzing comparative survival among patients stratified by autoantibody, we recently showed a similar survival among anti-SRP, -Jo-1 and -Mi-2 patients (36). However, survival may be very different from clinical improvement in a therapeutic trial. In contrast to autoAb positive subjects, those with no definable myositis autoAbs had a worse outcome, suggesting that possessing an autoAb may predict a favorable prognosis--even in autoAb subsets known to have a worse prognosis (i.e. anti-SRP). Anti-SRP autoAb positive patients showed a similar improvement as “other” autoAbs, but worse than anti-Jo-1 and anti-Mi-2, and fractionally better than groups with no MAA. Although we grouped all anti-synthetase autoAbs together, the predominant autoAb was anti-Jo-1; thus, these results cannot be applied to non-Jo-1 autoAbs (e.g. anti-PL-7, anti-PL-12, etc). In fact, recent evidence suggests that patients with non-Jo-1 autoAbs have worse survival as compared to Jo-1 antibody patients (36). Anti-MDA5 and anti-HMGCoA are 2 newly characterized myositis autoAbs that were not measured in the RIM trail and these patients might have been included in the no-autoAb group. Many PM patients were autoAb negative and there may be a concern that some of these patients represent PM mimics (e.g. IBM and adult muscular dystrophy) leading to a poor outcome of this group (37). However, a 3-member adjudication committee of myositis experts including a neurologist/neuropathologist reviewed the medical records and muscle biopsy to insure that PM mimics were excluded from the RIM trial. Despite these limitations, the current study is the first to comprehensively demonstrate that autoAbs are major predictive factors of clinical improvement in myositis patients treated with B-cell depleting therapy.
The current study also showed that lower global damage predicts clinical improvement. Previously published studies have shown that muscle damage in myositis is a poor prognostic marker (10). All 3 measures of damage (muscle damage and atrophy and physician global damage) were strongly associated in univariate analyses with poor clinical improvement. Physician global damage had the strongest association with improvement and was the only damage variable that remained in the final multivariable model, as muscle damage and atrophy did not substantially add to the prediction of improvement. Importantly, lower damage predicted a favorable outcome early in the course of the study as the association with improvement decreased after week 20. The reasons for this are not clear, although it would seem plausible to postulate that patients with more damage at baseline demonstrate a delayed improvement.
This is the first study that directly demonstrated a better outcome in patients with JDM compared with adult myositis. A Korean trial showed superior survival and clinical outcomes in JDM patients compared to adult DM (38), while other studies reported better long-term survival of JDM (20, 39). An older age at onset has been recognized as a poor prognostic marker for survival in myositis patients (19, 20, 40) and younger patients have higher remission rates (41). However, we specifically demonstrated that the JDM subgroup and not the age at diagnosis, predicted a favorable and more rapid clinical improvement in the final model. Also, these results in JDM patients are not attributable to a shorter disease duration. Similar to the observation with lower damage, JDM predicted a favorable outcome early in the course of the study as the association with improvement decreases after week 20. JDM patients have been shown to have lower myositis related damage as compared to adult DM/PM patients (42), however, as we demonstrated the chances of improvement were much higher for JDM patients even after adjusting for the global damage. JDM was associated with substantial improvement early in the trial compared to either DM or PM adult myositis, however, these individual differences were not statistically significant for the available sample sizes. DM and PM adult myositis subsets had similar DOI-free experiences and combined for all analyses. The difference in DOI-free survival of the JDM patient with combined adult myositis subtype (PM and DM) was similar to the differences with adult DM or PM separately.
It is important to recognize that in our analyses, univariate and multivariable analyses were employed to predict the association with clinical improvement in adult and juvenile myositis. This should not be interpreted as an overall predictor of response to rituximab since all patients received rituximab in the RIM trial. Thus, it is difficult to conclude whether the identified predictive factors are valid for myositis patients in general or only those treated with B cell depletion. Nevertheless, given that rituximab is a B-cell depleting agent which may directly inhibit autoAb production, it is plausible that the favorable results in myositis autoAb producing patients as compared to those without autoAbs are at least partly due to rituximab.
In summary, we found certain autoAbs to be the strongest predictive markers of clinical improvement in a cohort of ritixumab-treated myositis patients. Patients with anti-synthetase autoAbs (predominantly anti-Jo-1) and anti-Mi-2 had a better outcome and the absence of a myositis autoAb was associated with a worse outcome. We also found that myositis disease-associated damage and specifically muscle damage were markers of a poor clinical outcome while JDM predicted a better outcome as compared to adult DM or PM. However, low myositis damage and JDM were associated with more rapid improvement only early in the course of the study. We believe these findings improve our understanding of the pathogenesis of myositis and provide important guidelines for the design of future IIM clinical trials. Perhaps, future myositis clinical trials should be design to analyze separately the JDM vs. adult myositis groups, balance the global damage and account for autoantibodies in the analysis.
Acknowledgments
Supported by the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases contract N01-AR-4-2273), Genentech Inc., the Intramural Program of the NIH (National Institute of Environmental Health Sciences), and by a General Clinical Research Center/Clinical and Translational Science Award (M01-RR-023940/UL1-RR-033179) to the University of Kansas Medical Center.
We thank Diane Koontz and Sherrie Pryber as project managers for the RIM trial and acknowledge our research laboratory specialists Noreen Fertig and Zengbiao Qi.
APPENDIX A: RIM STUDY GROUP MEMBERS
Members of the RIM Study Group (countries, principal investigators, and centers) are as follows. In Canada (pediatric sites): Brian Feldman (Hospital for Sick Children, Toronto, Ontario) and Adam Huber (IWK Health Centre, Halifax, Nova Scotia). In the Czech Republic (adult site): Jiří Vencovský and Herman Mann (Institute of Rheumatology, Prague). In Sweden (adult site): Ingrid E. Lundberg (Karolinska Institutet, Stockholm). In the US (adult sites): Richard Barohn, Mazen Dimachkie, and Kevin Latinis (University of Kansas Medical Center, Kansas City), Lorinda Chung and David Fiorentino (Stanford University, Palo Alto), Leslie Crofford (University of Kentucky, Lexington), Mary Cronin (Medical College of Wisconsin, Milwaukee), Stephen DiMartino (Hospital for Special Surgery, New York), Barri Fessler (University of Alabama at Birmingham), Michael Harris-Love (Washington DC Veterans Affairs Medical Center), Sharon Kolasinski (University of Pennsylvania, Philadelphia), Todd Levine (Phoenix Neurological Associates), Galina Marder (North Shore–LIJ, New York), Richard Martin and Aaron Eggebeen (adult and pediatric site: Michigan State University, Grand Rapids), Frederick Miller (National Institute of Environmental Health Sciences, NIH, Bethesda), Pushpa Narayanaswami and Seward B. Rutkove (Beth Israel Deaconess Medical Center/Harvard Medical School, New York), Chester Oddis, Dana Ascherman, Rohit Aggarwal, David Lacomis, and Christopher Bise (University of Pittsburgh), Nancy Olsen and Andreas Reimold (University of Texas Southwestern Medical Center at Dallas), Elena Schiopu, Kristine Phillips, and James Seibold (University of Michigan, Ann Arbor), Khema Sharma (University of Miami), Swamy Venturupalli and Michael Weisman (Cedars-Sinai Medical Center, University of California at Los Angeles), and Steven Ytterberg (Mayo Clinic, Rochester). In the US (pediatric sites): Susan Kim (Children’s Hospital of Boston), Tzielan Lee (Stanford University, Palo Alto), Daniel Lovell (Cincinnati Children’s Hospital), C. Egla Rabinovich (Duke University Medical Center, Durham), Ann Reed (Mayo Clinic, Rochester), Lisa Rider (National Institute of Environmental Health Sciences, NIH, Bethesda), Rafael Rivas-Chacon (Miami Children’s Hospital), and David Sherry (The Children’s Hospital of Philadelphia).
Footnotes
ClinicalTrials.gov identifier: NCT00106184.
Dr. Aggarwal has received consulting fees and/or honoraria from Questcor and aTyr pharma (less than $10,000) for service on the Advisory Board. Dr. Oddis has served as an expert witness concerning appropriateness of rituximab therapy in a patient with myositis. Dr. Reed has received consulting fees and/or honoraria from Genentech (less than $10,000) for service on the Genentech Advisory Board. Dr. Levesque has received consulting fees, speaking fees, and/or honoraria from Genentech (less than $10,000) and has received research support from Genentech. Dr. Barohn has received consulting fees, speaking fees, and/or honoraria from Genzyme, Grifols, Novartis, and MedImmune (less than $10,000 each). Dr. Feldman has received consulting fees, speaking fees, and/or honoraria from Novartis (less than $10,000).
References
- 1.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts) The New England journal of medicine. 1975;292(8):403–7. doi: 10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 2.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) The New England journal of medicine. 1975;292(7):344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 3.Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Current Rheumatol Report. 2003;5(6):425–30. doi: 10.1007/s11926-003-0052-2. [DOI] [PubMed] [Google Scholar]
- 4.Koga T, Fujikawa K, Horai Y, Okada A, Kawashiri SY, Iwamoto N, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford) 2012;51(7):1278–84. doi: 10.1093/rheumatology/ker518. [DOI] [PubMed] [Google Scholar]
- 5.Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology (Oxford) 2012;51(5):800–4. doi: 10.1093/rheumatology/ker408. [DOI] [PubMed] [Google Scholar]
- 6.Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70(6):360–74. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Joffe MM, Love LA, Leff RL, Fraser DD, Targoff IN, Hicks JE, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. American Journal of Medicine. 1993;94(4):379–87. doi: 10.1016/0002-9343(93)90148-i. [DOI] [PubMed] [Google Scholar]
- 8.Mathiesen P, Hegaard H, Herlin T, Zak M, Pedersen FK, Nielsen S. Long-term outcome in patients with juvenile dermatomyositis: a cross-sectional follow-up study. Scandanavian Journal of Rheumatology. 2012;41(1):50–8. doi: 10.3109/03009742.2011.608376. [DOI] [PubMed] [Google Scholar]
- 9.Ravelli A, Trail L, Ferrari C, Ruperto N, Pistorio A, Pilkington C, et al. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Research (Hoboken) 2010;62(1):63–72. doi: 10.1002/acr.20015. [DOI] [PubMed] [Google Scholar]
- 10.Sanner H, Kirkhus E, Merckoll E, Tollisen A, Roisland M, Lie BA, et al. Long-term muscular outcome and predisposing and prognostic factors in juvenile dermatomyositis: A case-control study. Arthritis Care Research (Hoboken) 2010;62(8):1103–11. doi: 10.1002/acr.20203. [DOI] [PubMed] [Google Scholar]
- 11.Rider LG, Giannini EH, Brunner HI, Ruperto N, James-Newton L, Reed AM, et al. International consensus on preliminary definitions of improvement in adult and juvenile myositis. Arthritis Rheumatism. 2004;50(7):2281–90. doi: 10.1002/art.20349. [DOI] [PubMed] [Google Scholar]
- 12.Rider LG, Giannini EH, Harris-Love M, Joe G, Isenberg D, Pilkington C, et al. Defining Clinical Improvement in Adult and Juvenile Myositis. Journal of Rheumatology. 2003;30(3):603–17. [PubMed] [Google Scholar]
- 13.Sultan SM, Allen E, Oddis CV, Kiely P, Cooper RG, Lundberg IE, et al. Reliability and validity of the myositis disease activity assessment tool. Arthritis Rheumatism. 2008;58(11):3593–9. doi: 10.1002/art.23963. [DOI] [PubMed] [Google Scholar]
- 14.Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis and rheumatism. 2013;65(2):314–24. doi: 10.1002/art.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu YE, Co DO. Juvenile dermatomyositis: immunopathogenesis, role of myositis-specific autoantibodies, and review of rituximab use. Pediatric dermatology. 2011;28(4):357–67. doi: 10.1111/j.1525-1470.2011.01501.x. [DOI] [PubMed] [Google Scholar]
- 16.Feldman BM, Wang E, Willan A, Szalai JP. The randomized placebo-phase design for clinical trials. Journal of Clinical Epidemiology. 2001;54:550–7. doi: 10.1016/s0895-4356(00)00357-7. [DOI] [PubMed] [Google Scholar]
- 17.Miller FW, Rider LG, Chung YL, Cooper R, Danko K, Farewell V, et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2001;40(11):1262–73. doi: 10.1093/rheumatology/40.11.1262. [DOI] [PubMed] [Google Scholar]
- 18.Rider LG, Koziol D, Giannini EH, Jain MS, Smith MR, Whitney-Mahoney K, et al. Validation of manual muscle testing and a subset of eight muscles for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Research (Hoboken) 2010;62(4):465–72. doi: 10.1002/acr.20035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Airio A, Kautiainen H, Hakala M. Prognosis and mortality of polymyositis and dermatomyositis patients. Clinical Rheumatology. 2006;25(2):234–9. doi: 10.1007/s10067-005-1164-z. [DOI] [PubMed] [Google Scholar]
- 20.Danko K, Ponyi A, Constantin T, Borgulya G, Szegedi G. Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 2004;83(1):35–42. doi: 10.1097/01.md.0000109755.65914.5e. [DOI] [PubMed] [Google Scholar]
- 21.Torres C, Belmonte R, Carmona L, Gomez-Reino FJ, Galindo M, Ramos B, et al. Survival, mortality and causes of death in inflammatory myopathies. Autoimmunity. 2006;39(3):205–15. doi: 10.1080/08916930600622603. [DOI] [PubMed] [Google Scholar]
- 22.Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheumatic Disease Clinics of North America. 2002;28(4):859–90. viii. doi: 10.1016/s0889-857x(02)00032-7. [DOI] [PubMed] [Google Scholar]
- 23.Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. New England Journal of Medicine. 1993;329(27):1993–2000. doi: 10.1056/NEJM199312303292704. [DOI] [PubMed] [Google Scholar]
- 24.Muscle Study G. A randomized, pilot trial of etanercept in dermatomyositis. Annal of Neurology. 2011;70(3):427–36. doi: 10.1002/ana.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bronner IM, van der Meulen MF, de Visser M, Kalmijn S, van Venrooij WJ, Voskuyl AE, et al. Long-term outcome in polymyositis and dermatomyositis. Annals of Rheumatic Disease. 2006;65(11):1456–61. doi: 10.1136/ard.2005.045690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida S, Akizuki M, Mimori T, Yamagata H, Inada S, Homma M. The precipitating antibody to an acidic nuclear protein antigen, the Jo-1, in connective tissue diseases. A marker for a subset of polymyositis with interstitial pulmonary fibrosis. Arthritis and rheumatism. 1983;26(5):604–11. doi: 10.1002/art.1780260505. [DOI] [PubMed] [Google Scholar]
- 27.Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. Journal of Clinal Investigation. 1990;85(2):468–75. doi: 10.1172/JCI114461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis and rheumatism. 2007;56(9):3125–31. doi: 10.1002/art.22865. [DOI] [PubMed] [Google Scholar]
- 29.Rohit Aggarwal CVO, Bandos Andriy, Goudeau Danielle, Koontz Diane, Zenbiao Qi, Reed Ann M, Ascherman Dan P, Levesque Marc C. Effect of B Cell Depletion Therapy with Rituximab On Myositis Associated Antibody Levels in Idiopathic Inflammatory Myopathy. Arthritis & Rheumatism. 2012 Oct;64(10 Suppl):S325. [Google Scholar]
- 30.Wakasugi K, Schimmel P. Highly differentiated motifs responsible for two cytokine activities of a split human tRNA synthetase. Journal of Biology and Chemistry. 1999;274(33):23155–9. doi: 10.1074/jbc.274.33.23155. [DOI] [PubMed] [Google Scholar]
- 31.Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science. 1999;284(5411):147–51. doi: 10.1126/science.284.5411.147. [DOI] [PubMed] [Google Scholar]
- 32.Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. Journal of Experimental Medicine. 2002;196(6):781–91. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hengstman GJ, Vree Egberts WT, Seelig HP, Lundberg IE, Moutsopoulos HM, Doria A, et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Annals of Rheumatic Disease. 2006;65(2):242–5. doi: 10.1136/ard.2005.040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Archives of Dermatology. 2011;147(4):391–8. doi: 10.1001/archdermatol.2011.52. [DOI] [PubMed] [Google Scholar]
- 35.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis and rheumatism. 2004;50(1):209–15. doi: 10.1002/art.11484. [DOI] [PubMed] [Google Scholar]
- 36.Aggarwal R, Cassidy E, Fertig N, Koontz DC, Lucas M, Ascherman DP, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Annals of Rheumatic Disease. 2013 doi: 10.1136/annrheumdis-2012-201800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Meulen MF, Bronner IM, Hoogendijk JE, Burger H, van Venrooij WJ, Voskuyl AE, et al. Polymyositis: an overdiagnosed entity. Neurology. 2003;61(3):316–21. doi: 10.1212/wnl.61.3.316. [DOI] [PubMed] [Google Scholar]
- 38.Na SJ, Kim SM, Sunwoo IN, Choi YC. Clinical characteristics and outcomes of juvenile and adult dermatomyositis. Journal of Korean Medical Science. 2009;24(4):715–21. doi: 10.3346/jkms.2009.24.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 2013;92(1):25–41. doi: 10.1097/MD.0b013e31827f264d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marie I, Hatron PY, Levesque H, Hachulla E, Hellot MF, Michon-Pasturel U, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine. 1999;78(3):139–47. doi: 10.1097/00005792-199905000-00001. [DOI] [PubMed] [Google Scholar]
- 41.Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B, et al. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. The Journal of rheumatology. 2001;28(10):2230–7. [PubMed] [Google Scholar]
- 42.Rider LG, Lachenbruch PA, Monroe JB, Ravelli A, Cabalar I, Feldman BM, et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis and rheumatism. 2009;60(11):3425–35. doi: 10.1002/art.24904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sultan SM, Allen E, Oddis CV, Kiely P, Cooper RG, Lundberg IE, et al. Reliability and validity of the myositis disease activity assessment tool. Arthritis and rheumatism. 2008;58(11):3593–9. doi: 10.1002/art.23963. [DOI] [PubMed] [Google Scholar]