

Abstract
JC virus (JCV) lytically infects the oligodendrocytes in the central nervous system in a subset of immunocompromized patients and causes the demyelinating disease, progressive multifocal leukoencephalopathy. JCV replicates and assembles into infectious virions in the nucleus. However, understanding the molecular mechanisms of its virion biogenesis remains elusive. In this report, we have attempted to shed more light on this process by investigating molecular interactions between large T antigen (LT-Ag), Hsp70 and minor capsid proteins, VP2/VP3. We demonstrated that Hsp70 interacts with VP2/VP3 and LT-Ag; and accumulates heavily in the nucleus of the infected cells. We also showed that VP2/VP3 associates with LT-Ag through their DNA binding domains resulting in enhancement in LT-Ag DNA binding to Ori and induction in viral DNA replication. Altogether, our results suggest that VP2/VP3 and Hsp70 actively participate in JCV DNA replication and may play critical roles in coupling of viral DNA replication to virion encapsidation.
Keywords: Polyomavirus, JCV, SV40, BKV, Large T antigen, Capsid protein, VP2, VP3, Transcription, DNA replication, Progressive multifocal leukoencephalopathy
Introduction
JC virus (JCV) is a small, double-stranded human DNA virus that causes a fatal demyelinating disease of the human brain, known as progressive multifocal leukoencephalopathy (PML), in a subset of immunocompromized individuals by specifically and lytically infecting oligodendrocytes – the myelin producing cells of the central nervous system (CNS).
Like many other DNA viruses, JCV replicates and assembles into infectious virions in the nucleus. Upon translation, JCV capsid proteins enter the nucleus to build the different layers of the virions. During this process, the viral genome and histones form the inner core and two capsid proteins, VP2 and VP3 tightly wrap around the viral genome. Maturation of the virions is thought to be finalized by a regulated assembly process of the third capsid protein, VP1, which forms the outer layer of the infectious particles. However, the mechanistic details of virion assembly of JCV remain elusive. One of the key players in this assembly process is thought to be the host chaperone system, including the heat shock protein 70 (Hsp70). Heat shock proteins are rapidly induced by viral infection (Beere and Green, 2001) and are frequently recruited by numerous viruses (Mayer, 2005) to facilitate the various stages of the viral life cycle including genome replication (Chromy et al., 2003), transcription (Devireddy et al., 2000), cell entry (Guerrero et al., 2002) and virion assembly (Napuli et al., 2003). Without the regulatory activities of heat shock proteins, viruses cannot complete their replication cycle (Mayer, 2005).
The primary function of the heat shock proteins is to assist the proper folding of newly synthesized proteins, to prevent their aggregation and to direct damaged proteins to the ubiquitinproteasome system for degradation. Hsp70 consists of two distinct functional domains: (i) the C-terminal substrate binding domain, which mediates the target binding and refolding of the substrates and (ii) the N-terminal ATP hydrolysis (ATPase) domain, which facilitates the release of the client protein upon ATP hydrolysis (Vogel et al., 2006). The heat shock family of proteins requires a diverse group of co-chaperones for their function, including (i) J-domain co-chaperones (Young et al., 2004), (ii) the nucleotide exchange factor co-chaperones which catalyze the release of ADP that is required for the completion of Hsp70 ATPase cycle (e.g. Bag1, Hsp110 and HspBP1) and (iii) tetratricopeptide repeat (TPR)-domain co-chaperones (Hop, Chip) which are required for the combinatorial assembly of Hsp70 and Hsp90 complexes to mediate the stability of the Hsp90 client proteins (Mayer, 2005).
The critical importance of Hsp70 family in the replication of many viruses has been previously established (Chromy et al., 2003; Devireddy et al., 2000; Georgopoulos, 1977; Georgopoulos et al., 1972; Guerrero et al., 2002; Liberek et al., 1988; Liu et al., 1998; Mayer, 2005; Napuli et al., 2003), including SV40, BKV, and PyV (Chromy et al., 2003). Besides many other functions of Hsp70, there are reports describing the involvement of Hsp70 system in virion morphogenesis as well, where the interaction of Hsp70 with capsid proteins (VP1, VP2 and VP3) of mouse polyomavirus (PyV) was demonstrated (Chromy et al., 2003). Purified VP1 and VP3 can assemble into virion-like structures in the presence of prokaryotic Hsp70 (DnaK, DnaJ and GrpE) chaperones in an ATP-dependent manner. Such an assembly was also demonstrated using a functional mammalian Hsp70 when the J-domain of SV40 LT-Ag was used as co-chaperone for Hsp70 (Chromy et al., 2003).
The early open reading frame of JCV, BK virus (BKV) and Simian virus-40 (SV40) encodes primarily two regulatory proteins, large T antigen (LT-Ag) and small t antigen (Sm t-Ag), which are produced by the alternative splicing of a singly precursor early mRNA (An et al., 2012; White et al., 1992). The production of the additional small regulatory proteins originating from the viral early transcripts has also been reported for several polyomaviruses, including a 17 kD T protein for SV40 (Zerrahn et al., 1993), a tiny T protein for PyV (Riley et al., 1997), a 17–20 kD T protein for BKV (Abend et al., 2009) and three T′ proteins for JCV (Bollag et al., 2000; Prins and Frisque, 2001). The precise function of these small proteins is not known. However, one of the unifying structural features of all the polyomavirus early proteins is to have common sequences at their respective N-terminal region, which contains a co-chaperone J-domain regardless of their size. The J-domain shows significant sequence homology to the DnaJ family of chaperons, which are conserved from bacteria to human. This domain strongly interacts with the C-terminal region of host Hsp70 in a ATP-dependent manner (Sullivan et al., 2001; Sullivan and Pipas, 2002) and plays important regulatory roles in determining the substrate specificity of the Hsp70 family of proteins during viral DNA replication and cell transformation (Nevins, 1998; Zalvide et al., 1998). Moreover, this domain also contains a conserved HPD loop, which is essential for its co-chaperone activity (Kelley, 1999). Biochemical studies have shown that the J-domain of T antigen functions as cochaperone in multiple assays, which include stimulation of the binding activity of LT-Ag to Hsp70 (Campbell et al., 1997; Sawai and Butel, 1989; Sullivan et al., 2001), promotion of the release of bound substrate (Srinivasan et al., 1997) and ATPase activity of bovine Hsp70 (Srinivasan et al., 1997). Furthermore, mutational analysis of the J-domain of SV40 LT-Ag (L19F, P28S) revealed that this domain may also be involved in virion assembly (Spence and Pipas, 1994a; Spence and Pipas, 1994b).
In this report, we investigated molecular interactions taking place between JCV LT-Ag, Hsp70 and the JCV minor capsid proteins, VP2 and VP3. We also characterized a functional consequence of a specific interaction between LT-Ag and capsid proteins (VP2/VP3) using deletion mutants of VP2/VP3 by replication assays. Our data demonstrated that Hsp70 interacts with capsid proteins, VP2/VP3, and LT-Ag; and accumulates heavily in the nucleus of the infected cells, suggesting a role for them in virion biogenesis. Mapping studies revealed a strong interaction between the capsid proteins (VP2/VP3) and LT-Ag, mediated by the DNA binding domain of each capsid protein (aa 281–344) (Gasparovic et al., 2006; Huang et al., 2003). This interaction resulted in a marked enhancement in the DNA binding activity of LT-Ag to Ori. Our data also show that the DNA binding domain of each capsid protein is sufficient for the observed enhancement, indicating a possible cooperation between this region and LT-Ag in JCV virion biogenesis. Finally, the functional consequence of the interaction between capsid proteins and LT-Ag was also tested by viral DNA replication assays using specific deletion mutants of the capsid proteins. Results revealed that the efficiency of the LT-Ag-mediated viral DNA replication is significantly elevated in the presence of VP2 and the DNA binding domain of VP2/VP3 is sufficient for this enhancement, which confirms the functional interaction between capsid proteins and LT-Ag. Collectively, these findings suggest that JCV minor capsid proteins and the host Hsp70, actively participate in LT-Ag-mediated viral DNA replication and may play essential roles on coupling of the viral DNA replication to the encapsidation process.
Results
Bacterial heat shock protein, DnaK, co-purifies with JCV capsid protein, VP3
The polyomaviridae family of viruses is small, non-enveloped, icosahedral DNA viruses including JCV, BKV, SV40 and PyV Both the viral capsid proteins and viral genome assemble into the highly organized infectious particles in the nucleus, but the mechanism of the assembly process is largely unknown. X-ray crystallography studies on PyV (Liddington et al., 1991) and monkey polyomavirus, SV40 (Griffith et al., 1992) revealed that the outer shell of each virion is composed of 72 pentamers of the major capsid protein, VP1, which is arranged in T=7 icosahedral lattice and inner structures are made up of two minor capsid proteins, VP2 and VP3 and viral genome. There are numerous reports in the literature, indicating that the host heat shock protein, Hsp70, plays a critical role in this highly regulated viral assembly process during the maturation steps of these viruses.
To investigate the mechanisms of JCV virion biogenesis, JCV capsid proteins, including VP3, was fused to GST, expressed in bacteria, affinity purified and analyzed by SDS-PAGE followed by Coomassie blue staining. As shown in Fig. 1A, full-length VP3 (GST-VP3) migrated around ∼41 kDa (lane 3). Interestingly, we also observed an additional protein band migrating closer to ∼70 kDa, the identity of which was determined to be bacterial heat shock protein, DnaK, by proteomic sequencing studies. These findings demonstrate that DnaK strongly interacts with JCV VP3 and therefore co-purifies with it but not with GST alone indicating a specific interaction between DnaK and VP3.
Fig. 1.
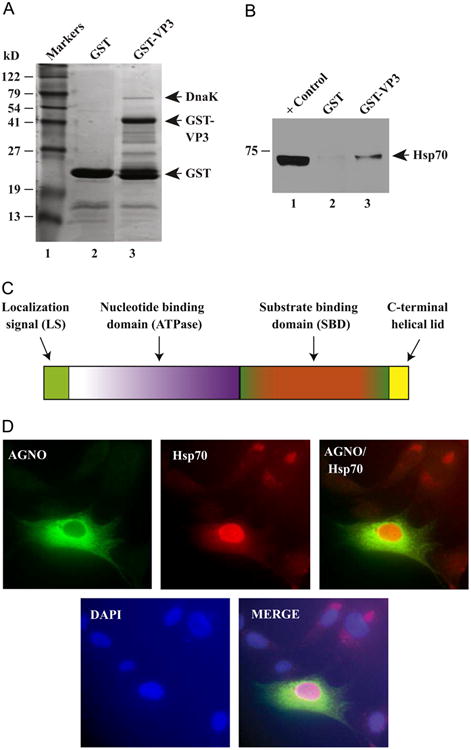
Interaction of JCV VP3 with heat shock protein, Hsp70. (A) SDS-PAGE analysis of bacterially expressed GST and JCV VP3 fused to GST (GST-VP3). Both GST alone and VP3 cloned into the pGEX1λt vector were expressed in Escherichia coli, affinity purified and analyzed on a SDS-10% PAGE followed by Coomassie blue staining. The positions of bacterial heat shock protein, DnaK, GST and GST-VP3 are indicated on the gel by arrows. (B) VP3 interacts with human Hsp70 in GST pull-down assays. Whole-cell extracts (0.3 mg) prepared from U-87MG cells were incubated with either GST alone (lane 2) or GST-VP3 (lane 3) and immobilized on GSH-Sepharose 4B affinity resin. After extensive washing, proteins interacting with GST or GST-VP3 were analyzed by western blotting using anti-Hsp70 antibody (W2) for detection of Hsp70 as described in Materials and methods. In lane 1, 20 μg aliquot of whole cell extract from U-87MG was loaded as positive control. (C) A graphical representation of Hsp70 protein with its subdomains. (D) Immunocytochemical analysis of Hsp70 in infected SVG-A cells. The cells were transfected with JCV Mad-1 WT and at day 5 posttranfection/infection, fixed with cold acetone and blocked with 5% BSA (prepared in PBST) for 2 h. Cells were then incubated with a combination of α-Agno (rabbit polyclonal, 1:200 dilution) (Del Valle et al., 2002) and anti-Hsp70 antibody (W2) (mouse monoclonal, 1:100 dilution) overnight. After washing with PBST three times with (10 min intervals), cells were incubated with the combination of FITC-conjugated anti-rabbit goat and Rhodamine-conjugated anti-mouse goat secondary antibodies for 45 min. Cells were finally washed with PBST three times with 10 min intervals, mounted with mounting media and examined under a fluorescence microscope as described under Materials and methods.
Host Hsp70 interacts with capsid protein, VP3
We, next, investigated whether Hsp70 also interacts with the viral capsid protein, VP3, by a powerful biochemical technique, GST-pull down assay. To assess this, bacterially expressed GST or GST-VP3 was immobilized on GSH-Sepharose 4B beads and incubated with whole-cell extracts prepared from U-87MG cells as described in Materials and methods and in the figure legend. Proteins bound to GST or GST-VP3 were analyzed by western blotting using an anti-Hsp70 antibody. As shown in Fig. 1B, Hsp70 strongly interacts with the viral capsid protein, VP3 (lane 3). A similar interaction between Hsp70 and GST alone was not detectable (lane 2), indicating the specificity of association between Hsp70 and VP3 and also supporting our findings from GST-VP3 bacterial expression studies, where we observed the co-purification of bacterial heat shock protein, DnaK with VP3 (Fig. 1A).
Hsp70 is induced by JCV infection and accumulates in the nucleus of the infected cells
JCV encodes a limited number of regulatory proteins and thus the success of virus proliferation depends on recruitment of host cellular components and chaperons for its own replication and virion assembly. Hsp70 chaperons are the central components of the cellular chaperon network, which are frequently recruited by viruses. Hsp70 chaperons display a cytoplasmic/nuclear distribution pattern in cells under unstressed conditions. We next examined the cellular distribution of Hsp70 during the viral replication cycle by immunocytochemistry. JCV Mad-1 WT strain was transfected/infected into SVG-A cells and, at 5th day posttranfection/infection, cells were processed for immunocytochem-istry using anti-Agno (rabbit polyclonal) antibody (Del Valle et al., 2002) and anti-Hsp70 antibody (W2) (mouse monoclonal); and examined under a fluorescence microscope as described in Materials and methods. As previously described (Saribas et al., 2012), viral regulatory agnoprotein, used as a positive control in this viral infection experiment, mostly accumulates around the perinuclear region of the infected cells with small amounts localized to the nucleus (Saribas et al., 2012). Hsp70, on the other hand, exhibited an elevated level of expression in the nucleus of the infected cells compared to that of the cytoplasmic detection, demonstrating that Hsp70 is induced by JCV infection and accumulates in the nucleus at relatively high levels (Fig. 1D), which is consistent with the previous findings with SV40 infection cases (Li et al., 2009).
JCV LT-Ag interacts with Hsp70 in vitro and in vivo
LT-Ag, a major regulatory protein of JCV, is involved in viral DNA replication (Lynch and Frisque, 1990, 1991; Lynch et al., 1994; Tavis and Frisque, 1991), viral gene transcription (Khalili et al., 1987; Lashgari et al., 1989; Safak et al., 2001) and cell transformation (Gordon et al., 1998; Krynska et al., 1997, 1999; Tretiakova et al., 1999); and produced by the viral early coding region. It shows significant sequence homology to the LT-Ag proteins from SV40 and BKV, with the greatest divergence occurring within the carboxy-terminal region (Fanning and Knippers, 1992; Frisque et al., 1984). The JCV early coding region also encodes several small regulatory proteins by an alternative splicing of the early precursor RNA, including small t-Antigen (Sm t-Ag) (Frisque et al., 1984) and T′proteins (T′135, T′136 and T′165) (Bollag et al., 2006, 2000; Frisque et al., 2003). Similarly, small regulatory proteins were also encoded by the early coding regions of the SV40, BKV and PyV, including 17kT protein (Zerrahn et al., 1993), a truncated T (Abend et al., 2009) and tiny T (Riley et al., 1997).
Several functions of the LT-Ag are regulated in part by discrete functional domains of the protein, most of which are located within the amino-terminal portion of the protein (Fig. 2A). For example, three independently acting transforming functions that involve adenovirus E1A conserved region 1 (Cr1) motif (E/DXXLXE/DLXX/I), a Cr2 motif which contains a binding domain (LXCXE) for the pRB family of proteins and a J domain motif (HPDKGG) (Kelley and Landry, 1994) which is found in the DnaJ family of molecular chaperones that interacts with the DnaK family of proteins (Kelley and Landry, 1994). Interaction of J domain of SV40 LT-Ag with the host chaperone, Hsp70, was also demonstrated (Zalvide et al., 1998).
Fig. 2.
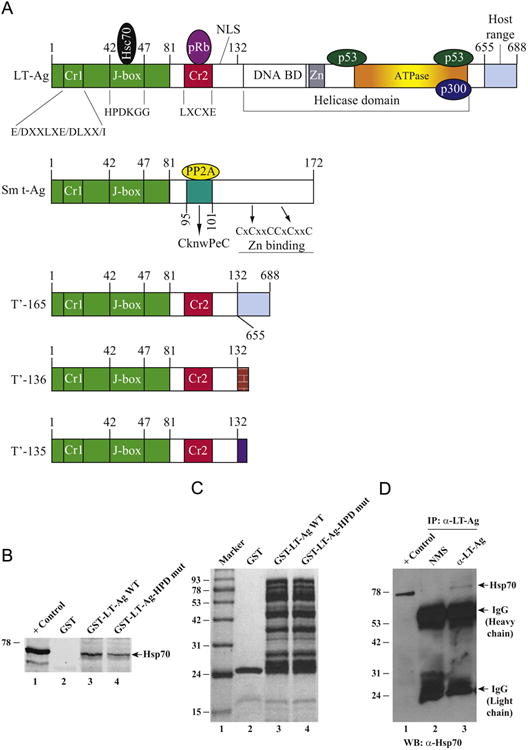
(A) Graphical presentation of JCV early proteins, LT-Ag, Sm t-Ag, and T′ proteins (T′-135, T′-136 and T′-165) with several functional domains, including Cr1, J-box and Cr2 domains (Bollag et al., 2000; Prins and Frisque, 2001; Sariyer et al., 2004; Sullivan et al., 2001; Sullivan and Pipas, 2002). (B) Interaction of Hsp70 with JCV LT-Ag in GST pull-down assays. Whole-cell extracts (0.3 mg) prepared from U-87MG cells were incubated with either GST alone (2 μg, lane 2) or GST-LT-Ag (2 μg, full length) (lane 3) or GST-LT-Ag (HPD to AAA) mutant (2 μg) (lane 4), immobilized on the GSH-Sepharose 4B affinity resins. After extensive washing with the washing buffer, proteins interacting with GST or GST-LT-Ag were analyzed by western blotting using anti-Hsp70 antibody (W2) for detection of Hsp70 as described in Materials and methods using Odyssey CLX infrared imaging system (Li-COR). In lane 1, 20 μg of whole cell extract from U-87MG was loaded as positive control. (C) Analysis of bacterially expressed and affinity purified GST, GST-JCV LT-Ag (full length) and GST-LT-Ag (HPD to AAA) mutant by SDS-10% PAGE followed by Coomassie blue staining. (D) Hsp70 coimmunoprecipitates with LT-Ag. Three hundred micrograms of whole-cell extracts prepared from hamster glial cells, HJC-15b (Raj et al., 1995), which constitutively express JCV LT-Ag and Hsp70, were immunoprecipitated with either normal mouse serum (NMS, 3 μg/lane) (lane 2) or anti-LT-Ag [mouse mAb (PAb416) (lane 3)] antibodies in lysis buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 0.5% NP-40. Immunocomplexes were washed with lysis buffer, resolved on a SDS–10% PAGE and analyzed by western blotting to detect Hsp70 using an anti-Hsp70 (W2) antibody. IP, immunoprecipitation; WB, western blotting. In lane 1, 20 μg protein prepared from HJC-15b cells were loaded as a positive (+) control. The positions of immunoglobulin heavy and light chains; and Hsp70 were indicated by arrows.
One of the unifying structural features of all the JCV early proteins is the presence of the commonly shared motifs located within their amino-terminus, including Cr1 and J-domain motifs (Fig. 2A), indicating that the common regions of these proteins are involved in similar functions including binding to the host heat shock protein, Hsp70. This was investigated by a GST pull-down assay to demonstrate an interaction between JCV LT-Ag and Hsp70. GST or GST-LT-Ag immobilized on GSH-Sepharose 4B beads was incubated with whole-cell extracts prepared from U-87MG cells as described in Materials and methods. After extensive washing of the beads with binding buffer, the proteins bound to GST or GST-LT-Ag were analyzed by western blotting using an anti-Hsp70 antibody. As shown in Fig. 2B, Hsp70 was visibly detected on the blot with an Hsp70 antibody when GST-LT-Ag is incubated with extracts (lane 3) but not with the beads that contained GST alone (Fig. 2B, lane 2). These results indicate a specific association of Hsp70 with LT-Ag. In parallel, to analyze the contribution of 42HPD44 motif of LT-Ag to the interaction between LT-Ag and Hsp70, this motif was mutated to 42AAA44 and GST-pulldown assays were repeated as described for WT LT-Ag. Results from these assays demonstrated that the strength of the interaction between LT-Ag and Hsp70 significantly reduced (> 50%) compare to that of WT (compare lane 4 with 3, Fig. 2B), indicating the importance of HPD motif in interaction between LT-Ag and Hsp70. Fig. 2C demonstrates the analysis of GST, GST-LT-Ag and GST-LT-Ag HPD to AAA mutant proteins by SDS-PAGE followed by coomassie blue staining.
In parallel, in vivo interaction of Hsp70 with LT-Ag was also investigated by a coimmunoprecipitation assay using whole cell extracts prepared from HJC-5b cells, which constitutively express both JCV LT-Ag and Hsp70 as described under Fig. 2D legend. This in vivo assay clearly demonstrated that Hsp70 coprecipitates with LT-Ag when a specific anti-LT-Ag antibody is used in immunoprecipitation assay (Fig. 2D, lane 3) but not with a nonspecific NMS (lane 2). These findings also validate our protein–protein interaction studies by GST pulldown assays shown in Fig. 2B.
VP2 and VP3 associate with LT-Ag
The minor capsid proteins are nuclear proteins with their own nuclear localization signals (Shishido-Hara et al., 2000), similar to those of BKV (Shishido-Hara et al., 2000), SV40 (Clever and Kasamatsu, 1991) and PyV (Chang et al., 1992) VP2/VP3 proteins. These two proteins are encoded by a single mRNA species with two ribosomal entry sites and share a 224 amino acid common sequence region (Fig. 3A). X-ray diffraction data to obtain an electron-density map of the inside of SV40 virions showed that these two proteins locate within the inner layers of a virion along with the viral minichromosome (Griffith et al., 1992). These structural studies showed that the minor capsid proteins must come in contact first with the viral DNA during the encapsidation process. In fact, DNA binding studies using JCV (Huang et al., 2003) and SV40 (Clever et al., 1993) VP2/VP3 proteins demonstrated that two Lys and Arg-rich cluster regions (Lys319 to Lys324 and Lys332 to Ser344, for JCV Mad-1) located at the far C-terminus regions of these proteins (Fig. 3B) play an important role in DNA binding activity of these proteins. Amino acid comparison studies showed that the C-terminal region of JCV, BKV and SV40 VP2/VP3 proteins show high sequence homology. However, there is an 8 amino acid (V/LSRGGSSQK) differential region, located towards the C-terminal region of BKV and SV40 VP2/VP3, where they are missing in JCV VP2/VP3 (Fig. 3B). It is possible that these sequences may play a differential role in encapsidation process of the viral DNA into the capsids in these viruses.
Fig. 3.
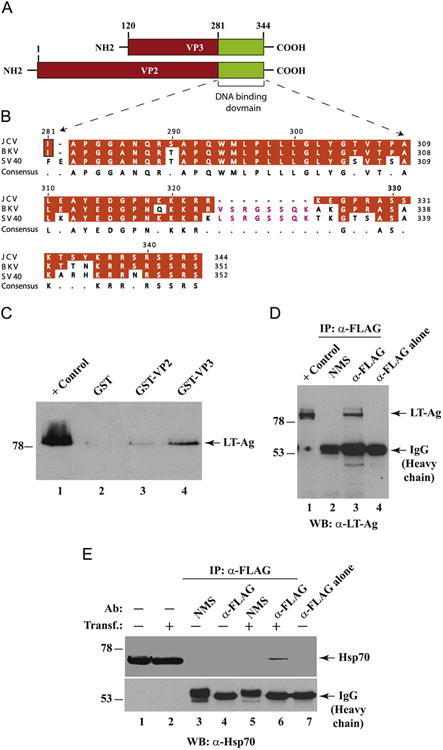
Interaction of JCV LT-Ag with JCV VP2 and VP3 in GST pull-down assays. (A) Graphical representation of JCV Mad-1 VP2/VP3 coding region. Putative DNA binding domain of both capsid proteins, VP2 and VP3 was indicated. (B) Comparison of the amino acids from the putative DNA binding domains of VP2 for JCV, BKV and SV40 using DNASTAR Lasergene software (Wisconsin, Madison). (C) JCV LT-Ag interacts with JCV VP2 and VP3. Bacterially expressed GST (lane 2) or GST-VP2 (lane 3) or GST-VP3 (lane 4) was immobilized on GSH-Sepharose 4B beads and incubated with whole-cell extract prepared from hamster glial cells [HJC-15b, (Raj et al., 1995)] constitutively expressing JCV LT-Ag. Nonbinding proteins were removed from the beads by extensive washing, and proteins interacting with GST alone or GST-VP2 or GST-VP3 were separated on SDS-10% PAGE and analyzed by western blotting using an anti-LT-Ag antibody (Ab-2, monoclonal). In lane 1, whole-cell extracts from HJC-15b cells were loaded as a migration control (10 μg/lane). (D) JCV capsid protein, VP2, coimmunoprecipitates with LT-Ag. Three hundred micrograms of whole-cell extracts prepared from HJC-15b cells transfected with an 3xFLAG-tagged JCV VP2 expression plasmid [p3xFLAG-CMV™-7.1-JCV-VP2 (1–344)] were immunoprecipitated with either normal mouse serum (NMS, 3 μg/lane) (lane 2) or anti-FLAG antibody (3 μg/lane) overnight at 4 °C in lysis buffer containing 0.5% NP-40. Immunocomplexes were washed with lysis buffer, resolved on a SDS–10% PAGE and analyzed by western blotting for detection of LT-Ag using an anti-LT-Ag (PAb416) (lane 3) antibody. In lane 1, 20 μg protein prepared from HJC-15b cells were loaded as a positive (+) control. In lane 4, anti-FLAG antibody alone (3 μg/lane) was subjected to immunoprecipitation as a negative control. The positions of immunoglobulin heavy chain and LT-Ag were indicated by arrows. (E) JCV VP2 interacts with Hsp70 in vivo. In parallel to the in vivo interaction studies described for LT-Ag and Hsp70 in panel D, a possible protein-protein interaction between JCV VP2 and Hsp70 was also investigated by a coimmunoprecipitation assay using whole cell extracts (300 μg/lane) prepared from HJC-15b cells untransfected (lanes 3 and 4) or transfected with 3xFLAG-tagged VP2 expression plasmid (lanes 5 and 6). Coimmunoprecipitation conditions were the same as described for panel D. Ab: Antibody, Transf.: Transfection. In lane 1 and 2, 20 μg protein were separately loaded from untransfected and transfected cells respectively as controls. In lane 7, anti-FLAG antibody (3 μg/lane) was loaded as a negative control. Note that after protein transfer onto the membrane, immunoblot was trimmed into two small strips and incubated with anti-Hsp70 (W2 antibody) separately to avoid a competition between IgGs resulting from antibodies used in immunoprecipitation and Hsp70 for binding to anti-Hsp70 antibody.
Since encapsidation process for JCV takes place in the nucleus of the infected cells and Hsp70 binds to both LT-Ag and the capsid proteins, we reasoned that there might be an association between LT-Ag and JCV capsid proteins as well. This possibility was investigated by a protein–protein interaction assay. For this, whole cell extracts prepared from HJC-15b cells, constitutively expressing JCV LT-Ag (Raj et al., 1995) were incubated with either GST or GST-VP2 or GST-VP3 immobilized on GSH-Sepharose 4B beads as described in the legend of Fig. 3. After extensive washing of the resins with binding buffer, the proteins bound to GST or GST-VP2 or GST-VP3 were analyzed by western blotting using an anti-LT-Ag antibody. As shown in Fig. 3C, we detected a weak and a strong signal for LT-Ag on the western blot when whole cell extracts were incubated GST-VP2 and GST-VP3 respectively (Fig. 3C, lanes 3 and 4), but no such a signal was detected with resins that adsorbed GST alone (lane 2). These results demonstrate a specific interaction between LT-Ag and VP2 and LT-Ag and VP3.
In order to further investigate whether interaction between JCV capsid protein, VP2, and LT-Ag also occurs in vivo, we performed coimmunoprecipitation assays by using whole cell extracts prepared from HJC 15b cells, which are either untransfected or transfected with a FLAG-tagged VP2 expression vector and by using appropriate antibodies as described under respective figure legend (Fig. 3D). As shown in Fig. 3D, LT-Ag coprecipitates with VP2 (lane 3) when a specific (anti-FLAG antibody) but not nonspecific NMS (lane 2) antibody is used for immunoprecipitation, which is consistent with our findings from in vitro GST-pulldown assays (Fig. 3C). In parallel, an in vivo interaction between VP2 and Hsp70 was also examined by coimmunoprecipitation assays as shown in Fig. 3E legend. In agreement with our in vitro GST-pulldown assays (Fig. 3C), results clearly demonstrated that VP2 also associates with Hsp70 (Fig. 3E, lane 6) in vivo. Findings from these in vivo studies also strongly suggest a triple protein complex formation between VP2/VP3, LT-Ag and HSp70 in JCV infected cells as illustrated in Fig. 9 as a model.
Fig. 9.

A Model representing the proposed replication-encapsidation coupling for JCV. Hsp70 first associates with the capsid proteins (VP2/VP3) and tethers them to LT-Ag either in the cytoplasm (I) or in the nucleus (II) at the replication centers. This also allows interaction of VP2/VP3 with LT-Ag (i). Interaction of VP2/VP3 with LT-Ag then induces a conformational change in LT-Ag (ii) resulting in a marked increase in the binding activity of LT-Ag to the Ori (iii). Newly replicated viral DNA is immediately captured by VP2/VP3 capsid proteins, initiating the coupling of the viral DNA replication to encapsidation (iv and v). Finally, nucleocapsids mature as VP1 wraps around them (vi) and virions are released. In addition to aforementioned interaction model between Hsp70, (VP2/VP3) and LT-Ag, other possible orders of interactions are also equally likely. For example, Hsp70/LT-Ag interaction may occur first either in the cytoplasm or in the nucleus, and then VP2/VP3 is added to the complex; or (VP2/VP3)/LT-Ag complex can form first, and then Hsp70 is added to the complex either in the cytoplasm or in the nucleus.
JCV VP2 and VP3 enhance LT-Ag binding to JCV origin of DNA replication, Ori
Being localized to the nucleus and interacting with LT-Ag (Fig. 3C) suggested that the JCV structural proteins, VP2 and VP3 may also play functional roles in JCV replication cycle perhaps by influencing the DNA binding activity of LT-Ag to Ori. We explored this possibility in vitro by employing a band shift assay. For this, we used recombinant proteins (bacterial produced GST, GST-VP2 and GST-VP3; and baculovirus produced LT-Ag) and a double-stranded nucleotide probe, which encompasses 5064–5115 n uc leotide region of J CV (JCV Mad- 1 numbering) which completely overlaps with BS I (binding site I) and IR (inverted repeat) regions of JCV Ori respectively as shown in Fig. 4A (Lynch and Frisque, 1990). As expected, LT-Ag formed complexes with Ori sequences (Fig. 4B, lane 2). The multiple banding patterns that we observed are most likely due to the interaction of monomeric and different multimeric forms of LT-Ag with DNA (Borowiec et al., 1990; Fanning and Knippers, 1992).
Fig. 4.
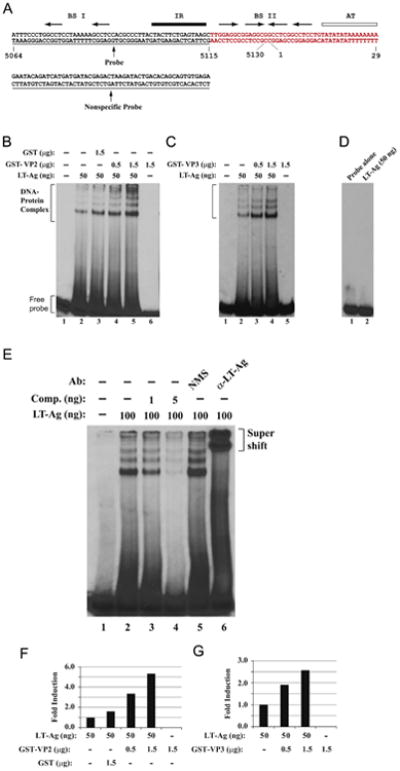
JCV VP2 and VP3 enhance LT-Ag binding to Ori DNA sequences. (A) Double-stranded nucleotide structure of the putative JCV core origin of replication. Arrows indicate the positions of JCV LT-Ag recognition sites. The positions of binding sites I (BS I) and II (BSII) are also indicated. The inverted repeat region (IR) is underlined and the A+T-rich tract (AT) is identified by an open bar. Position of the probe used in the band shift assays is marked by a thin line which encompasses the nucleotides of 5064–5115 (JCV Mad-1, NC_001699). (B and C) Band shift assays. The double-stranded nucleotide encompassing nt 5064–5115 Mad-1 was end-labeled with γ-[32P]-dATP (40,000 cpm/lane) and was incubated with different amount and combinations of the baculovirus-produced JCV LT-Ag, and bacterially produced GST or GST-VP2 or GST-VP3 as indicated on the respective figures. DNA-protein complexes were then separated on a 6% native polyacrylamide gel (PAGE) and visualized by autoradiography. In lane 1, probe alone was loaded on the gel. (D) Incubation of LT-Ag with a nonspecific DNA probe. In parallel to Fig. 4B, a γ-[32P]-ATP-labeled double stranded nonspecific DNA probe (see the sequence for it on panel A) was incubated with baculovirus-produced JCV LT-Ag (50 ng/lane) and formed DNA-protein complexes, if any, were then separated on a 6% native polyacrylamide gel (PAGE) and visualized by autoradiography. (E) Competitive band shift and antibody super-shift assay. Labeled probe was incubated with fixed amount of LT-Ag (100 ng/lane) as indicated plus with different amounts of unlabeled oligonucleotide (Comp.= Competitor) (1 ng and 5 ng, lanes 3 and 4 respectively). In addition, the reaction mixture was also incubated with either normal mouse serum (NMS) (2 μg) or α-LT-Ag (2 μg, Ab-2) antibody as indicated. In lane 1, probe alone was loaded on the gel. DNA-protein-antibody complexes were separated on a 6% PAGE under non-denaturing conditions and analyzed by autoradiography. (F and G) Quantitative analysis of the “DNA-protein complexes” on panels B and C by a semi-quantitative densitometry method using ImageJ program (NIH) and bar graph presentation of the results in arbitrary units in fold. The DNA binding efficiency of LT-Ag in the presence of VP2 or VP3 was expressed relative to that of LT-Ag binding to Ori alone.
Next, we analyzed whether VP2 and VP3 influence the DNA binding activity of LT-Ag to its target sequences on the Ori probe. Interestingly, simultaneous addition of the increasing concentrations of recombinant GST-VP2 (Fig. 4B, lanes 4 and 5) or GST-VP3 (Fig. 4C, lanes 3 and 4) to the reaction mixture significantly enhanced the binding activity of LT-Ag to Ori in a dose-dependent manner, although neither VP2 (Fig. 4B, lane 6 and F) nor VP3 (Fig. 4C, lane 5 and G) alone show any binding activity to Ori, indicating that VP2/VP3 induces LT-Ag binding to DNA while not binding to DNA itself in the absence of LT-Ag. However, addition of both capsid proteins, VP2 and VP3 to the reaction mixture did not alter the banding pattern or mobility of the LT-Ag/Ori complexes other than enhancing the binding activity of LT-Ag, suggesting that both capsid proteins interact with LT-Ag off the DNA and this interaction may result in a conformational change on LT-Ag which subsequently leads to more efficient binding of LT-Ag to its target sequences (Fig. 4B and C). Under identical reaction conditions, the specificity of the VP2/VP3-mediated enhancement of LT-Ag binding to DNA was tested using the GST protein in the binding reactions as well (Fig. 4B, lane 3). In this regard, addition of GST to the binding reaction did not result in a considerable level of induction on LT-Ag binding to DNA (Fig. 4B, lane 3 and F) compared to that observed for GST-VP2 (Fig. 4B, lanes 4 and 5, and F). This suggests that the binding activity of LT-Ag to its target sequences is specifically stimulated by VP2 and VP3 but not GST (Fig. 4B, compare lane 3 with lane 5). Note that in one of our recent reports, we also demonstrated that GST alone did not interact with the same probe at all (Saribas et al., 2012). It is also important to note here that JCV VP2/VP3 was previously reported to have a DNA binding activity (Huang et al., 2003) but we did not observe any DNA activity by these proteins using a specific JCV Ori sequence as a probe. This suggests two possibilities: (1) Either these proteins were not able to bind to DNA under our experimental conditions or (2) the signal sequences that are required for these proteins bind to are located at different locations on the JCV minichromo-some. Additionally, we also investigated the sequence specific binding activity of LT-Ag to Ori by including a nonspecific double stranded probe in our DNA binding assays. As shown in Fig. 4D, LT-Ag does not bind to a nonspecific DNA probe under our DNA binding assays, confirming the specific binding activity of LT-Ag to Ori (Fig. 3D, lane 2). Collectively, these results demonstrate that the binding activity of JCV LT-Ag to its target sequences within Ori is specifically enhanced by the JCV minor capsid proteins, VP2 and VP3, which is a similar case where we have also recently reported an enhanced binding activity of LT-Ag to Ori in the presence of JCV agnoprotein (Saribas et al., 2012).
We further examined the specificity of interaction of LT-Ag with the probe by competitive band shift and antibody super-shift assays. The Ori probe was incubated with LT-Ag protein in the absence or presence of 50- and 250-fold molar excesses of unlabeled competitor DNA (Fig. 4E, lanes 3 and 4). It was observed that, wild-type Ori DNA efficiently competes with the labeled Ori probe for LT-Ag binding (Fig. 4E, lanes 3 and 4). Reaction mixture was also incubated with either a specific antibody directed against LT-Ag or with normal mouse serum (NMS) as indicated. While α-LT-Ag antibody super-shifted the protein–DNA complexes (compare lane 5 with 6), the normal mouse serum (NMS) did not have a noticeable effect on the migration pattern of LT-Ag-Ori complexes. As a result, both the competitive band shift and the antibody super-shift assays confirm the specificity of LT-Ag binding to Ori.
DNA binding domains of VP2 and VP3 are important for their interaction with LT-Ag
In the next series of studies, we attempted to identify the region(s) of VP2/VP3 necessary for the interaction with LT-Ag. For this purpose, series of amino-terminal and carboxy-terminal deletion mutants of VP2/VP3 were created as GST fusion proteins and incubated with whole cells extracts prepared from HJC-15b, constitutively expressing JCV LT-Ag. The bound complexes were resolved by western blotting as described in Materials and methods. As shown in Fig. 5A, three C-terminal deletion mutants (aa 1–119, aa 1–280 and aa 120–280) which do not contain the putative DNA binding domain of VP2/VP3 showed no interaction with LT-Ag (Fig. 5A, lanes 2–4). However, two N-terminal deletion mutants, which encompasses the previously designated DNA binding domain of JCV VP2/VP3 (aa 120–344 and aa 281–344) exhibited a strong binding activity toward LT-Ag (Fig. 5A, lanes 5 and 6). Even the DNA binding domain of both capsid proteins (aa 281–344) is sufficient for this interaction, indicating that VP2/VP3 interacts with LT-Ag through their short C-terminal DNA binding domains. These results along with those from Fig. 3C are summarized in Fig. 5C. Bacterially produced GST-VP2 and the deletion mutants of VP2 fused to GST or GST alone were analyzed by SDS-PAGE followed by Coomassie staining in Fig. 5B. Also note that bacterial heat shock protein, DnaK, also co-purifies with GST-VP2 and its several deletion mutants (Fig. 5B) suggesting that human heat shock protein 70, Hsp70, may also interact with JCV VP2 protein as observed for JCV VP3 (Fig. 1B).
Fig. 5.
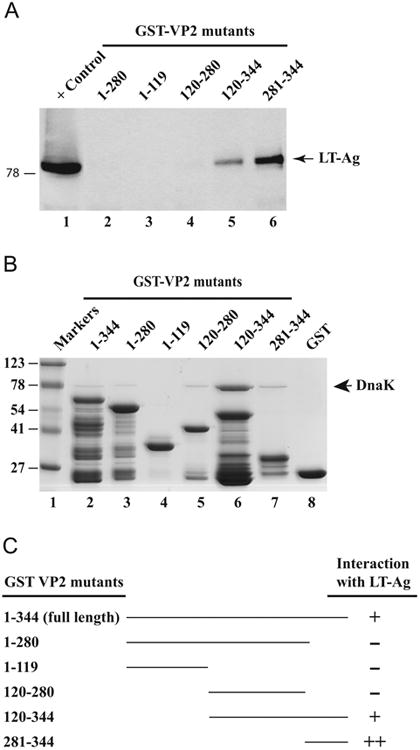
Localization of the domain(s) of VP2 that interacts with LT-Ag. (A) GST-VP2, and N-terminal and C-terminal VP2 deletion mutants immobilized on GSHSepharose 4B beads were incubated with whole-cell extract prepared from HJC-15b cells (Raj et al., 1995) for 2 h at 4 °C as described in Material and methods. The Sepharose beads were washed extensively and proteins interacting with GST or GST-VP2 or GST-VP2 mutants were separated on a SDS-10% PAGE and analyzed by western blotting with an anti-LT-Ag antibody (Ab-2, monoclonal). (B) SDS-PAGE analysis of GST, GST-VP2 and, GST-VP2 N-terminal and C-terminal deletion mutants. (C) Summary of the results obtained from in vitro mapping assays (from Fig. 3C and Fig. 5A). The abilities of VP2 and its deletion mutants to interact with LTAg are shown on the right (+, specific interaction; -, no interaction, ++, enhanced interaction). The positions of molecular weight markers (in kilodaltons, kD) are shown to the left of panel B.
Amino acids (aa 281–344) encompassing the DNA binding domain of VP2/VP3 are sufficient to enhance the LT-Ag binding to Ori
Next, we identified the region of VP2/VP3 responsible for the enhancement of LT-Ag binding to Ori. Full length (FL) VP2 and two deletion mutants, one interacts with LT-Ag (aa 281–344) and the other does not (aa 1–280) were expressed as GST fusion proteins in bacteria, affinity purified, dialyzed against DNA binding buffer as described in Materials and methods and used in a band shift assay. A recombinant JCV LT-Ag alone was incubated with a double-stranded probe (Fig. 6B, lane 2). A labeled DNA probe plus recombinant LT-Ag was also incubated in combination with purified GST protein alone (lane 3) or with GST-VP2 (lane 4, 1–344 FL) or VP2 deletion mutants fused to GST (lanes 6 and 8) as indicated. DNA–protein complexes were then separated on a 6% native non-denaturing gel and analyzed by autoradiography. As shown in Fig. 6B, consistent with the observations from Fig. 4B, LT-Ag binds to Ori (lane 2) and this binding was significantly stimulated by VP2 (lane 4 and Fig. 6C). However, the addition of GST alone or that of an N-terminal deletion mutant of VP2 (aa 1–280) to the binding mixture did not show any appreciable effect on the enhancement of LT-Ag binding to Ori DNA (Fig. 6B, lane 6 and C). In contrast, a deletion mutant that retained the DNA binding domain of VP2/VP3 amino acids (aa 281–344) significantly stimulated the binding activity of LT-Ag to the Ori, even more strongly than full length VP2 (Fig. 6B, lane 8, and C). These findings demonstrate that the DNA binding domain of VP2/VP3 is sufficient for the enhancement of the LT-Ag binding to Ori, which is consistent with our findings from the mapping studies (Fig. 5A, lane 6).
Fig. 6.
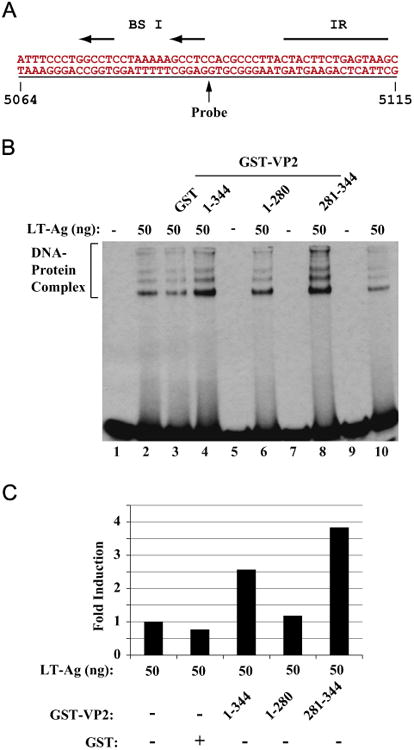
The putative DNA binding domain of VP2/VP3 is important for induced DNA binding activity of LT-Ag to Ori DNA. Labeled probe (A) was incubated either with LT-Ag alone (50 ng, lane 2) or LT-Ag in combination with GST (1.5 μg, lane 3) or LTAg in combination with GST-VP2 full length (1.5 μg lane 4) or LT-Ag plus VP2 deletion mutants fused to GST (1.5 μg each, lanes 6 and 8) as indicated. DNAprotein complexes were separated on a 6% PAGE and analyzed by autoradiography as described in the legend for fig. 4B (B). In lane 1, probe alone was loaded. (C) Quantitative analysis of the “DNA-protein complexes” on panel B by a semiquantitative densitometry method using ImageJ (NIH) and bar graph presentation of the results in arbitrary units in fold. Protein concentrations for GST and GST-VP2 and GST-VP2 mutants are the same as described for panel B. The DNA binding efficiency of LT-Ag in the presence of VP2 or its mutants was expressed relative to that of LT-Ag binding to Ori alone.
Effect of VP2 on LT-Ag-mediated JCV DNA replication
The observed positive effect of VP2 on DNA binding activity of LT-Ag to Ori suggested that VP2 may also exert a regulatory effect on LT-Ag-mediated viral DNA replication. To investigate this possibility, we performed DpnI replication assays (Sami Saribas et al., 2013). A replication-competent plasmid containing the JCV origin of DNA replication was introduced alone or in combination with a VP2 and LT-Ag expression plasmids into U-87MG cells. At 72 h posttransfection the low molecular weight DNA was isolated and digested with BamHI/DpnI restriction enzymes. BamHI linearizes the input (transfected) and replicated DNA; and DpnI is known to specifically digest out the transfected DNA (bacterially produced and methylated input DNA) while keeping the newly replicated viral DNA intact (Hirt, 1967). DNA samples were then analyzed by Southern blotting as described in Materials and methods. As shown in Fig. 7A, as expected, in the presence of LT-Ag, a plasmid containing JCV regulatory region of replication (a replication competent plasmid, pBLCAT3-Mad-1L) was replicated at detectable level (lane 3). However, in the presence of increasing amount of a VP2 expression plasmid, a substantial increase in LT-Ag-mediated viral DNA replication was observed in a dose-dependent manner (lanes 4 and 5, ∼2.5 and ∼6-fold respectively). VP2 alone showed no ability to induce viral DNA replication (lane 6). Taken together, these results demonstrate that VP2 can positively affect LT-Ag-mediated viral DNA replication and functionally plays a role in JCV life cycle even though it was previously described as a structural protein (Frisque et al., 1984) and participates in JCV virion biogenesis. Fig. 7B demonstrates the analysis of the detectable level of expression of the 3XFLAG-tagged VP2 (full-length, aa 1–344) by western blotting. Fig. 7C shows equal level of LT-Ag expression in transfectants from the replication assay (Fig. 7A) confirming that the induced level of replication of JCV genome observed in transfectants in Fig. 7A (lanes 4 and 5) is due to the positive effect of VP2 expression on LT-Ag mediated replication.
Fig. 7.
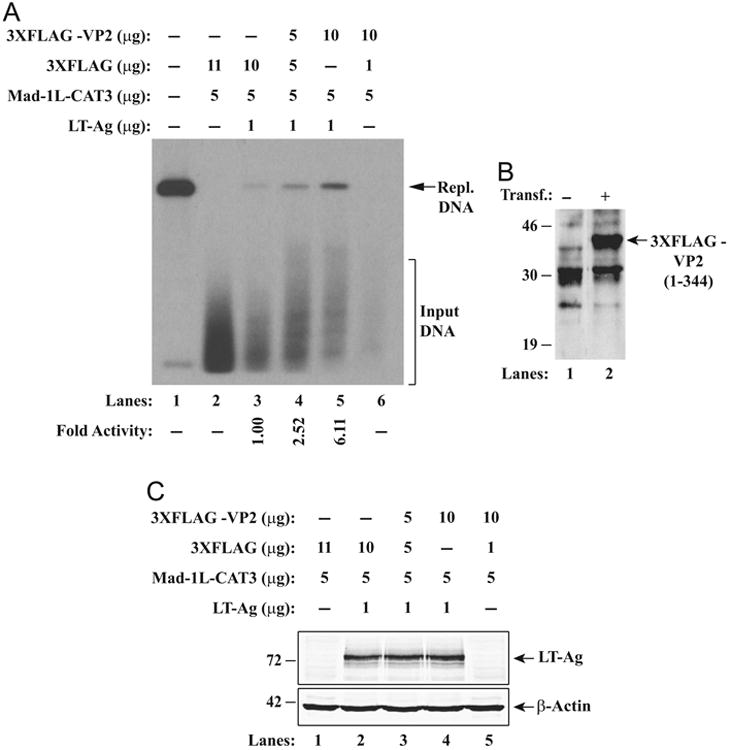
Effect of VP2 on LT-Ag-mediated JCV DNA replication. (A) DpnI assay. Replication assays were carried out as previously described (Safak et al., 2001). Briefly, a replication-competent plasmid, pBLCAT3-Mad-1L (late), was transfected alone or in combination with expression vectors into U-87MG (0.4 × 106 cells/60 mm tissue culture dish) cells as described in materials and methods. At 72 h posttransfection, the low-molecular-weight DNA containing both input and replicated viral DNA was isolated using QIAprep spin columns (QIAGEN) (Ziegler et al., 2004), digested with BamHI and DpnI enzymes, resolved on a 1% agarose gel and analyzed by Southern blotting as described in Materials and methods. The amounts of plasmids used in transfections are indicated on the figure. The newly replicated DNA is indicated by a labeled arrow (Repl. DNA=Replicated DNA). The replication efficiency of the pBLCAT3-Mad-1L plasmid was quantitated by a semi-quantitative densitometry method using ImageJ (NIH) and presented in arbitrary units in fold at the bottom side of the figure. The replication efficiency of pBLCAT3-Mad-1Lin the presence of LT-Ag was compared with the other data points. (B) Transient expression analysis of 3XFLAG-CMV 7.1-VP2 plasmid in U-87MG cells by western blotting using an anti-FLAG antibody as described in materials and methods. The position of the 3XFLAG-tagged VP2 protein is indicated by a labeled arrow. Transf.: Transfection. (C) Analysis of JCV LT-Ag expression in nuclear extracts prepared from replication assays in panel A. In parallel to preparing samples for DNA replication assays, nuclear extracts prepared from the transfectants were analyzed for expression levels of JCV LT-Ag and β-actin for as a gel loading control.
Amino acids spaning 281–344 region ofVP2 confirms the cooperation between LT-Ag and VP2 in LT-Ag mediated viral DNA replication
To further assess the functional interaction between VP2/VP3 and LT-Ag, we examined the effect of deletion mutant VP2 proteins which have retained or lost their binding activity with LT-Ag on JCV replication in glial cells (U-87MG). We chose a mutant protein [VP2 (aa 1–280)] with no binding activity to LT-Ag and a mutant protein [VP2 (aa 281–344)] with a strong binding activity to LT-Ag in GST pull-down assay (Fig. 5A). The mutant VP2 (aa 1–280) that does not contain the DNA binding domain of the protein, showed no appreciable stimulatory effect on the LT-Ag-mediated viral DNA replication (Fig. 8A, lanes 4 and 5). However, a mutant VP2 (aa 281–344) that retains the DNA binding domain and exhibits a strong interaction activity towards LT-Ag significantly induced the LT-Ag-mediated viral DNA replication in a dose-dependent manner (Fig. 8D, lanes 4 and 5), consistent with our results obtained from full length VP2 replication studies (Fig. 7A). Collectively, the data from these replication studies utilizing deletion mutants of VP2, one which physically and functionally interact with LT-Ag [VP2 (aa 281–344)] and one which lacks such activities [VP2 (aa 1–280)], further confirm the significance of an interaction between VP2/VP3 and LT-Ag in regulation of JCV replication. Of note, the stable expression of VP2 mutants used in replication assays was investigated by western blotting (Fig. 8B and E) and therefore the absence of functional interaction between LT-Ag and VP2 (1–280) mutant may not be attributed to its lack of expression in transfected cells. In addition, in Fig. 8C and F, the relatively unaltered expression level of LT-Ag in replication assays (Fig. 8A and D respectively) was demonstrated by western blotting.
Fig. 8.
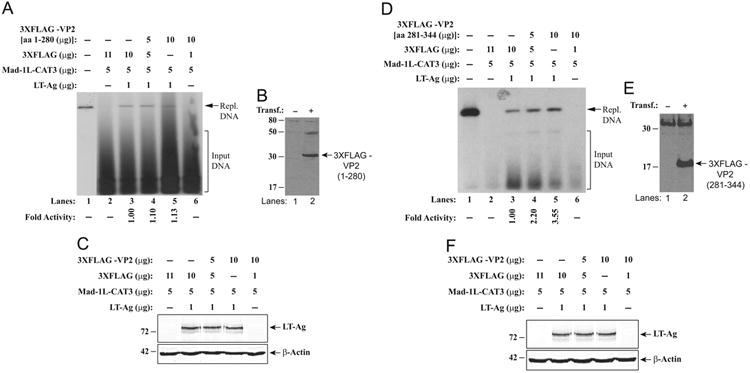
Effect of VP2 mutants on LT-Ag-mediated JCV DNA replication. (A and D) DpnI assay. Replication assays were carried out as described for Fig. 7A. The amounts of plasmids used in transfections are indicated on the figure. The newly replicated DNA is indicated by a labeled arrow (Repl. DNA=Replicated DNA). The replication efficiency of the pBLCAT3-Mad-1L plasmid was quantitated by a semi-quantitative densitometry method using ImageJ (NIH) and presented in arbitrary units in fold at the bottom side of each figure. The replication efficiency of pBLCAT3-Mad-1L in the presence of LT-Ag was compared with the other data points. (B and E) Transient expression analysis of p3XFLAG-CMV™-7.1-JCV VP2 mutant plasmids (aa 1–280 and aa 281–344) in U-87MG cells by western blotting using an anti-FLAG antibody as described in materials and methods. The position of the FLAG-tagged VP2 (1–280) and that of FLAG-tagged VP-2 (281–344) on each blot are each indicated by a labeled arrow. (C and F) Analysis of JCV LT-Ag expression in nuclear extracts prepared from transfectants by western blotting as described for Fig. 7C. Blots were also probed for detection of β-actin using an anti-β actin antibody as control.
Discussion
JCV replicates and assembles into infectious virions in the nucleus. However, the molecular mechanism of this process is not fully understood. In this report, we attempted to shed more light on this process by analyzing the interactions between viral minor capsid proteins, VP2/VP3, LT-Ag and a host chaperone system protein, Hsp70. We demonstrated that Hsp70 interacts with LT-Ag (Fig. 2B) and viral structural protein, VP3 (Fig. 1B) and heavily accumulates in the nucleus of the infected cells (Fig. 1D). It is known that Hsp70 interacts with the J domain (co-chaperone domain) of LT-Ag (Sullivan et al., 2000, 2001; Sullivan and Pipas, 2002). Note that all early regulatory proteins of JCV contain this J domain (Fig. 2A), indicating that they all contribute to the JCV replication cycle. We also demonstrated that viral structural proteins, VP2/VP3 enhance the DNA binding activity of LT-Ag to Ori through their C-terminal DNA binding domains (Fig. 6B). It appears that this enhancement leads to an increase in the level of LT-Ag mediated viral DNA replication (Figs. 7A and 8D). Collectively, these results suggest a novel hypothesis in which Hsp70 mediates an interaction between LT-Ag and the viral capsid proteins and plays a role in coupling of viral DNA replication to the virion encapsidation process as illustrated in a model presented in Fig. 9. According to this model, upon translation, Hsp70 interacts with the viral capsid proteins, VP2 and VP3, in cytoplasm brings them to the replication centers in the nucleus and anchors them to the N-terminus J-domain of LT-Ag, thereby they may also mediate an interaction between capsid proteins and LT-Ag. This interaction then induces a conformational change on LT-Ag structure, resulting in an enhancement in its DNA binding activity and therefore its ability to replicate DNA. The newly replicated viral genome is then captured by VP2/VP3 through their DNA binding domains, initiating a nucleation process during the initial stages of virion maturation. This then leads to the coupling of the viral DNA replication to the virion encapsidation events. Finally, VP1 is added to the last layer of a virion and the maturation process is completed and virions are released.
The DNA binding domain of VP2/VP3 was previously characterized for JCV (Huang et al., 2003) and SV40 (Clever et al., 1993). Amino acid comparison studies (Fig. 3B) revealed that the C-terminal region of VP2/VP3, encompassing the DNA binding domain of each protein, exhibits high homology between JCV and BKV than that of between JCV and SV40. This comparison also revealed that the C-terminal regions VP2/VP3 of SV40 and BKV contain an extra and highly conserved 8 amino acid sequence (VSRGSSQK for BKV and LSRGSSQK for SV40) than that of JCV. It is known that JCV is a relatively slow growing virus than BKV and SV40; and thus the presence or absence of these additional amino acid sequences may play a consequential role on the efficiency of the encapsidation process of each virus, which ultimately affects, in part, the propagation rate of these viruses.
Our DNA binding studies demonstrated that capsid proteins, VP2/VP3, significantly stimulate the DNA binding activity of LT-Ag to Ori without directly interacting with DNA (Fig. 4B and C), which is similar to a case where we have recently reported an induced binding activity of LT-Ag in the presence of JC agnoprotein (Saribas et al., 2012). This is also a novel finding suggesting that JCV capsid proteins may be involved in viral DNA replication through interaction with LT-Ag. However, these proteins do not seem to form an apparent ternary complex with LT-Ag/DNA complex under our band shift assay conditions, which is apparent from the lack of the alteration in the migration pattern of DNA–protein complexes upon addition of capsid proteins into the binding mixture (Fig. 4B and C). These findings suggest the following possibilities: (i) one possibility is that interaction of capsid proteins with LT-Ag takes place off the DNA but induces a conformational change on LT-Ag protein, resulting in a more efficient DNA binding. The capsid proteins were then most likely liberated from LT-Ag/capsid protein complexes before or after LT-Ag binds to DNA (Fig. 4B and C). This is, at least, one of the reasons that capsid proteins are not the part of LT-Ag/DNA complex. (ii) Another possibility is that the interaction between capsid proteins and LT-Ag/DNA complex is weak and therefore is unstable under our gel shift conditions. Nonetheless, the ability of one protein to influence the DNA binding capacity of another one has been well-established. There are previously reported similar interactions in the literature, including the stimulation of the serum response factor binding to DNA by Phox1 (Grueneberg et al., 1992), induction of YB-1 binding to the 23-bp element of JCV archetype by LT-Ag (Safak et al., 1999), the induction of Tst-1 binding to DNA by HMG-1/Y (Leger et al., 1995) and enhancement of LT-Ag binding to Ori by JCV agnoprotein (Saribas et al., 2012). We also noticed that although JCV minor capsid proteins contain a DNA binding domain, we did not detect a noticeable DNA binding activity by both capsid proteins in our DNA binding assays, suggesting that either our DNA binding assay conditions are not favorable for such a binding or the DNA fragment that was used as a probe in our band shift assays does not contain a DNA signal sequence for binding of the minor capsid proteins.
Mapping studies demonstrated that JCV minor capsid proteins, VP2/VP3 interact with LT-Ag through their DNA binding domains (aa 281–344) (Fig. 5A) and this region is also sufficient to induce LT-Ag binding Ori (Fig. 6B). This region also was shown to stimulate the LT-Ag-mediated viral DNA replication (Fig. 8D). All these findings suggest that the DNA binding domain of the capsid proteins also plays functional roles either in JCV DNA replication or in virion encapsidation or in both processes. It should be noted here that we, first time, report a functional role for these two structural proteins of JCV, VP2 and VP3, in viral life cycle. Further investigations are required to elucidate the details of the functional roles of these proteins in JCV biology.
Viruses are intracellular parasites and recruit host cellular components for their own replication, protein synthesis and virion assembly. During the course of virion production, a large number viral proteins are synthesized in relatively short period of time, some of which require proper protein folding in order to be functional and host chaperones are required for this process. In addition, in order to create a favorable environment for their propagation, and avoid premature host cell death, viruses need to interfere with cellular processes including premature induction of apoptosis, signal transduction and cell cycle regulation pathways. Many host chaperones including Hsp70s are involved in control of these cellular processes and some viruses reprogram their host environment by interfering with the chaperone activity of the host proteins. Hsp70 chaperones are the central component of the cellular chaperone network and are being frequently recruited by numerous DNA and RNA viruses (Mayer, 2005). Without their involvement, viruses cannot complete their replication cycle (Mayer, 2005). Hsp70s are potentially involved in all phases of the viral life cycle including, cell entry (Guerrero et al., 2002), viral transcription (Devireddy et al., 2000), virions disassembly, transfer of viral genome into the nucleus, replication of the viral genome (Chromy et al., 2003), morphogenesis of the virion particles (Mayer, 2005; Napuli et al., 2003).
Heat shock families of proteins are critically important for the replication cycle of many prokaryotic and eukaryotic viruses (Mayer, 2005). In fact, two of the chaperone systems in Escherichia coli Hsp70 (DnaK, DnaJ and GrpE) and Hsp60 (GroEL and GroES) were originally discovered as the host factors essential for growth of prokaryotic (bacterial) viruses, bacteriophage λ and T4 (Georgopoulos, 1977; Georgopoulos et al., 1972), where it was demonstrated that the E. coli bacteriophage λ protein λP sequesters the E. coli DNA helicase DnaB and recruits it to the origin of the replication of λ bacteriophage and where it interacts with the four dimers of λO assembled at Ori λ to form a multimeric complex (Liberek et al., 1988). In the case of eukaryotic viral infections, it was shown that DNA binding activity of the human papillomavirus-11 (HPV-11) DNA helicase E1 to DNA was stimulated by Hsp70, HdJ1 and HdJ2 in a ATP-dependent manner (Liu et al., 1998). Hsp70 system also was shown to be necessary for the replication of the SV40 genome. The N-terminus of SV40 LT-Ag, like JC virus and BK virus LT-Ag, contains a signature motif for J-domain, which is important for recruiting Hsp70 to the replication centers and modulating the activity of LT-Ag. There are also reports describing the involvement of Hsp70 system in virion morphogenesis where the interaction of Hsp70 with capsid proteins, VP1, VP2 and VP3 of PyV was demonstrated (Chromy et al., 2003). Purified VP1 and VP3 was shown to assemble into virion-like structures in the presence of prokaryotic Hsp70 (DnaK, DnaJ and GrpE) chaperones in an ATP-dependent manner. Such an assembly was also demonstrated using a mammalian Hsp70 when the J-domain of SV40 LT-Ag was used as a co-chaperone (Chromy et al., 2003).
Understanding the molecular mechanisms of the JCV virion maturation remains elusive. The host heat shock proteins, including Hsp70, appear to play important regulatory roles in the JCV virion maturation process, which is consistent with our findings from protein–protein interaction studies data and published reports (Mayer, 2005). We will further characterize the molecular mechanisms of the involvement of Hsp70 in JCV virion maturation particularly in coupling of JCV replication to virion encapsidation process, which will eventually help us develop effective therapeutic strategies against JCV infections in affected individuals.
Materials and methods
Cell lines
U-87 MG is a cell line that was derived from human malignant gliomas (ATCC HTB-14) (Ponten and Macintyre, 1968). SVG-A is a human cell line established by transformation of primary human fetal glial cells with an origin-defective SV40 mutant (Major et al., 1985). These transformed cells (SVG-A) do not express either SV40 viral capsid proteins (VPs) or agnoprotein, but express SV40 large T antigen (LT-Ag). HJC-15b is a hamster glial cell line constitutively expressing JCV LT-Ag (Raj et al., 1995). These cell lines were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics [penicillin/streptomycin (100 μg/ml), ciprofloxacin (10 μg/ml]. They were maintained at 37 °C in a humidified atmosphere supplemented with 7% CO2.
Plasmid constructs
The cloning of JCV LT-Ag into a bacterial expression vector, pGEX1λT, was previously described (Safak et al., 2001). HPD to AAA mutations were made on pGEX1λT-LT-Ag by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit (Cat#: 210516, Agilent Technologies) and the plasmid was designated as pGEX1λ-LT-Ag (HPD to AAA). The full-length of JCV VP2 (1–344) and VP3 [VP2 (120–344)]; and the deletion mutants of each protein were cloned into a bacterial expression vector, pGEX1λT (GE Health Care, Cat#: 27-4805-01), at BamHI/EcoRI sites by PCR-based cloning using appropriate PCR primers. The resulting plasmids were designated as pGEX1λT-VP2 (1–344), pGEX1λT-VP3 (1–244), pGEX1λT-VP2 (1–280), pGEX1λT-VP2 (1–119), pGEX-1λT-VP2 (120–280), pGEX1λT-VP2 (120–344), pGEX1λT-VP2 (281–344). The full length of JCV VP2 (aa 1–344) and VP3 (aa 1–244) and deletion mutants of VP2 (aa 1–280 and aa 281–344) were cloned into a eukaryotic expression vector (p3XFLAG-CMV™-7.1, Sigma, Cat#: E7533) at BamHI and EcoRI sites. Each clone was tagged at the N-terminus with a 3X-FLAG peptide (MDYKDHDGDYKDHDIDYKDDDDK) and designated as p3XFLAG-CMV™-7.1-JCV VP2 (1–344), p3XFLAG-CMV™-7.1-JCV VP3 (1– 244), p3XFLAG-CMV™-7.1-JCV VP2 (1–280) and p3XFLAG-CMV™-7.1-VP2 (281–344). JCV LT-Ag was also cloned into a eukaryotic expression vector [pcDNA3.1(+), Invitrogen, Cat#: V790-20] at EcoRI site and designated as pcDNA3.1(+)–JCV LT-Ag, which expresses both LT-Ag and Sm t-Ag. The integrity of all plasmids was verified by DNA sequencing.
Expression and purification of recombinant proteins
Bacterial expression of Glutathione-S-Transferase (GST) alone and GST-fusion proteins were previously described (Safak et al., 2001). Briefly, a 100 ml overnight culture of E. coli DH5α cells transformed with plasmids expressing either GST alone or full length VP2/VP3 fused to GST [(GST-VP2 (1–344) and GST-VP3 (1–244)] or different deletion mutants of VP2/VP3 fused to GST [GST-VP2 (1–280), GST-VP2 (1–119), GST-VP2 (120–280), GST-VP2 (120–344), GST-VP2 (281–344)] were first diluted 1:10 in fresh Luria-Bertani (LB) broth in 1 L supplemented with ampicillin (100 μg/ml) and grown at 37 °C until at an optical density (at 595 nm) of 0.5. Bacterial cultures were then induced with 0.3 mM isopropyl-β-thiogalactopyranoside (IPTG) and incubated for an additional 2 h at 28 °C. Bacterial cells were harvested by centrifugation at 4 °C and pellets were resuspended in 20–40 ml of PENT lysis buffer containing 20 mM Tris–HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40 (NP-40) supplemented with a cocktail of protease inhibitors (Sigma, Cat #: P8465). Bacterial cells were first sonicated and clear cell lysates were prepared by centrifugation at 20,000 × g. Lysates were then incubated with 200 μl of 50% Glutathione (GSH)-Sepharose 4B beads (GE Health Care, Cat#: 17-0756-05) in phosphate buffered saline (1 × PBS) overnight at 4 °C. GST fusion proteins bound to beads were purified by three cycles of washing and centrifugation with 5 ml of PENT lysis buffer, eluted with GSH elution buffer (50 mM Tris– HCl, 10 mM reduced GSH, pH 8.0), dialyzed overnight in band shift assay sample buffer [12 mM HEPES (pH 7.9), 4 mM Tris–HCl (pH 7.5), 60 mM KCl, 5 mM MgCl2 and 1.0 mM DTT].
JCV LT-Ag was expressed in Spodeptera frugiperda (Sf9) insect cells using a baculovirus expression system and affinity purified as previously described (Bollag et al., 1996). Finally, the protein quality of each protein after purification was determined by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining.
Electrophoretic mobility shift assays (EMSA)
EMSA were carried out as previously described (Kim et al., 2003; Sadowska et al., 2003; Saribas et al., 2012). Briefly, a doublestranded synthetic oligonucleotide corresponding to the part of JCV Mad-1 origin (nt 5064–5115) was end-labeled with [γ-32P] ATP using T4 polynucleotide kinase and gel purified. Baculovirus-produced and purified JCV LT-Ag or bacterially produced and affinity purified GST alone or GST-VP2/VP3(full length or their mutants) were incubated with labeled probe (40,000-cpm/lane) in different combinations, as described in the respective figure legends in 50 μl reaction volume, in binding buffer containing 1.0 μg poly(dI-dC), 12 mM HEPES (N-2-hydroxyethylpipezine-N-2-ethanesulfonic acid) (pH 7.9), 4 mM Tris–HCl (pH 7.5), 60 mM KCl, 5 mM MgCl2 and 1.0 mM DTT. The reaction mixtures were incubated at 4 °C for 45 min to allow the formation of DNA–protein complexes. The complexes were then fractionated on a 6% polyacrylamide gel in 0.5 × TBE [1 × TBE is 89 mM Tris–HCl (pH 8.0), 89 mM boric acid, and 2 mM EDTA (pH 8.0)]. The gel was dried and DNA–protein complexes were detected by autoradiography. Detailed experimental conditions for all band shift assays are described in the respective figure legends.
GST affinity chromatography (GST pulldown) and coimmunoprecipitation assays
GST pull-down assay was performed as previously described (Safak et al., 2001). Briefly, 2 μg of either GST alone, or GST-VP2 wt (full length) or JCV VP2 deletion mutants fused to GST, immobilized on GSH-Sepharose 4B beads were incubated with 0.3 mg of whole-cell extract prepared from hamster glial cells (HJC-15b) constitutively expressing JCV LT-Ag (Raj et al., 1995), for 2 h at 4 °C in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.5% NP-40. The protein-complexed beads were washed extensively with lysis buffer and resolved by SDS-10% PAGE followed by western blot analysis using anti-LT-Ag antibody [Ab-2, mouse mAb (PAb416), EMD Millipore, Cat#: DP02-200UG]. Amersham ECL prime western blot detection system (Cat#: RPN2232) was used to develop western blots. Ab-2 monoclonal antibody was raised against SV40 LT-Ag but cross-reacts with JCV LT-Ag. Similarly, GST pull-down assays were also performed to examine an interaction between GST-JCV LT-Ag (2 μg fusion protein) and human heat shock protein Hsp70 (0.3 mg of whole cell extract prepared from U-87 MG) and that between GST-JCV VP3 (2 μg fusion protein) and Hsp70 (0.3 mg of whole cell extract prepared from U-87 MG) as described above using anti-Hsp70 antibody (W2) (Santa Cruz, Cat#: SC-24). In vivo interaction of Hsp70 with LT-Ag and that of capsid protein, VP2, with both LT-Ag and Hsp70 were examined by coimmunoprecipitation assays as described under respective figure legends.
Indirect immunofluorescence microscopy
Indirect immunofluorescence microscopy studies were performed as previously described (Sadowska et al., 2003; Sariyer et al., 2011). Briefly, SVG-A cells were transfected/infected with pBluescript KS (+)–JCV Mad-1 WT DNA already linearized by BamHI restriction enzyme digestion and at 5th day posttransfection/infection, cells were seeded at subconfluency on polylysinecoated glass chamber slides (Thermo Scientific, Cat#:177380). The next day, cells were washed twice with 1 × PBS and fixed in ice-cold acetone. Fixed cells were then blocked with 5% bovine serum albumin in 1 × PBS for 2 h, and incubated with two different antibody combinations (anti-JCV Agno primary polyclonal rabbit antibody (1–200 dilution), Del Valle et al., 2002) plus anti-Hsp70 monoclonal mouse antibody (Santa Cruz, Cat#: SC-24, 1:100 dilution) overnight. Antibody dilution and incubation were performed in incubation buffer (PBST=[1 × PBS-0.01% Tween 20] buffer. Cells were then washed three times with PBST buffer for 10 min intervals and subsequently incubated with a Rhodamineconjugated goat anti-mouse plus fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibodies (BD Biosciences) for 45 min. Cell were finally washed with incubation buffer three times at 10 min intervals, mounted using Vectashield mounting media (Vector Laboratories Inc., Cat#: H-1200) and examined under a fluorescence microscope (Nikon eclipse TE300; objectives: 40 × /1.3 oil and eyepiece: 10×; operating software: Slidebook 5.0.) for detection of JCV agnoprotein and Hsp70.
Replication assays
Replication assays were carried out as previously described (Safak et al., 2001). Briefly, a replication-competent plasmid, pBLCAT3-Mad-1 L, containing the regulatory region of the Mad-1 strain of JCV in late orientation (L), was introduced alone or in combination with eukaryotic expression vectors, pcDNA3.1(+) JCV LT-Ag, p3XFLAG-CMV™-7.1-JCV VP2, p3XFLAG-CMV™-7.1-JCV VP2 (1–280) and p3XFLAG-CMV™-7.1-VP2 (281–34), into U-87 MG (0.4 × 106 cells/60 mm tissue culture dish) cells with the calcium phosphate (CaPO4) precipitation method (Kim et al., 2003). Plasmid concentrations used in transfections are indicated in the respective figure legends, and the total amount of DNA transfected into the cells was normalized with appropriate empty DNA vectors. Cells were washed with 1 × PBS at 3 h posttransfection and the medium was replenished. At 72 h posttransfection, the low-molecular-weight DNA containing both input and replicated viral DNA was isolated using QIAprep® small spin columns (QIAGEN) as previously described (Ziegler et al., 2004), digested with BamHI and DpnI enzymes, resolved on a 1% agarose gel and analyzed by Southern blotting. DpnI enzyme specifically digests transfected and bacterially methylated DNA and leaves the newly replicated DNA intact. Probes for Southern blots were prepared using pBLCAT3-Mad-1 L (late) plasmid as a template in a ready-prime random labeling reaction (Cat#: 300385, Agilent Technologies Inc).
Preparation of whole-cell and nuclear extracts for western blotting
Whole cell extracts were prepared as described previously (Safak et al., 2001). Briefly, U-87 MG cells (1 × 106) were lysed in a lysis buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 0.5% NP-40) on a racking platform at 4 °C for 20 min. Cell lysate was then cleared by centrifugation at 15,000× for 5 min at 4 °C and stored at −70 °C until use. Nuclear extracts were prepared as previously described (Kim et al., 2003). Briefly, U-87 MG cells (1 × 106 cells/100 mm culture dish) were separately transfected with a VP2 and its deletion mutant expression plasmids [p3XFLAG-CMV™-7.1-JCV VP2, p3XFLAG-CMV™-7.1-JCV VP2 (1–280) and p3XFLAG-CMV™-7.1-VP2 (281–34)] by CaPO4 method and second day posttranfection, nuclear extracts were prepared by a modification of the miniextract protocol, as described earlier (Schreiber et al., 1989). Transfected and untransfected cells were harvested by trypsinization, washed once with complete DMEM and twice with 1 × PBS, and transferred to an Eppendorf tube. The cells were then resuspended in cold hypotonic buffer [10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM ethylene glycol bis(β -aminoethyl ether)-N,N,N,N,-tetraacetic acid (EGTA), 1 mM dithiothreitol (DTT), and protease inhibitors Cat #: P8340, Sigma] and allowed to swell on ice. Cellular membranes were cleared by the addition of NP-40 (0.5% final concentration) and vortexing for 12 s in full speed. The nuclei were pelleted by centrifugation at 6,000 RPM (Eppendorf microcentrifuge), resuspended in cold extraction buffer containing 20 mM HEPES (pH 7.9), 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT and supplemented with a cocktail of protease inhibitors (Sigma, Cat#: P8340), and extracted at 4 °C for 15 min on a racking platform. The nuclear extracts were centrifuged at 15,000 RPM (Eppendorf microcentrifuge) for 5 min and the cleared supernatant was frozen at −70 °C until use. In parallel to preparing DNA samples for DNA replication assays, nuclear extracts were also prepared to analyze the expression level of LT-Ag from transfectants by western blotting as described above.
Western blotting
Expression plasmids [p3XFLAG-CMV™-7.1-JCV VP2 (1–344), p3XFLAG-CMV™-7.1-JCV VP2 (1–280) and p3XFLAG-CMV™-7.1-VP2 (281–344)] were transfected into U-87 MG cells by the CaPO4 precipitation method (Kim et al., 2003). At 48 h posttransfection, nuclear extracts were prepared from transfected and untransfected cells. Forty micrograms of the nuclear extract were analyzed for full length JCV VP2 by 10% SDS-PAGE followed by western blotting using anti-FLAG M2 antibody (Cat # 200470-21, Agilent Technologies Inc.). Expression of the deletion mutants of JCV VP2 (aa 1–280 and aa 281–344) was analyzed on a 4–20% NuPAGE® Bis-Tris gel (Life Technologies™) and followed by western blotting as described for JCV VP2 above using monoclonal anti-FLAG M2 antibody. JCV LT-Ag expression was also analyzed in nuclear extracts prepared from the transfectants used for DNA replication assays. Blots were probed with a primary anti-SV40 LT-Ag antibody (Ab-2, EMD Millipore, Cat #: DP02-200UG), which is also cross-reactive with JCV LT-Ag, and with secondary goat anti-mouse antibody (IRDYE 680 LT, LI-COR) and were analyzed using LI-COR Odyssey CLX instrument. Blots were also probed with a primary monoclonal anti-β-actin (Sigma, Cat #: A5441) and secondary goat anti-mouse antibody labeled with IRDYE 800 CW (LI-COR) and were analyzed on LI-COR Odyssey CLX instrument.
Acknowledgments
We would like to thank past and present members of the Department of Neuroscience and Center for Neurovirology for their insightful discussion and sharing of ideas and reagents. This work was made possible by grants awarded by NIH to MS.
References
- Abend JR, Joseph AE, Das D, Campbell-Cecen DB, Imperiale MJ. A truncated T antigen expressed from an alternatively spliced BK virus early mRNA. J Gen Virol. 2009;90(Pt 5):1238–1245. doi: 10.1099/vir.0.009159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An P, Saenz Robles MT, Pipas JM. Large T antigens of polyomaviruses: amazing molecular machines. Annu Rev Microbiol. 2012;66:213–236. doi: 10.1146/annurev-micro-092611-150154. [DOI] [PubMed] [Google Scholar]
- Beere HM, Green DR. Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001;11(1):6–10. doi: 10.1016/s0962-8924(00)01874-2. [DOI] [PubMed] [Google Scholar]
- Bollag B, Kilpatrick LH, Tyagarajan SK, Tevethia MJ, Frisque RJ. JC virus T′135, T′136 and T′165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol. 2006;12(6):428–442. doi: 10.1080/13550280601009553. [DOI] [PubMed] [Google Scholar]
- Bollag B, Mackeen PC, Frisque RJ. Purified JC virus T antigen derived from insect cells preferentially interacts with binding site II of the viral core origin under replication conditions. Virology. 1996;218(1):81–93. doi: 10.1006/viro.1996.0168. [DOI] [PubMed] [Google Scholar]
- Bollag B, Prins C, Snyder EL, Frisque RJ. Purified JC virus T and T′ proteins differentially interact with the retinoblastoma family of tumor suppressor proteins. Virology. 2000;274(1):165–178. doi: 10.1006/viro.2000.0451. [DOI] [PubMed] [Google Scholar]
- Borowiec JA, Dean FB, Bullock PA, Hurwitz J. Binding and unwinding – how T antigen engages the SV40 origin of DNA replication. Cell. 1990;60(2):181–184. doi: 10.1016/0092-8674(90)90730-3. [DOI] [PubMed] [Google Scholar]
- Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, Pipas JM, Silver PA, Roberts TM, Schaffhausen BS, DeCaprio JA. DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev. 1997;11(9):1098–1110. doi: 10.1101/gad.11.9.1098. [DOI] [PubMed] [Google Scholar]
- Chang D, Haynes JI, 2nd, Brady JN, Consigli RA. Identification of a nuclear localization sequence in the polyomavirus capsid protein VP2. Virology. 1992;191(2):978–983. doi: 10.1016/0042-6822(92)90276-u. [DOI] [PubMed] [Google Scholar]
- Chromy LR, Pipas JM, Garcea RL. Chaperone-mediated in vitro assembly of polyomavirus capsids. Proc Natl Acad Sci USA. 2003;100(18):10477–10482. doi: 10.1073/pnas.1832245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clever J, Dean DA, Kasamatsu H. Identification of a DNA binding domain in simian virus 40 capsid proteins Vp2 and Vp3. J Biol Chem. 1993;268(28):20877–20883. [PubMed] [Google Scholar]
- Clever J, Kasamatsu H. Simian virus 40 Vp2/3 small structural proteins harbor their own nuclear transport signal. Virology. 1991;181(1):78–90. doi: 10.1016/0042-6822(91)90472-n. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radhakrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Natl Cancer Inst. 2002;94(4):267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- Devireddy LR, Kumar KU, Pater MM, Pater A. BAG-1, a novel Bcl-2-interacting protein, activates expression of human JC virus. J Gen Virol. 2000;81(Pt 2):351–357. doi: 10.1099/0022-1317-81-2-351. [DOI] [PubMed] [Google Scholar]
- Fanning E, Knippers R. Structure and function of simian virus 40 large tumor antigen. Annu Rev Biochem. 1992;61:55–85. doi: 10.1146/annurev.bi.61.070192.000415. [DOI] [PubMed] [Google Scholar]
- Frisque RJ, Bollag B, Tyagarajan SK, Kilpatrick LH. T′ proteins influence JC virus biology. J Neurovirol. 2003;9(Suppl 1):15–20. doi: 10.1080/13550280390195270. [DOI] [PubMed] [Google Scholar]
- Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51(2):458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparovic ML, Gee GV, Atwood WJ. JC virus minor capsid proteins Vp2 and Vp3 are essential for virus propagation. J Virol. 2006;80(21):10858–10861. doi: 10.1128/JVI.01298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulos CP. A new bacterial gene (groPC) which affects lambda DNA replication. Mol Gen Genet. 1977;151(1):35–39. doi: 10.1007/BF00446910. [DOI] [PubMed] [Google Scholar]
- Georgopoulos CP, Hendrix RW, Kaiser AD, Wood WB. Role of the host cell in bacteriophage morphogenesis: effects of a bacterial mutation on T4 head assembly. Nat N Biol. 1972;239(89):38–41. doi: 10.1038/newbio239038a0. [DOI] [PubMed] [Google Scholar]
- Gordon J, Krynska B, Otte J, Houff SA, Khalili K. Oncogenic potential of human neurotropic papovavirus, JCV, in CNS. Dev Biol Stand. 1998;94:93–101. [PubMed] [Google Scholar]
- Griffith JP, Griffith DL, Rayment I, Murakami WT, Caspar DL. Inside polyomavirus at 25-A resolution. Nature. 1992;355(6361):652–654. doi: 10.1038/355652a0. [DOI] [PubMed] [Google Scholar]
- Grueneberg DA, Natesan S, Alexandre C, Gilman MZ. Human and Drosophila homeodomain proteins that enhance the DNA-binding activity of serum response factor. Science. 1992;257(5073):1089–1095. doi: 10.1126/science.257.5073.1089. [DOI] [PubMed] [Google Scholar]
- Guerrero CA, Bouyssounade D, Zarate S, Isa P, Lopez T, Espinosa R, Romero P, Mendez E, Lopez S, Arias CF. Heat shock cognate protein 70 is involved in rotavirus cell entry. J Virol. 2002;76(8):4096–4102. doi: 10.1128/JVI.76.8.4096-4102.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Huang YL, Wang M, Ou WC, Fung CY, Chen LS, Chang D. Analysis of DNA-binding activity of the JC virus minor capsid protein VP2. J Neurovirol. 2003;9(Suppl 1):21–24. doi: 10.1080/13550280390195289. [DOI] [PubMed] [Google Scholar]
- Kelley WL. Molecular chaperones: How J domains turn on Hsp70s. Curr Biol. 1999;9(8):R305–R308. doi: 10.1016/s0960-9822(99)80185-7. [DOI] [PubMed] [Google Scholar]
- Kelley WL, Landry SJ. Chaperone power in a virus? Trends Biochem Sci. 1994;19(7):277–278. doi: 10.1016/0968-0004(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Khalili K, Feigenbaum L, Khoury G. Evidence for a shift in 5′-termini of early viral RNA during the lytic cycle of JC virus. Virology. 1987;158(2):469–472. doi: 10.1016/0042-6822(87)90224-8. [DOI] [PubMed] [Google Scholar]
- Kim J, Woolridge S, Biffi R, Borghi E, Lassak A, Ferrante P, Amini S, Khalili K, Safak M. Members of the AP-1 family, c-Jun and c-Fos, functionally interact with JC virus early regulatory protein large T antigen. J Virol. 2003;77(9):5241–5252. doi: 10.1128/JVI.77.9.5241-5252.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krynska B, Gordon J, Otte J, Franks R, Knobler R, DeLuca A, Giordano A, Khalili K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem. 1997;67(2):223–230. doi: 10.1002/(sici)1097-4644(19971101)67:2<223::aid-jcb7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Krynska B, Otte J, Franks R, Khalili K, Croul S. Human ubiquitous JCV(CY) T-antigen gene induces brain tumors in experimental animals. Oncogene. 1999;18(1):39–46. doi: 10.1038/sj.onc.1202278. [DOI] [PubMed] [Google Scholar]
- Lashgari MS, Tada H, Amini S, Khalili K. Regulation of JCVL promoter function: transactivation of JCVL promoter by JCV and SV40 early proteins. Virology. 1989;170(1):292–295. doi: 10.1016/0042-6822(89)90381-4. [DOI] [PubMed] [Google Scholar]
- Leger H, Sock E, Renner K, Grummt F, Wegner M. Functional interaction between the POU domain protein Tst-1/Oct-6 and the high-mobility-group protein HMG-I/Y. Mol Cell Biol. 1995;15(7):3738–3747. doi: 10.1128/mcb.15.7.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PP, Itoh N, Watanabe M, Shi Y, Liu P, Yang HJ, Kasamatsu H. Association of simian virus 40 vp1 with 70-kilodalton heat shock proteins and viral tumor antigens. J Virol. 2009;83(1):37–46. doi: 10.1128/JVI.00844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberek K, Georgopoulos C, Zylicz M. Role of the Escherichia coli DnaK and DnaJ heat shock proteins in the initiation of bacteriophage lambda DNA replication. Proc Natl Acad Sci USA. 1988;85(18):6632–6636. doi: 10.1073/pnas.85.18.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddington RC, Yan Y, Moulai J, Sahli R, Benjamin TL, Harrison SC. Structure of simian virus 40 at 3.8-A resolution. Nature. 1991;354(6351):278–284. doi: 10.1038/354278a0. [DOI] [PubMed] [Google Scholar]
- Liu JS, Kuo SR, Makhov AM, Cyr DM, Griffith JD, Broker TR, Chow LT. Human Hsp70 and Hsp40 chaperone proteins facilitate human papillomavirus-11 E1 protein binding to the origin and stimulate cell-free DNA replication. J Biol Chem. 1998;273(46):30704–30712. doi: 10.1074/jbc.273.46.30704. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Identification of critical elements within the JC virus DNA replication origin. J Virol. 1990;64(12):5812–5822. doi: 10.1128/jvi.64.12.5812-5822.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Factors contributing to the restricted DNA replicating activity of JC virus. Virology. 1991;180(1):306–317. doi: 10.1016/0042-6822(91)90035-a. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Haggerty S, Frisque RJ. DNA replication of chimeric JC virussimian virus 40 genomes. Virology. 1994;204(2):819–822. doi: 10.1006/viro.1994.1600. [DOI] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci USA. 1985;82(4):1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP. Recruitment of Hsp70 chaperones: a crucial part of viral survival strategies. Rev Physiol Biochem Pharmacol. 2005;153:1–46. doi: 10.1007/s10254-004-0025-5. [DOI] [PubMed] [Google Scholar]
- Napuli AJ, Alzhanova DV, Doneanu CE, Barofsky DF, Koonin EV, Dolja VV. The 64-kilodalton capsid protein homolog of Beet yellows virus is required for assembly of virion tails. J Virol. 2003;77(4):2377–2384. doi: 10.1128/JVI.77.4.2377-2384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9(8):585–593. [PubMed] [Google Scholar]
- Ponten J, Macintyre EH. Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand. 1968;74(4):465–486. doi: 10.1111/j.1699-0463.1968.tb03502.x. [DOI] [PubMed] [Google Scholar]
- Prins C, Frisque RJ. JC virus T′ proteins encoded by alternatively spliced early mRNAs enhance T antigen-mediated viral DNA replication in human cells. J Neurovirol. 2001;7(3):250–264. doi: 10.1080/13550280152403290. [DOI] [PubMed] [Google Scholar]
- Raj GV, Gordon J, Logan TJ, Hall DJ, Deluca A, Giordano A, Khalili K. Characterization of glioma cells derived from polyomavirus-induced brain tumors in hamsters. Int J Oncol. 1995;7:801–808. doi: 10.3892/ijo.7.4.801. [DOI] [PubMed] [Google Scholar]
- Riley MI, Yoo W, Mda NY, Folk WR. Tiny T antigen: an autonomous polyomavirus T antigen amino-terminal domain. J Virol. 1997;71(8):6068–6074. doi: 10.1128/jvi.71.8.6068-6074.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowska B, Barrucco R, Khalili K, Safak M. Regulation of human polyomavirus JC virus gene transcription by AP-1 in glial cells. J Virol. 2003;77(1):665–672. doi: 10.1128/JVI.77.1.665-672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75(3):1476–1486. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Gallia GL, Ansari SA, Khalili K. Physical and functional interaction between the Y-box binding protein YB-1 and human polyomavirus JC virus large T antigen. J Virol. 1999;73(12):10146–10157. doi: 10.1128/jvi.73.12.10146-10157.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sami Saribas A, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M. Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology. 2013;443(1):161–176. doi: 10.1016/j.virol.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: evidence for its involvement in viral DNA replication. Virology. 2012;433(1):12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan K, Del Valle L, Khalili K, Safak M. Tumor induction by simina and human polyomaviruses. Cancer Ther. 2004;2:85–95. [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8(1):255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai ET, Butel JS. Association of a cellular heat shock protein with simian virus 40 large T antigen in transformed cells. J Virol. 1989;63(9):3961–3973. doi: 10.1128/jvi.63.9.3961-3973.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucl Acids Res. 1989;17(15):6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y, Hara Y, Larson T, Yasui K, Nagashima K, Stoner GL. Analysis of capsid formation of human polyomavirus JC (Tokyo-1 strain) by a eukaryotic expression system: splicing of late RNAs, translation and nuclear transport of major capsid protein VP1, and capsid assembly. J Virol. 2000;74(4):1840–1853. doi: 10.1128/jvi.74.4.1840-1853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence SL, Pipas JM. Simian virus 40 large T antigen host range domain functions in virion assembly. J Virol. 1994a;68(7):4227–4240. doi: 10.1128/jvi.68.7.4227-4240.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence SL, Pipas JM. SV40 large T antigen functions at two distinct steps in virion assembly. Virology. 1994b;204(1):200–209. doi: 10.1006/viro.1994.1524. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, McClellan AJ, Vartikar J, Marks I, Cantalupo P, Li Y, Whyte P, Rundell K, Brodsky JL, Pipas JM. The amino-terminal transforming region of simian virus 40 large T and small t antigens functions as a J domain. Mol Cell Biol. 1997;17(8):4761–4773. doi: 10.1128/mcb.17.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Cantalupo P, Pipas JM. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol Cell Biol. 2000;20(17):6233–6243. doi: 10.1128/mcb.20.17.6233-6243.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Gilbert SP, Pipas JM. ATP-dependent simian virus 40 T-antigen-Hsc70 complex formation. J Virol. 2001;75(4):1601–1610. doi: 10.1128/JVI.75.4.1601-1610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Pipas JM. T antigens of simian virus 40: molecular chaperones for viral replication and tumorigenesis. Microbiol Mol Biol Rev. 2002;66(2):179–202. doi: 10.1128/MMBR.66.2.179-202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Frisque RJ. Altered DNA binding and replication activities of JC virus T-antigen mutants. Virology. 1991;183(1):239–250. doi: 10.1016/0042-6822(91)90136-y. [DOI] [PubMed] [Google Scholar]
- Tretiakova A, Krynska B, Gordon J, Khalili K. Human neurotropic JC virus early protein deregulates glial cell cycle pathway and impairs cell differentiation. J Neurosci Res. 1999;55(5):588–599. doi: 10.1002/(SICI)1097-4547(19990301)55:5<588::AID-JNR6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Vogel M, Bukau B, Mayer MP. Allosteric regulation of Hsp70 chaperones by a proline switch. Mol Cell. 2006;21(3):359–367. doi: 10.1016/j.molcel.2005.12.017. [DOI] [PubMed] [Google Scholar]
- White FA, 3rd, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66(10):5726–5734. doi: 10.1128/jvi.66.10.5726-5734.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperonemediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5(10):781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Zalvide J, Stubdal H, DeCaprio JA. The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol. 1998;18(3):1408–1415. doi: 10.1128/mcb.18.3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerrahn J, Knippschild U, Winkler T, Deppert W. Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J. 1993;12(12):4739–4746. doi: 10.1002/j.1460-2075.1993.tb06162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler K, Bui T, Frisque RJ, Grandinetti A, Nerurkar VR. A rapid in vitro polyomavirus DNA replication assay. J Virol Methods. 2004;122(1):123–127. doi: 10.1016/j.jviromet.2004.08.012. [DOI] [PubMed] [Google Scholar]