

Abstract
Human papillomavirus (HPV) has evolved mechanisms that allow it to evade the human immune system. Studies have shown HPV-mediated suppression of activation of Langerhans cells (LC) is a key mechanism through which HPV16 evades initial immune surveillance. However, it has not been established whether high- and low-risk mucosal and cutaneous HPV genotypes share a common mechanism of immune suppression. Here, we demonstrate that LC exposed to capsids of HPV types 18, 31, 45, 11, (alpha-papillomaviruses) and HPV5 (beta-papillomavirus) similarly suppress LC activation, including lack of costimulatory molecule expression, lack of cytokine and chemokine secretion, lack of migration, and deregulated cellular signaling. In contrast, HPV1 (mu-papillomavirus) induced costimulatory molecule and cytokine upregulation, but LC migration and cellular signaling was suppressed. These results suggest that alpha and beta HPV genotypes, and partially a mu genotype, share a conserved mechanism of immune escape that enables these viruses to remain undetected in the absence of other inflammatory events.
Keywords: human papillomavirus, Langerhans cell, immune suppression, immune escape, antigen presentation, antigen presenting cell, migration
Introduction
Human papillomavirus (HPV) infection is causally associated with the development of benign and malignant lesions, including various types of cancer (Walboomers et al., 1999). Over 170 genotypes of HPV have been identified to date, each with preferential tropism for particular locations (i.e. mucosal versus cutaneous) and with varying propensities for causing cancer (i.e. high-risk versus low-risk) (Lizano, Berumen, and Garcia-Carranca, 2009). It is estimated that 15% of women with a high-risk HPV infection are unable to mount an effective immune response (Stanley, Pett, and Coleman, 2007), and viral clearance takes on average about one year (Winer et al., 2011), indicating that HPV possesses mechanisms to escape immune detection and clearance.
The life cycle of HPV occurs within the epithelium of the skin and mucosa and is dependent on the differentiation of epithelial cells, making in vitro studies challenging. As alternatives to native virions, multiple particle platforms have been developed, such as virus-like particles (VLPs) and pseudovirions (PsV), which both generate HPV particles that contain the major capsid protein L1 and minor capsid protein L2 (Kirnbauer et al., 1993; Roden et al., 1996; Zhou et al., 1991). Due to the location of HPV infection, the resident antigen presenting cells in the epithelium, Langerhans cells (LC), are the first immune cell to contact HPV and therefore are thought to be responsible for initiating an effective anti-viral immune response (Banchereau and Steinman, 1998). Upon recognition of a foreign antigen, LC undergo maturation, which consists of phenotypic and functional changes including up-regulation of MHC and co-stimulatory molecules, secretion of cytokines and chemokines, and migration to regional lymph nodes where T cell activation takes place (Cunningham, Carbone, and Geijtenbeek, 2008). However, we have previously demonstrated that human LC exposed to the high-risk genotype HPV16 do not become activated, suggesting a HPV16 immune escape mechanism specifically targeting LC (Fausch et al., 2002). We furthermore demonstrated that this HPV16 immune escape mechanism was dependent on the presence of the minor capsid protein L2 (Fahey et al., 2009). HPV actively changes the LC through deregulation of the PI3 kinase (PI3K)-Akt signaling pathway such that although they present HPV peptides on their MHC molecules to T cells; this presentation occurs in the absence of T cell co-stimulatory molecules and essential T cell activating cytokines (Fausch, Da Silva, and Kast, 2003; Fausch et al., 2005). The resulting immunological phenotype is an LC that can potentially induce T cell tolerance and/or inhibit subsequent T cell recognition of HPV.
Whether the LC-specific immune escape mechanism exhibited by HPV16 is also utilized by other HPV genotypes, including other high-risk mucosal genotypes, low-risk mucosal genotypes and cutaneous genotypes, has been an intriguing question. Because we had previously shown that the immune escape mechanism was dependent on the HPV16 L2 minor capsid protein (Fahey et al., 2009) and the L2 protein has highly conserved regions across multiple genotypes (Lowe et al., 2008; Yang et al., 2003), we hypothesized that other HPV genotypes besides HPV16 would similarly initiate immune escape by targeting antigen presentation by LC. To explore this hypothesis, we exposed human monocyte-derived LC to high-risk mucosal genotypes associated with malignancy (HPV16, HPV18, HPV31 and HPV45), a low-risk mucosal genotype associated with benign genital condylomas (HPV11) and cutaneous genotypes associated with skin lesions (HPV5) or hand and foot warts (HPV1), and subsequently assessed the phenotypic and functional immunologic characteristics of virus exposed LC.
Results
Phenotypic activation of LC after treatment with high- and low-risk mucosal HPV genotypes and cutaneous HPV genotypes
HPV particles representative of high-risk mucosal HPV types (HPV16, HPV18, HPV31, HPV45), a low-risk mucosal HPV type (HPV11), and cutaneous HPV types (HPV1, HPV5) were generated by overexpression of the L1 and L2 genes in either insect cells for virus-like particles (VLP) or in the 293TT human embryonic kidney cell line for pseudovirions (PsV). To determine the effect of different HPV genotypes on phenotypic maturation of LC, we assessed the expression of cell surface activation markers on LC after exposure to HPV16, HPV18, HPV11, HPV1 VLPs and HPV31, HPV45, HPV5 PsVs. In line with our previous findings, LC exposed to HPV16 expressed comparable levels of MHC class II, the costimulatory markers CD80 and CD86, and the maturation marker CD83 compared to untreated LC (Fig. 1). Similarly, LC exposed to HPV18, HPV31, HPV45, HPV11, and HPV5 also did not upregulate these cell surface activation markers. In contrast, HPV1 treated LC showed significant upregulation of all four surface molecules (p< 0.01 compared to untreated LC). LC treated with the positive control LPS, a potent TLR4 agonist, also upregulated the cell surface expression of all these activation markers. To rule out variations in LC activation due to the source of viral particle, we performed a direct comparison between HPV16 VLP and HPV16 PsV on LC activation and found no differences in the levels of LC activation markers (Fig. S1). Titration of HPV1 capsids resulted in reduced upregulation of surface markers MHC class II and CD86, although the levels of expression still remained above that of the untreated cells at the lowest dose tested, indicating that there is a dose effect for HPV1-induced LC activation (Fig. S2).
Fig. 1. High and low risk mucosal HPV genotypes and a cutaneous HPV genotype all suppress phenotypic activation of LC, in contrast to HPV1.

Human immature LC were left untreated, treated with LPS (positive control) or exposed to various HPV genotypes for 48 h at 37°C. Subsequently, cells were analyzed by flow cytometry for expression of MHC molecules, costimulatory markers, and maturation markers. Analysis was performed on total population of large cells (>75% of total population) which are >95% CD1a+ (indicative of LC phenotype). Shown are MHC class II, CD80, CD86, and CD83 markers. High-risk mucosal HPV genotypes treated on LC include HPV16, HPV18, HPV31, and HPV45. HPV11 is the representative low-risk HPV genotype. HPV5 and HPV1 are the representative cutaneous HPV genotypes. Fold change of surface marker expression was calculated based on mean fluorescence intensity (MFI) normalized against the untreated LC. Data is a representation of mean ± SD of LC from 8 healthy donors. (*p < 0.05, **p < 0.01).
Cytokine and chemokine secretion by LC after treatment with high- and low-risk HPV genotypes and cutaneous HPV genotypes
The cytokine and chemokine profile generated by activated antigen presenting cells plays an important role in directing the adaptive immune response. Therefore, to next assess functional activation of LC, we analyzed the levels of cytokines and chemokines secreted by LC after exposure to HPV16, HPV18, HPV31, HPV45, HPV11, HPV5 and HPV1. Secretion of IL-6, TNFα, MIP-1α and RANTES after exposure to all tested HPV genotypes, except for HPV1, was similar to untreated LC (Fig. 2). Consistent with the upregulation of activation markers observed above, LC stimulated with HPV1 demonstrated a significant increase in the secretion of MIP-1b and RANTES (p<0.05), and a moderate increase in IL-6 and TNFα, indicative of a pro-inflammatory immune response. For the majority of secreted immune mediators analyzed, LC exposed to the HPV particles maintained baseline secretion of pro-inflammatory cytokines and chemokines analyzed (Table S1), supporting the view that, in general, LC remain in an immature state after HPV encounter, particularly with non-cutaneous genotypes.
Fig. 2. Cytokine and chemokine secretion by LC are similar after treatment with high and low risk HPV genotypes and cutaneous HPV5, in contrast to HPV1.

Supernatant from LC were collected after 48 hours of treatment with LC only, LPS, high-risk (HPV16, HPV18 VLPs and HPV31, HPV45 PsV), low-risk (HPV11 VLP) or cutaneous genotypes (HPV5 PsV, HPV1 VLP). Cell supernatants were analyzed for a panel of 12 cytokines and chemokines (IL-1β, IFNα, IL-6, IL-8, IL-10, IL-12 p70, TNFα, IP-10, MCP-1, MIP-1α, MIP-1β, RANTES) using a Bio-plex suspension bead ELISA (BioRad, Hercules, CA). Shown are four analytes selected from the panel of twelve. Data represent the mean (± SEM) analyte concentration of 4 individual healthy donors tested. (*p < 0.05, **p<0.01).
Migration of LC in response to exposure with high- and low-risk HPV genotypes and cutaneous HPV genotype
Once activated, antigen presenting cells migrate to draining lymph nodes using the cell surface receptor CCR7, which binds to the lymph node associated chemokine CCL21 (Saeki et al., 1999). By utilizing a transwell migration assay with CCL21, we assessed the migratory capacity of LC after exposure to HPV16, HPV18, HPV31, HPV45, HPV11, HPV5 and HPV1. While exposure to LPS caused the LC to increase their migration towards CCL21, LC exposed to all of the HPV types, including HPV1, maintained a steady state migration similar to untreated LC (Fig. 3), suggesting that exposure to viral particles does not induce migration of local LC to lymphocyte-rich areas where immune reactions take place.
Fig. 3. Migration of LC does not change in response to exposure with high- and low-risk mucosal and cutaneous HPV genotypes.
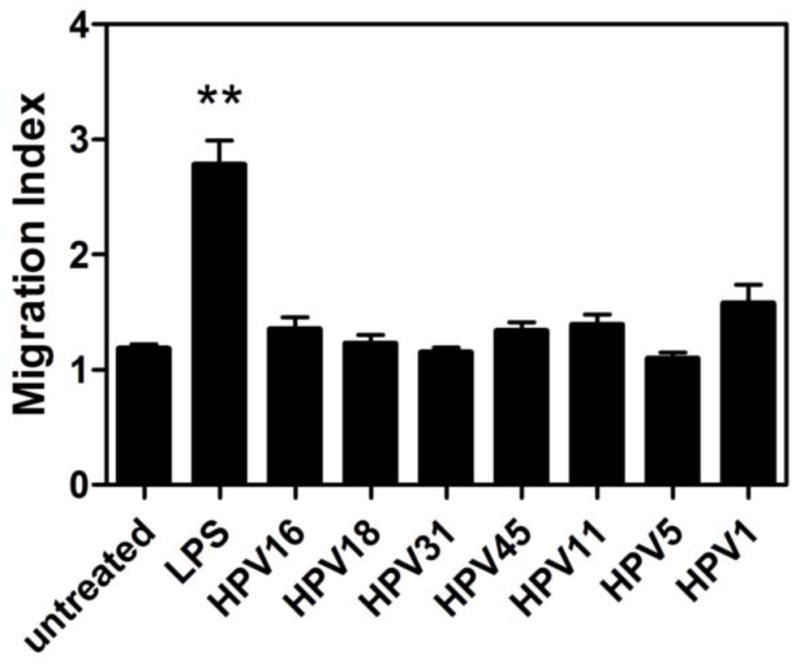
Human LC were treated with LPS or HPV genotypes indicated for 48 hours and then were analyzed for migration through a 5μm transwell insert to medium alone or medium supplemented with 250 ng/mL CCL21/SLC. After 4 h, cells migrating to the lower chamber containing chemokine were counted. Visualization of migrating cells by microscopy showed only large “LC-like” cells moving through the transwell membrane. Shown is the mean migration index calculated as the number of cells migrating to CCL21 over spontaneous migration (±SEM) of 6 healthy donors relative to untreated LC. No significant differences are observed between the untreated LC and LC exposed to different HPV genotypes. (**p < 0.01).
Deregulation in the Akt signaling pathway in LC after treatment with high- and low-risk mucosal HPV genotypes and cutaneous HPV genotypes
In our previous studies we demonstrated that the mechanism of HPV16-induced immune escape in LC occurred through the deregulation of the PI3K-Akt pathway, as shown by a decrease in phospho-Akt (p-Akt, Ser 473) (Fausch et al., 2005). To determine if this signaling pathway plays a role in immune escape of other HPV genotypes, LC were exposed to HPV16, HPV18, HPV31, HPV45, HPV11, HPV5 and HPV1 and subsequently assayed for the levels of p-Akt and total Akt. LC exposed to all HPV genotypes tested demonstrated a decrease in p-Akt compared to baseline p-Akt levels in untreated LC at 15 min and 30 min post exposure (Fig. 4A), indicating they share a common mechanism for suppressing LC signaling. The relative p-Akt concentration was quantified and normalized to GAPDH and confirms the decrease in p-Akt compared to untreated LC (Fig. 4B). The largest decreases in pAkt were observed after 15 min, whereas by 30 min levels of pAkt were returning to, but still below baseline. These results suggest that diverse HPV genotypes are able to cause a down regulation of signal transduction molecules in LC that normally control the transcription of immune response genes and cell survival and point to a potential mechanism for inhibiting the function of LC in the epidermal tissue during HPV infection.
Fig. 4. Disruption in the Akt signaling pathway in LC is similar after treatment with high- and low-risk mucosal HPV genotypes and cutaneous HPV genotypes.

(A) Cellular extracts from LC exposed to HPV genotypes for 15 min (top) or 30 min (bottom) at 37°C were separated by SDS-PAGE, transferred to PVDF and immunoblotted using antibodies to p-Akt (Ser473), total Akt, and GAPDH. Visualization was performed using the Odyssey Infrared Imaging System. (B) Protein bands were quantified using the Odyssey system software, normalized to total Akt and GAPDH expression, and the relative levels of pAkt (mean ± SEM) compared to untreated LC are shown from three individual experiments.
Discussion
LC, also known as epidermal dendritic cells (DC), only constitute 3–5% of nucleated cells in the epidermis, but are able to form an expansive cellular network through their extended dendrites that survey the epidermis for foreign antigens (Romani et al., 2003). Classically, activated LC are thought to migrate to the lymph nodes, present antigen and activate antigen-specific T-cells. However, recent studies have challenged the importance of LC in the initiation of skin immunity (Romani, Brunner, and Stingl, 2012). Studies in mice have identified a specialized subset of langerin+CD103+ dermal DC which cross-present antigen to CD8+ T cells when the antigen is exclusively expressed in keratinocytes (Bedoui et al., 2009; Henri et al., 2010). Furthermore, long-term effector/memory CD4+ T cells are induced only when dermal DC are present, although LC presenting epidermally expressed antigen can still stimulate CD4+ T cell proliferation (Shklovskaya et al., 2011). Selective depletion of langerin+ LC in mice results in enhanced contact hypersensitivity to haptens, suggesting that in the steady-state, when LC are present, epidermal immune responses are suppressed (Bobr et al., 2010; Kaplan et al., 2005). Human studies have shown, however, that LC isolated from human skin can cross-present antigen, prime naïve CD8+ T cells and preferentially expand antigen-specific cytotoxic T lymphocytes (CTLs) (Klechevsky et al., 2008). Notably, mouse skin and human skin differ quite substantially both anatomically and immunologically, and it is unclear whether a human equivalent exists to mouse langerin+CD103+ dermal DC. Investigation of APC subsets in human vaginal tissue reveals that vaginal APCs have distinct phenotypes and specialized functions in directing local immune responses which are different from other tissues and organs, including skin and gut (Duluc et al., 2013). LC isolated from human vaginal tissue have been shown to induce naïve CD8+ T cell proliferation and are able to induce CD8+ T cells expressing the cytokines IFNγ and TNFα. Overall, it is clear that human LC can contribute both to immune suppression as well as immune activation, the outcome being determined by the particular environment, the presence or absence of inflammation and the presence of pathogenic stimuli.
HPV infections may take many months to years to be cleared (Ho et al., 1998; Winer et al., 2011), even for non-oncogenic strains (Giuliano et al., 2002), suggesting that HPVs have acquired the ability to either remain hidden or suppress the immune system. Persistence of an HPV infection is the greatest risk factor in the development of HPV-related malignancies (Schlecht et al., 2001). Previously, we identified an HPV16 capsid-driven immune escape mechanism that suppresses the immunostimulatory function of LC (Fausch, Da Silva, and Kast, 2003; Fausch et al., 2002; Fausch et al., 2005). When HPV16, a high risk mucosal HPV genotype, comes into contact with LC, the result is that HPV16 actively prevents LC maturation into an activated antigen presenting cell (APC). The current study indicates that other HPV genotypes besides HPV16, such as high-risk (HPV18, HPV31, and HPV45), low-risk (HPV11) and a cutaneous (HPV5) genotype, also suppress LC activation through a similar mechanism. By inhibiting the maturation of the APC at the site of infection, these representative HPV genotypes prevent the induction of the HPV-specific adaptive immune response required for viral clearance. Human LC have been shown to internalize papillomavirus capsids from diverse genotypes, including animal papillomaviruses (Fausch, Da Silva, and Kast, 2005; Yan et al., 2004), suggesting that differential HPV antigen uptake is unlikely to be responsible for the effects we observed in this study. Notably, APC displaying peptides in the absence of both costimulation and pro-inflammatory cytokines have the ability to cause T cell anergy (Frauwirth, Alegre, and Thompson, 2000) and generate regulatory T cells (Dhodapkar and Steinman, 2002; Dhodapkar et al., 2001), the latter being increased in HPV-associated pre-neoplastic lesions and in cervical cancer compared to healthy tissue (Leong et al., 2010b; Scott et al., 2009; van der Burg et al., 2007).
To date, 170 genotypes of HPV have been sequenced and are clustered based on the major L1 capsid, the type of infection caused (whether mucosal or cutaneous), and their ability to either induce cancer or benign condylomas (Bernard, 2005; de Villiers, 2013). From the alpha-papillomavirus genus, HPV16 and HPV18 are most frequently associated with cervical cancer. Importantly, the four high risk genotypes analyzed in this study – HPV16, HPV18, HPV31, and HPV45 – are the most prevalent genotypes attributed to causing greater than 75% of all cervical squamous cell carcinoma (Smith et al., 2007). Low risk HPV genotypes from the alpha-papillomavirus group are associated with benign condylomas with the highest occurrence of genital warts primarily associated with HPV6 and laryngeal papillomas primarily associated with HPV11. Cutaneous mu-papillomavirus genotypes such as HPV1 are generally benign and associated with foot and hand warts. However, some cutaneous beta-papillomavirus genotypes such as HPV5 and HPV8 have been associated with epidermodysplasia verruciforms, a cutaneous neoplastic disease, and can also be associated with skin cancer in immune compromised individuals (Bernard, 2005).
Although papillomaviruses are distinct enough to be classified into different genera and have distinct life-cycle characteristics and disease associations, one characteristic is fairly consistent – which is that the majority, if not all, of HPVs tend to persist longer than the typical transient viral infection. Longitudinal natural history studies demonstrate that infection with high-risk oncogenic HPV types persist on average for greater than one year. The median duration of cervical infection in young women has been reported to be anywhere from 12–19 months for HPV16 and HPV31, and 9–15 months for HPV18 and HPV45 (Ho et al., 1998; Kulmala et al., 2007; Moscicki et al., 2004; Richardson et al., 2003). Low-risk types such as HPV6 and HPV11 persist on average for six months (Ho et al., 1998; Richardson et al., 2003). The ability of the high-risk types (HPV16,18,31,45) to persist longer than a low risk type (HPV11) despite them being equally capable of inducing LC dysfunction may be related to other virally mediated mechanisms contributing to immune escape such as differential modulation of keratinocyte gene expression, and antiviral and inflammatory cytokines (Kanodia, Fahey, and Kast, 2007). Similar to the mucosatropic alpha-papillomaviruses, our data suggest that even cutaneous HPV genotypes (HPV5 and partially HPV1) can suppress LC immune function. Clinical observations indicate that cutaneous warts in healthy individuals generally regress within 3–6 months with regression being accompanied by dense mononuclear cell infiltrates (Jablonska et al., 1997). However, asymptomatic subclinical infection with cutaneous HPV types is very prevalent and some studies indicate cutaneous HPVs, such as HPV5, can persist for months to years (de Koning et al., 2007; de Koning et al., 2009; Hazard et al., 2007). It is interesting to note that LC exposed to HPV1 secreted increased levels of MIP-1b and RANTES (Fig. 2), two chemokines that are chemoattractant towards CD4+ T cells, B cells, DC, monocytes and polymorphonuclear cells. If LC become activated when HPV1-producing lesions become sufficiently large to provide enough virion stimulation to LC, secretion of these chemokines may be sufficient to cause mononuclear cell infiltration to biologically promote wart regression.
Related to the potential functional impact on LC suppression by the HPV capsid proteins of diverse genotypes that we observe, it has also been shown by Leong et. al. that LC numbers and E-cadherin expression in HPV infected tissue are reduced in lesions infected with alpha-, gamma- and mu-papillomaviruses, but not the beta-papillomavirus types indicating that other effects on LC dysfunction are conserved across diverse HPV genera (Leong et al., 2010a; Leong et al., 2010b). Moreover, keratinocytes infected with high-risk HPV16 can block the differentiation of monocytes into LC in a contact dependent manner (Iijima et al., 2013). These observations, together with our data, suggest that a consequence of early HPV infection by a majority of genotypes from diverse genera is to dampen the immune response via suppression of LC that are initially present in the epidermis of healthy tissue. Following infection of keratinocytes, HPV gene expression results in further host immune suppression by causing a reduction in the LC adjacent to infected cells, promoting immune evasion and favoring viral persistence (Kanodia, Fahey, and Kast, 2007).
In our study, a significant amount of capsid was required to observe partial LC activation by HPV1. When we titrated the HPV1 capsids and decreased the capsid:LC ratio, we did observe that the upregulation of surface markers was decreased. This observation suggests that low HPV1 infectious doses would be even less likely to trigger an antiviral immune reaction, also contributing to preventing immune recognition despite the fact that some cutaneous lesions and warts can become sizable and produce large amounts of infectious particles. In natural infections, it is not known how many viral particles are required to alert epidermal LC to the presence of viral pathogen. However, a significant observation is that some cutaneous HPV infections result in epithelial tissue which is less cornified, leading to more fragile cell envelopes that can easily be broken, releasing high concentrations of virions that can encounter surrounding LC in the infected individual (Brown and Bryan, 2000). During person to person transmission, virions can be delivery in concentrated packages by desquamated cornified cells, releasing a focal amount of virions on a small area of epithelium (Bryan and Brown, 2001), which would enhance the likelihood of LC of the uninfected person coming into contact with a concentrated amount of virions.
Our study reveals that, in general, diverse genotypes (mucosal, cutaneous, high-risk, and low-risk) tend to interact with human LC similarly to cause immune suppression. This finding may allude to the utilization of similar cell surface receptors on LCs and epithelial cells, though productive infection is not expected to take place in myeloid-derived immune cells. Early events of papillomavirus infection are believed to use shared mechanisms based on studies demonstrating similar infection susceptibility of mucosal and skin tissues with diverse HPV genotypes (Handisurya et al., 2012). Additionally, efficiency of infection, based on reporter gene transduction, was the same for human and animal papillomavirus types, suggesting that downstream post cell entry events determine papillomavirus species and tissue tropism rather than initial capsid interactions and virus entry (Handisurya et al., 2012). It is therefore surprising that in our study certain properties of LC were different when exposed to the HPV1 cutaneous genotype. Although LC migration and Akt signaling were not demonstrably affected, high dose HPV1-exposed LC did become partially activated through upregulation of surface molecules and secretion of cytokines involved in adaptive immunity, suggesting that, at least for HPV1, the host response is more reactive to this papillomavirus genotype compared to the other genotypes that were used at the same capsid concentration. Whether these partially activated LC are able to induce HPV1-specific T cells in vivo cannot be determined from our study.
As described earlier, the LC-specific immune escape mechanism of HPV16 was shown to be dependent on the presence of the L2 protein, because capsids composed solely of the HPV16 L1 major capsid protein are capable of activating LC, whereas HPV16 capsids containing L2 do not (Fahey et al., 2009). Capsids composed solely of the HPV6b L1 protein have also been shown to activate LC (Yan et al., 2004). The L2 protein has several conserved regions in its amino acid sequence across genotypes such as L2(13-31) and L2(108-120) which have previously been implicated in cell surface entry and binding events, respectively (Kawana et al., 2001; Lowe et al., 2008; Yang et al., 2003). Therefore, suppression of LC across diverse genotypes may be due to the conserved nature of the L2 protein sequence. Recently, our laboratory has demonstrated that the HPV16 L2(108-120) region binds to the S100A10 subunit of the annexin A2 heterotetramer (A2t) on epithelial cells (Woodham et al., 2012). Further, the interaction between HPV16 L2 and A2t was shown to contribute to HPV16 internalization and infection, particularly the L2(108-111 ) region (Woodham et al., 2012). Importantly, all of the particles used in this study contained the L2 protein of each genotype (Fig. S3), resulting in use of capsids identical to physiologically relevant wild-type capsids. Whether this conserved L2 region of all genotypes mediates the demonstrated immune escape mechanism, through binding to A2t on LC, is an active area of research in our group. Supporting this idea, we have found that HPV16 capsids are taken up by LC and functionally suppress LC maturation through an interaction with A2t in an L2-dependent manner (Woodham et al., 2014). Notably, the L2 proteins from HPV1 and HPV5 contain an extra 34 and 45 amino acids, respectively, compared to HPV16 L2, whereas the L2 proteins from HPV18, 31 and 45 show more sequence conservation to HPV16. Interestingly, when key amino acids in the L2(108-111) region are aligned, the HPV1 L2 sequence is the most different from HPV16 and the consensus sequence of the region between the papillomavirus genotypes tested in this study (Fig. 5). This observation suggests that amino acid divergence in this key A2t-binding region may be responsible for differential immunological outcomes post interaction with LC surface receptors. Additional studies will be needed to explore whether sequence differences in the L2 protein between different HPV genotypes affect their ability to initiate LC immune suppression.
Fig. 5. Alignment and consensus sequence of HPV L2 Proteins to HPV16 L2 (aa 108–111).
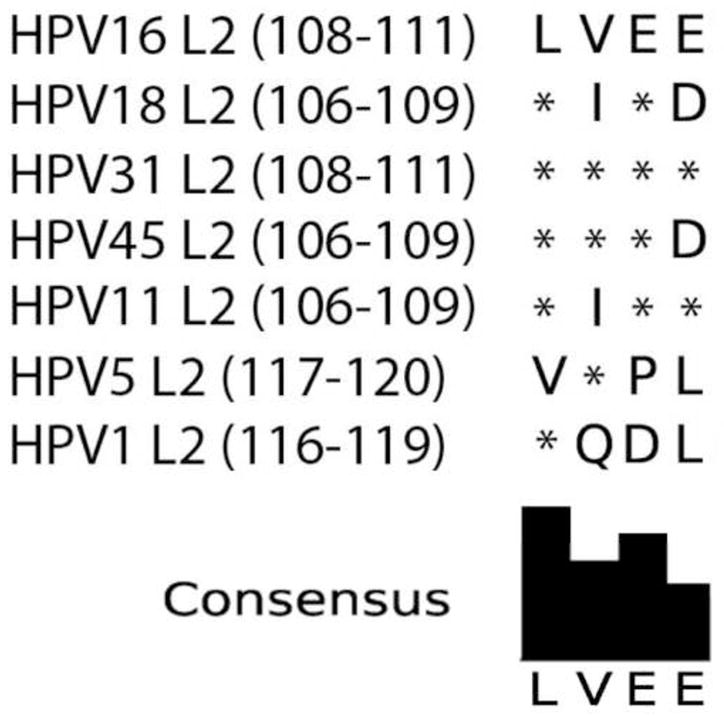
The sequence of amino acids (108–120) on HPV 16 L2 was shown to bind to epithelial cells and is conserved between high risk HPV genotypes. L2(108-111) are key residues mediating binding of L2 to the annexin A2/S100A10 receptor complex. The L2 protein sequences of HPV18, HPV31, HPV45, HPV11, HPV5 and HPV1 were compared against HPV16. HPV16, HPV18, HPV31, and HPV45 show protein sequence similarity. HPV1 shows only one conserved amino acid compared to HPV16 in the analyzed region. Sequences were individually aligned against HPV16 using the NCBI tool COBALT (COnstraint-Based multiple protein ALignment Tool). The consensus sequence is shown below the alignment. Asterisks indicate sequence conservation.
Papillomavirus capsid binding to the cell surface is associated with epithelial cell signaling, even before the majority of virus is internalized. One study showed that several high risk HPV genotypes and bovine papillomavirus induce tyrosine kinase and PI3K signaling events resulting in downstream signaling to Akt, which the authors proposed was a result of virus binding to integrins (Fothergill and McMillan, 2006). In contrast, whereas in epithelial cells the PI3K pathway is activated, we have previously shown that HPV16 disconnects PI3K signaling from downstream Akt signaling and downregulates MAPK signaling in LC (Fausch et al., 2005). The observation that all of the HPV genotypes tested here appear to suppress Akt signaling in LC extend these findings to other HPV genotypes, pointing to a common mechanism of immune evasion through deregulated cellular signaling.
While this study evaluated a variety of HPV genotypes from representative classes, we have not ruled out that genotypes not tested here may behave differently. HPVs have evolved a variety of mechanisms for immune escape and likely utilize a combination of approaches to subvert the immune system and maintain persistent infections. However, this study suggests a conserved mechanism of immune escape through targeting LC amongst high-risk, low-risk and at least one cutaneous HPV genotype, and furthers our understanding of the interaction between divergent HPV types and the human immune system.
Materials and Methods
Generation of human Langerhans Cells
Human Langerhans cells (LC) were derived as described previously and express phenotypic markers characteristic of skin-derived LC (Langerin+, E-cadherin+, CD11c+, CD1a+) (Fig. S1) (Fausch et al., 2002; Geissmann et al., 1998). These studies were approved by the University of Southern California Institutional Review Board and informed consent was obtained from all donors.
HPV pseudovirions and virus-like particles
HPV1, HPV11, HPV16, and HPV18 VLP were produced using a recombinant baculovirus expression system in Trichoplusia ni (Hi-5) insect cells as previously described and purified using sucrose cushion and cesium chloride density gradient ultracentrifugation (Fausch et al., 2002; Kirnbauer, 1992). HPV5, HPV16, HPV18, HPV31, and HPV45 pseudovirions were generated in 293TT cells and purified by optiprep (iodixinol) density gradient ultracentrifugation as described previously using the adapted protocol for bulk replication-incompetent PsV production (Buck and Thompson, 2007). Plasmid constructs (p5sheLL, p16sheLL, p18sheLL, p31sheLL, p45sheLL) were kindly provided by Dr. John Schiller (National Cancer Institute). Capsids were subjected to SDS-PAGE and Western blot analyses to confirm the presence of L1 and L2 proteins (Fig. S3), while transmission electron microscopy confirmed the presence of intact particles. Coomassie Blue staining following SDS-PAGE was performed to determine viral stock purity and standardize the concentration of L1 content of the VLP and PsV preparations. Capsid preparations were negative for endotoxin contamination as tested with an E-toxate (Limulus amebocyte lysate (LAL) assay kit (Sigma-Aldrich, St. Louis, MO).
LC Activation
Immature LC were either left untreated, treated with 5 μg LPS (Sigma-Aldrich), or 5 μg HPV VLP or PsV/106 cells. LC were incubated for 1 h at 37°C, mixed occasionally, then incubated at 37°C for an additional 48 h at a cell concentration of 1 × 105 LC/mL. Supernatants were collected for cytokine analysis and cells were harvested, washed, stained for surface markers or isotype controls, The following human antibodies were purchased from BD Biosciences (San Jose, CA): HLA-DP/DQ/DR-FITC, CD40-PE, CD86-FITC, CD1a-PE, CD197 (CCR7), goat anti-rat IgG-PE, goat anti-mouse IgG1-FITC, rat IgG2a isotype control, mouse IgG1-FITC, mouse IgG1-PE, mouse IgG2b-FITC. The following human antibodies were purchased from Biolegend (San Diego, CA): HLA-ABC-FITC, CD80-FITC, CD83-PE, CD207-PE (Langerin), CD324-PE (E-cadherin), Human TruStain FcX (Fc receptor blocking solution). Samples were analyzed by flow cytometry on a Cytomics FC500 flow cytometer using CXP software (Beckman Coulter, Indianapolis, IN).
Secreted cytokines and chemokines were analyzed using the Bio-Plex Suspension Array System (Bio-Rad, Hercules, CA) using a Milliplex MAP human cytokine/chemokine kit containing detection reagents for interferon (IFN)α, interleukin (IL)-1β, IL-6, IL-8, IL-10, IL-12p70, Inducible Protein 10 (IP-10), tumor necrosis factor (TNF)α, monocyte chemotactic protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, and RANTES (EMD Millipore, Billerica, MA).
Migration assay
Chemokine directed migration of LC was carried out using 24-well Transwell plates with 5 μm-pore-size polycarbonate filters (Corning Costar, Tewksbury MA). Media was added to the lower chamber containing either 250 ng/ml recombinant human CCL21 (R&D Systems, Minneapolis, MN) or medium alone to control for spontaneous migration. LC untreated or treated as described in the LC activation assay were added to the upper chamber and incubated for 4 hrs at 37°C. The cells that migrated to the lower chamber were harvested and counted using a Z1 Beckman Coulter particle counter. CCL21-dependent migration was calculated as the ratio of cells that migrated with CCL21 to cells that migrated spontaneously, termed migration index.
Immunoblotting
LC were treated as described in the activation assay at 37 °C for 15 min. Cellular extracts were prepared using the Mammalian Protein Extraction Reagent (Thermo Scientific, Rockford, IL). Equal concentrations of cell lysates were electrophoresed on 10% NuPage Novex Bis-Tris gels (Life Technologies) and transferred to PVDF membranes. Immunoblotting was performed using p-Akt, Akt and GAPDH antibodies (Cell Signaling Technologies, Danvers, MA) and species specific infrared dye labeled secondary antibodies (Thermo Scientific). Visualization and quantification of bands was performed using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Supplementary Material
(A) Flow cytometric gating strategy and phenotypic characterization of LC exposed to HPV capsids. Large viable cells are analyzed as indicated on left and phenotypically express markers characteristic of human Langerhans cells: Langerin, E-cadherin, CD1a and CD11c (black histograms, isotype control shown in gray). (B) Expression of surface activation markers after exposure to HPV16 capsids. LC were untreated (left column), exposed to HPV16 L1L2 capsids made in insect cells (HPV16 VLP, middle column), or exposed to HPV16 L1L2 capsids made in 293TT cells (HPV16 PSV, right column). Immature LC express moderate levels of MHC class I and II; high levels of the LC marker CD1a; low levels of costimulatory molecules CD40, CD80 and CD86; and low to undetectable levels of the maturation marker CD83 and the chemokine receptor CCR7. Data are representative of four different healthy donors.
Research Highlights.
HPV16 evades host defense through inactivating antigen-presenting Langerhans cells
HPV18, HPV31, HPV45, HPV11 and HPV5 also suppress Langerhans cell activation
HPV1, a mu-papillomavirus, activates Langerhans cells at high viral titers
Mucosal and cutaneous papillomaviruses share a common mechanisms of immune escape
Acknowledgments
We thank John Schiller and Chris Buck for providing pseudovirion plasmids for the HPV genotypes used in this study. We thank Neil Christensen, John Schiller, and Robert Garcia for providing L1 and L2 antibodies used in immunoblots. This research was supported by Public Health Service grant R01 CA074397 from the National Cancer Institute (to W.M.K). The project described was supported in part by award number P30CA014089 from the National Cancer Institute (to Norris Comprehensive Cancer Center). Contributions from the Netherlands American Foundation and the Karl H. and Ruth M. Balz Trust are also gratefully acknowledged. W. Martin Kast holds the Walter A. Richter Cancer Research Chair. Cytokine multiplex assays were run with the assistance of the USC Norris Comprehensive Cancer Center Beckman Center for Immune Monitoring. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Diane M. Da Silva, Email: ddasilva@usc.edu.
Carly A. Movius, Email: c.a.movius@gmail.com.
Adam B. Raff, Email: adam.raff@gmail.com.
Heike E. Brand, Email: hbrand@usc.edu.
Joseph G. Skeate, Email: skeate@usc.edu.
Michael K. Wong, Email: Mike.Wong@med.usc.edu.
W. Martin Kast, Email: Martin.Kast@med.usc.edu.
References
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, Brooks AG, Heath WR. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10(5):488–95. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- Bernard HU. The clinical importance of the nomenclature, evolution and taxonomy of human papillomaviruses. J Clin Virol. 2005;32(Suppl 1):S1–6. doi: 10.1016/j.jcv.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Bobr A, Olvera-Gomez I, Igyarto BZ, Haley KM, Hogquist KA, Kaplan DH. Acute ablation of Langerhans cells enhances skin immune responses. J Immunol. 2010;185(8):4724–8. doi: 10.4049/jimmunol.1001802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Bryan JT. Abnormalities of cornified cell envelopes isolated from human papillomavirus type 11-infected genital epithelium. Virology. 2000;271(1):65–70. doi: 10.1006/viro.2000.0317. [DOI] [PubMed] [Google Scholar]
- Bryan JT, Brown DR. Transmission of human papillomavirus type 11 infection by desquamated cornified cells. Virology. 2001;281(1):35–42. doi: 10.1006/viro.2000.0777. [DOI] [PubMed] [Google Scholar]
- Buck CB, Thompson CD. Curr Protoc Cell Biol. John Wiley & Sons; 2007. Production of papillomavirus-based gene transfer vectors. [DOI] [PubMed] [Google Scholar]
- Cunningham AL, Carbone F, Geijtenbeek TB. Langerhans cells and viral immunity. Eur J Immunol. 2008;38(9):2377–85. doi: 10.1002/eji.200838521. [DOI] [PubMed] [Google Scholar]
- de Koning MN, Struijk L, Bavinck JN, Kleter B, ter Schegget J, Quint WG, Feltkamp MC. Betapapillomaviruses frequently persist in the skin of healthy individuals. J Gen Virol. 2007;88(Pt 5):1489–95. doi: 10.1099/vir.0.82732-0. [DOI] [PubMed] [Google Scholar]
- de Koning MN, Weissenborn SJ, Abeni D, Bouwes Bavinck JN, Euvrard S, Green AC, Harwood CA, Naldi L, Neale R, Nindl I, Proby CM, Quint WG, Sampogna F, ter Schegget J, Struijk L, Wieland U, Pfister HJ, Feltkamp MC. Prevalence and associated factors of betapapillomavirus infections in individuals without cutaneous squamous cell carcinoma. J Gen Virol. 2009;90(Pt 7):1611–21. doi: 10.1099/vir.0.010017-0. [DOI] [PubMed] [Google Scholar]
- de Villiers EM. Cross-roads in the classification of papillomaviruses. Virology. 2013 doi: 10.1016/j.virol.2013.04.023. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM. Antigen-bearing immature dendritic cells induce peptide-specific CD8(+) regulatory T cells in vivo in humans. Blood. 2002;100(1):174–7. doi: 10.1182/blood.v100.1.174. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193(2):233–8. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duluc D, Gannevat J, Anguiano E, Zurawski S, Carley M, Boreham M, Stecher J, Dullaers M, Banchereau J, Oh S. Functional diversity of human vaginal APC subsets in directing T-cell responses. Mucosal Immunol. 2013;6(3):626–38. doi: 10.1038/mi.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey LM, Raff AB, Da Silva DM, Kast WM. A major role for the minor capsid protein of human papillomavirus type 16 in immune escape. J Immunol. 2009;183(10):6151–6. doi: 10.4049/jimmunol.0902145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausch SC, Da Silva DM, Kast WM. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Res. 2003;63(13):3478–3482. [PubMed] [Google Scholar]
- Fausch SC, Da Silva DM, Kast WM. Heterologous papillomavirus virus-like particles and human papillomavirus virus-like particle immune complexes activate human Langerhans cells. Vaccine. 2005;23(14):1720–1729. doi: 10.1016/j.vaccine.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Fausch SC, Da Silva DM, Rudolf MP, Kast WM. Human papillomavirus virus-like particles do not activate Langerhans cells: a possible immune escape mechanism used by human papillomaviruses. J Immunol. 2002;169(6):3242–3249. doi: 10.4049/jimmunol.169.6.3242. [DOI] [PubMed] [Google Scholar]
- Fausch SC, Fahey LM, Da Silva DM, Kast WM. Human papillomavirus can escape immune recognition through Langerhans cell phosphoinositide 3-kinase activation. J Immunol. 2005;174(11):7172–7178. doi: 10.4049/jimmunol.174.11.7172. [DOI] [PubMed] [Google Scholar]
- Fothergill T, McMillan NA. Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology. 2006;352(2):319–28. doi: 10.1016/j.virol.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Frauwirth KA, Alegre ML, Thompson CB. Induction of T cell anergy in the absence of CTLA-4/B7 interaction. J Immunol. 2000;164(6):2987–93. doi: 10.4049/jimmunol.164.6.2987. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Prost C, Monnet JP, Dy M, Brousse N, Hermine O. Transforming growth factor beta1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J Exp Med. 1998;187(6):961–6. doi: 10.1084/jem.187.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano AR, Harris R, Sedjo RL, Baldwin S, Roe D, Papenfuss MR, Abrahamsen M, Inserra P, Olvera S, Hatch K. Incidence, prevalence, and clearance of type-specific human papillomavirus infections: The Young Women’s Health Study. J Infect Dis. 2002;186(4):462–9. doi: 10.1086/341782. [DOI] [PubMed] [Google Scholar]
- Handisurya A, Day PM, Thompson CD, Buck CB, Kwak K, Roden RB, Lowy DR, Schiller JT. Murine skin and vaginal mucosa are similarly susceptible to infection by pseudovirions of different papillomavirus classifications and species. Virology. 2012;433(2):385–94. doi: 10.1016/j.virol.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazard K, Karlsson A, Andersson K, Ekberg H, Dillner J, Forslund O. Cutaneous human papillomaviruses persist on healthy skin. J Invest Dermatol. 2007;127(1):116–9. doi: 10.1038/sj.jid.5700570. [DOI] [PubMed] [Google Scholar]
- Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, Devilard E, Viret C, Azukizawa H, Kissenpfennig A, Malissen B. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med. 2010;207(1):189–206. doi: 10.1084/jem.20091964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med. 1998;338(7):423–428. doi: 10.1056/NEJM199802123380703. [DOI] [PubMed] [Google Scholar]
- Iijima N, Goodwin EC, Dimaio D, Iwasaki A. High-risk human papillomavirus E6 inhibits monocyte differentiation to Langerhans cells. Virology. 2013;444(1–2):257–62. doi: 10.1016/j.virol.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska S, Majewski S, Obalek S, Orth G. Cutaneous warts. Clin Dermatol. 1997;15 (3):309–19. doi: 10.1016/s0738-081x(96)00170-8. [DOI] [PubMed] [Google Scholar]
- Kanodia S, Fahey LM, Kast WM. Mechanisms used by human papillomaviruses to escape the host immune response. Curr Cancer Drug Targets. 2007;7(1):79–89. doi: 10.2174/156800907780006869. [DOI] [PubMed] [Google Scholar]
- Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23(6):611–20. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Kawana Y, Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. Human papillomavirus type 16 minor capsid protein l2 N-terminal region containing a common neutralization epitope binds to the cell surface and enters the cytoplasm. J Virol. 2001;75 (5):2331–6. doi: 10.1128/JVI.75.5.2331-2336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirnbauer R, Taub J, Greenstone H, Roden R, Dürst M, Gissmann L, Lowy DR, Schiller JT. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. Journal of Virology. 1993;67(12):6929–36. doi: 10.1128/jvi.67.12.6929-6936.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirnbauer RBF, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A. 1992;89(24):12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, Briere F, Chaussabel D, Zurawski G, Palucka AK, Reiter Y, Banchereau J, Ueno H. Functional specializations of human epidermal langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29(3):497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmala SM, Shabalova IP, Petrovitchev N, Syrjanen KJ, Gyllensten UB, Johansson BC, Syrjanen SM. Type-specific persistence of high-risk human papillomavirus infections in the New Independent States of the former Soviet Union Cohort Study. Cancer Epidemiol Biomarkers Prev. 2007;16(1):17–22. doi: 10.1158/1055-9965.EPI-06-0649. [DOI] [PubMed] [Google Scholar]
- Leong CM, Doorbar J, Nindl I, Yoon HS, Hibma MH. Deregulation of E-cadherin by human papillomavirus is not confined to high-risk, cancer-causing types. Br J Dermatol. 2010a;163(6):1253–63. doi: 10.1111/j.1365-2133.2010.09968.x. [DOI] [PubMed] [Google Scholar]
- Leong CM, Doorbar J, Nindl I, Yoon HS, Hibma MH. Loss of epidermal Langerhans cells occurs in human papillomavirus alpha, gamma, and mu but not beta genus infections. J Invest Dermatol. 2010b;130(2):472–80. doi: 10.1038/jid.2009.266. [DOI] [PubMed] [Google Scholar]
- Lizano M, Berumen J, Garcia-Carranca A. HPV-related carcinogenesis: basic concepts, viral types and variants. Archives of medical research. 2009;40(6):428–34. doi: 10.1016/j.arcmed.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Lowe J, Panda D, Rose S, Jensen T, Hughes WA, Tso FY, Angeletti PC. Evolutionary and structural analyses of alpha-papillomavirus capsid proteins yields novel insights into L2 structure and interaction with L1. Virology Journal. 2008;5:150. doi: 10.1186/1743-422X-5-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscicki AB, Ellenberg JH, Farhat S, Xu J. Persistence of human papillomavirus infection in HIV-infected and -uninfected adolescent girls: risk factors and differences, by phylogenetic type. J Infect Dis. 2004;190(1):37–45. doi: 10.1086/421467. [DOI] [PubMed] [Google Scholar]
- Richardson H, Kelsall G, Tellier P, Voyer H, Abrahamowicz M, Ferenczy A, Coutlee F, Franco EL. The natural history of type-specific human papillomavirus infections in female university students. Cancer Epidemiol Biomarkers Prev. 2003;12(6):485–90. [PubMed] [Google Scholar]
- Roden RB, Greenstone HL, Kirnbauer R, Booy FP, Jessie J, Lowy DR, Schiller JT. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. Journal of virology. 1996;70(9):5875–5883. doi: 10.1128/jvi.70.9.5875-5883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani N, Brunner PM, Stingl G. Changing views of the role of Langerhans cells. J Invest Dermatol. 2012;132(3 Pt 2):872–81. doi: 10.1038/jid.2011.437. [DOI] [PubMed] [Google Scholar]
- Romani N, Holzmann S, Tripp CH, Koch F, Stoitzner P. Langerhans cells - dendritic cells of the epidermis. Apmis. 2003;111(7–8):725–40. doi: 10.1034/j.1600-0463.2003.11107805.x. [DOI] [PubMed] [Google Scholar]
- Saeki H, Moore AM, Brown MJ, Hwang ST. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. Journal of Immunology (Baltimore, Md : 1950) 1999;162(5):2472–2475. [PubMed] [Google Scholar]
- Schlecht NF, Kulaga S, Robitaille J, Ferreira S, Santos M, Miyamura RA, Duarte-Franco E, Rohan TE, Ferenczy A, Villa LL, Franco EL. Persistent human papillomavirus infection as a predictor of cervical intraepithelial neoplasia. JAMA: The Journal of the American Medical Association. 2001;286(24):3106–3114. doi: 10.1001/jama.286.24.3106. [DOI] [PubMed] [Google Scholar]
- Scott ME, Ma Y, Kuzmich L, Moscicki AB. Diminished IFN-gamma and IL-10 and elevated Foxp3 mRNA expression in the cervix are associated with CIN 2 or 3. Int J Cancer. 2009;124(6):1379–83. doi: 10.1002/ijc.24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shklovskaya E, O’Sullivan BJ, Ng LG, Roediger B, Thomas R, Weninger W, Fazekas de St Groth B. Langerhans cells are precommitted to immune tolerance induction. Proc Natl Acad Sci U S A. 2011;108(44):18049–54. doi: 10.1073/pnas.1110076108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Lindsay L, Hoots B, Keys J, Franceschi S, Winer R, Clifford GM. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer. 2007;121(3):621–32. doi: 10.1002/ijc.22527. [DOI] [PubMed] [Google Scholar]
- Stanley MA, Pett MR, Coleman N. HPV: from infection to cancer. Biochemical Society Transactions. 2007;35(Pt 6):1456–1460. doi: 10.1042/BST0351456. [DOI] [PubMed] [Google Scholar]
- van der Burg SH, Piersma SJ, de Jong A, van der Hulst JM, Kwappenberg KM, van den Hende M, Welters MJ, Van Rood JJ, Fleuren GJ, Melief CJ, Kenter GG, Offringa R. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A. 2007;104(29):12087–92. doi: 10.1073/pnas.0704672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Winer RL, Hughes JP, Feng Q, Xi LF, Cherne S, O’Reilly S, Kiviat NB, Koutsky LA. Early natural history of incident, type-specific human papillomavirus infections in newly sexually active young women. Cancer Epidemiol Biomarkers Prev. 2011;20(4):699–707. doi: 10.1158/1055-9965.EPI-10-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodham AW, Da Silva DM, Skeate JG, Raff AB, Ambroso MR, Brand HE, Isas JM, Langen R, Kast WM. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE. 2012;7(8):e43519. doi: 10.1371/journal.pone.0043519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodham AW, Raff AB, Raff LM, Da Silva DM, Yan L, Skeate JG, Wong MK, Lin YG, Kast WM. Suppression of Langerhans cell maturation by human papillomavirus type 16: a novel role for the annexin A2 heterotetramer. 2014. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Peng J, Jabbar IA, Liu X, Filgueira L, Frazer IH, Thomas R. Despite differences between dendritic cells and Langerhans cells in the mechanism of papillomavirus-like particle antigen uptake, both cells cross-prime T cells. Virology. 2004;324(2):297–310. doi: 10.1016/j.virol.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Yang R, Day PM, Yutzy WH, Lin KY, Hung CF, Roden RB. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J Virol. 2003;77(6):3531–3541. doi: 10.1128/JVI.77.6.3531-3541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Sun XY, Stenzel DJ, Frazer IH. Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology. 1991;185(1):251–257. doi: 10.1016/0042-6822(91)90772-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Flow cytometric gating strategy and phenotypic characterization of LC exposed to HPV capsids. Large viable cells are analyzed as indicated on left and phenotypically express markers characteristic of human Langerhans cells: Langerin, E-cadherin, CD1a and CD11c (black histograms, isotype control shown in gray). (B) Expression of surface activation markers after exposure to HPV16 capsids. LC were untreated (left column), exposed to HPV16 L1L2 capsids made in insect cells (HPV16 VLP, middle column), or exposed to HPV16 L1L2 capsids made in 293TT cells (HPV16 PSV, right column). Immature LC express moderate levels of MHC class I and II; high levels of the LC marker CD1a; low levels of costimulatory molecules CD40, CD80 and CD86; and low to undetectable levels of the maturation marker CD83 and the chemokine receptor CCR7. Data are representative of four different healthy donors.