

Abstract
Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant neurodegenerative disease that results from a pathogenic glutamine-repeat expansion in the protein ataxin-1 (ATXN1). Although the functions of ATXN1 are still largely unknown, there is evidence to suggest that ATXN1 plays a role in regulating gene expression, the earliest process known to go awry in SCA1 mouse models. In this study, we show that ATXN1 reduces histone acetylation, a post-translational modification of histones associated with enhanced transcription, and represses histone acetyl transferase-mediated transcription. In addition, we find that depleting the Leucine-rich Acidic Nuclear Protein (LANP)—an ATXN1 binding inhibitor of histone acetylation—reverses aspects of SCA1 neuritic pathology.
Keywords: LANP, pp32, ANP32-A, Spinocerebellar ataxia type 1, SCA1, Neurite outgrowth
Introduction
SCA1 is an autosomal dominant disorder characterized by progressive motor incoordination. Its genetic basis lies in a CAG trinucleotide repeat expansion in the coding region of the ATXN1 gene, that in turn results in a glutamine-repeat expansion in the protein ataxin-1 (ATXN1) (Opal and Zoghbi, 2002; Orr et al., 1993). As such, SCA1 is similar to eight other polyglutamine expansion diseases, of which Huntington disease is the most common (Orr and Zoghbi, 2007). As a group these disorders share key features: a delayed onset, a tendency for the glutamine-repeat tract to expand over generations (causing more severe disease and earlier onset), and a relatively circumscribed neuronal degeneration despite widespread expression of the relevant mutant protein. For instance in SCA1, Purkinje cells are the first neurons to be affected. This accounts for motor incoordination or ataxia as the presenting symptom (Orr and Zoghbi, 2007; Wells and Warren, 1998). With time other neuronal groups degenerate, particularly select neuronal groups in the brainstem. It is from brainstem dysfunction that patients eventually succumb, since they can no longer protect their airway (Orr and Zoghbi, 2007; Wells and Warren, 1998). There is as yet no treatment to cure this relentless disease; hence the dire need for understanding pathogenic mechanisms so as to inspire avenues for therapy.
It is still unclear which toxic pathways are triggered by mutant ATXN1. However, there are significant clues—particularly those gleaned from key experiments performed using genetically engineered mice. Most illustrative perhaps are the findings that ATXN1 null mice do not display ataxia, whereas ATXN1 transgenic mice and knock-in mice mirror features of the human phenotype, most visibly the ataxia (Burright et al., 1995; Watase et al., 2002). These results suggest that the incoordination characteristic of SCA1 is for the most part caused by a gain of toxic ATXN1 function. The subsequent observation that mice overexpressing expanded ATXN1 with a mutant nuclear localization signal fail to exhibit neurodegeneration argues for a central role for ATXN1's nuclear properties in pathogenesis (Klement et al., 1998). One nuclear pathway likely to be involved is transcription, given that ATXN1 regulates transcription in concert with transcriptional modulators, and that mutant ATXN1 causes alterations in gene expression in animal models (Cvetanovic et al., 2007; Lam et al., 2006; Serra et al., 2006; Tsai et al., 2004; Tsuda et al., 2005). In fact, in mouse models, transcriptional derangements well precede other pathologic and behavioral features of SCA1 (Gatchel et al., 2008; Klement et al., 1998; Lin et al., 2000; Serra et al., 2004). Recently, we have found that rescuing the levels of at least one gene directly downregulated by expanded ATXN1—the neurotrophic/angiogenic factor VEGF—ameliorates SCA1 toxicity (Cvetanovic et al., 2011). This result adds further weight to the notion that transcriptional deficits play a role in pathogenesis. Here we wished to investigate the mechanism by which ATXN1 modulates transcription, particularly focusing on the role of ATXN1 binding corepressors. Specifically, we tested whether modulating the levels of ATXN1-binding corepressor LANP (also known as pp32, ANP32-A), a protein known to inhibit histone acetylation by virtue of a histone masking mechanism (Matilla and Radrizzani, 2005; Opal et al., 2003; Seo et al., 2001, 2002) affects ATXN1's ability to repress transcription and modulate the SCA1 phenotype.
Materials and methods
Cell culture and cell lines
The rat pheochromocytoma PC12 cell line was grown in Dulbecco's modified Eagle's medium containing 8% horse serum and 8% fetal bovine serum (FBS) (Kular et al., 2010). PC12 cells were induced to differentiate and extend neurites by the addition of 100 ng/ml Nerve Growth Factor (NGF; Sigma) and by reducing the serum in the media (1% FBS and no horse serum).
Human epithelial carcinoma (HeLa) and mouse neuroblastoma Neuro2A (N2A) cells were grown in Dulbecco's modified Eagle's medium (supplemented with 10% (v/v) fetal bovine serum, 0.1 mM MEM non-essential amino acids, 1 mM sodium pyruvate, 100 units/ml penicillin G, and 100 mg/ml streptomycin) under standard tissue culture conditions as suggested by ATCC.
Transfections and neurite outgrowth assays
To assay neuritic outgrowth, PC12 cells were seeded onto laminin (1 μg/ml; Roche Diagnostics) coated coverslips at a density of 175,000 cells/well in 6 well dishes. Plated cells were co-transfected with 2 μg GFP vector, wild-type (2Q) and mutant ATXN1 (84Q) tagged with GFP with or without siRNA targeting LANP using Lipofectamine 2000 (Invitrogen). Knockdown of LANP was achieved by transfecting small interfering RNA (siRNA) at a concentration of 100 nM (Dharmacon Smart Pool; also see Kular et al., 2009). For optimal knockdown, transfections were performed on two consecutive days. Nerve growth factor (NGF, 100 ng/ml Sigma) was added to cells each day after transfection. Two days after the second transfection, the cells were fixed and stained with primary anti-tubulin antibody (T-8660; Sigma) to mark neurites. Immunofluorescence staining was performed as described and images were captured on a Zeiss Axiovert microscope (Kular et al., 2009). Cells displaying processes longer than one-cell body length were counted as cells with neurites. The length of the longest neurite was measured using the Length option tool in Axiovision software. Only transfected cells (i.e. cells displaying expression of green fluorescence plasmids) were included in the analyses. In the experiment shown, more than 100 cells were counted per condition. Statistical analysis was performed using the one way ANOVA with post Bonferroni testing.
Histone acetylation analyses
HeLa and PC12 cells (plated at 1×106 per 10 cm dish) were transfected with 2 μg of GFP-labeled ATXN1 (2Q or 84Q) or GFP as a control. One day after transfection, the transfected HeLa cells were sorted using a fluorescent activated cell sorter (Beckman Coulter) and subsequently lysed in 100 μl of histone preparation buffer (10 mM Hepes, pH 8.0, 10 mM KCl, 1.5 mM MgCl2, 1.5 mM PMSF, 0.5 mM DTT and 0.2 M HCl). PC12 cells were co-transfected with control or LANP targeting Smart Pool siRNA (100 nM, Dharmacon) and treated with 100 ng/ml of NGF. Transfection and NGF treatment were repeated the following day and after 48 h the cells were lysed. The lysates were centrifuged and histones were extracted from the supernatant by incubating it overnight with 8 volumes of acetone at −20 °C. After additional centrifugation, the pellets were resuspended in dH2O. The data is representative of three independent experiments.
Histones were isolated from cerebellar lysates in a similar manner by lysing one cerebellum in 2 ml of histone preparation buffer. The rest of the histone extraction procedure was as described above. The lysates were examined using Western blotting with acetylated H3 (Millipore), actin (Sigma), or total H3 (Millipore) antibodies. The intensity of the bands was quantified by densitometry using the Chemidoc XRS Imaging system and QuantityOne software (Biorad). The data is the average intensity of nine pairs of wild type and SCA1 adult mice or three pairs of LANP null and wild-type. All pairs were gender matched littermates (between six-months of age and a year). Statistical analysis was performed using the paired t-test.
CBP-Gal4 luciferase assays
Luciferase assays were performed using the Dual-Luciferase Reporter Assay System as described (Cvetanovic et al., 2007). pRL-CMV, a CMV promoter-driven renilla luciferase reporter construct (Promega), was included in all transfections to control for experimental variability arising from differences in transfection efficiency and cell number. N2A cells were plated in 24 well plates at 1.5–4×104 cells/well and transfected 24 h later using Effectene Transfection Reagent (Qiagen). Each transfection contained a total of 305 ng of DNA, which included 5 ng of pRL-CMV, 100 ng of Gal4-Luciferase reporter, 100 ng CBP-Gal4, and up to 100 ng of ATXN1 (2Q or 84Q) or LANP. Empty vector pcDNA3 was used to control for equal amount of DNA in each transfection. Data for the titration experiment is representative of three independent experiments. Data for the experiment with the synergistic effect is representative of eight independent experiments. In each independent experiment, all transfections were performed at least in duplicate and each transfection lysate was also assayed twice. Results were calculated as the ratio of firefly luciferase activity/renilla luciferase activity and were normalized to the relative basal activity generated by control samples.
Rotarod analysis
The rotarod assay was performed on 13 weeks old mice; there was no difference in performance based on gender; hence the data was pooled comparing mice that were age-matched. Mice were placed on the rotating rod (Ugo Basile) which accelerates from 4 rpm to 40 rpm for the first 5 min followed by constant speed of 40 rpm (Kular et al., 2010). The time it takes for a mouse to fall off the rotating rod is noted to a maximum of 10 min (the duration of each run). Average of four trials on four consecutive days is plotted. Statistical analysis was performed using two-way ANOVA.
Pathological analysis of mice
Cerebella from one year old mice were fixed with 4% paraformaldehyde, incubated in 30% sucrose overnight and embedded in Optimal Cutting Temperature (OCT) media. Cerebella were sectioned to obtain 40 μm para-sagittal slices (starting from midline) using a cryostat and the sections were subsequently stained with the relevant antibodies as described (Cvetanovic et al., 2011). Briefly, sections were permeabilized by incubating in 0.3% triton-X-100 for 10 min and blocked for 3 h in 5% normal donkey serum. Sections were stained using anti-calbindin (Sigma) and anti-VGLUT2 (Millipore) antibodies for three days and washed three times using TBST. Alexa-594 labeled secondary antibody (Invitrogen) staining was used to visualize calbindin and VGLUT2. Sections were mounted with Vectashield with DAPI (Vector laboratories) and the images of the lobules were captured using Leica laser scanning confocal microscopy or Zeiss Axiovert microscope. We systematically compared images across all the lobules in parasagittal sections equidistant from the midline across all experimental genotypes. The sections were evaluated in a blinded fashion. Width of the molecular layer was obtained by using Leica Application Suite Advanced Fluorescence software (LAS AF Lite software). An average of more than 10 values from 4 cerebellar sections is plotted. Mean fluorescence intensity was calculated from a 25 μm optical slab generated from a z-stack. Statistical analysis was performed using the paired t-test.
Results
Expanded ATXN1 inhibits histone acetylation
Transcriptional deficits represent the earliest pathological change in SCA1, with a majority of the misregulated genes showing downregulation (Lin et al., 2000; Serra et al., 2004). Transcriptional repression correlates closely with histone hypoacetylation (Shahbazian and Grunstein, 2007). Given that ATXN1 binds corepressors that modulate histone acetylation, we asked whether there is detectable histone hypoacetylation in the context of SCA1 pathology.
For these experiments, we used the SCA1 knock-in mouse model. This mouse expresses one copy of expanded ATXN1 (with 154 glutamines/Q) at endogenous levels, and is the most precise model of SCA1 toxicity to date, displaying a robust pathological and behavioral phenotype (Watase et al., 2002). In these mice, using Western blotting techniques on cerebellar lysates, we observed a significant decrease in the acetylation of histone H3—one of the core histones whose acetylation is typically increased with transcriptional activation (Shahbazian and Grunstein, 2007) (Fig. 1a).
Fig. 1.
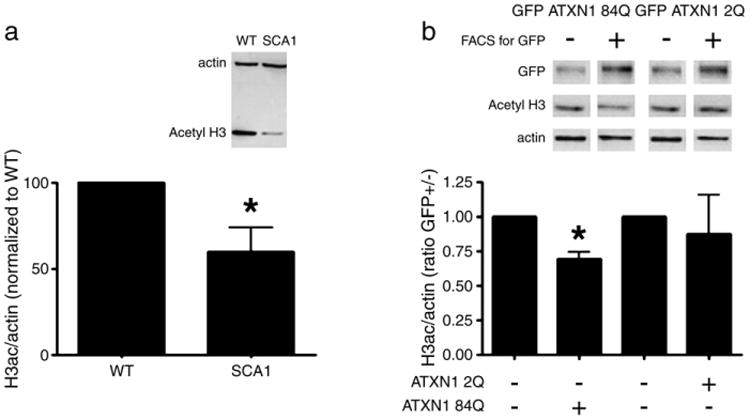
ATXN1 affects histone acetylation. a) The level of histone acetylation in cerebellar lysates of SCA1 mice is compared to wild-type littermates by staining with an antibody specific to acetylated histone H3. Staining with an actin-specific antibody serves as a loading control. The intensity of the lanes is quantified by densitometry where the histograms show the mean value of densitometry with error bars showing SE. n=9 pairs of adult mice. p=0.0347 using paired two-tailed t-test. b) Histone acetylation is decreased in cells expressing mutant ATXN1. HeLa cells transiently transfected with mutant ATXN1 84Q-GFP or wild-type ATXN1 2Q-GFP were sorted for GFP positive (transfected) and GFP negative (untransfected) cells. Western blot with an anti-GFP antibody shows the efficiency of sorting. Western blot with an anti-actin antibody serves as a loading control. Data is representative of three independent experiments. p=0.0122 for ATXN1 84Q (with asterisk) and p=0.3471 for ATXN1 2Q using paired two tailed t-test.
In order to examine whether ATXN1 expression affects H3 acetylation in cell-based experiments, we overexpressed Green-Fluorescent Protein (GFP) tagged-mutant ATXN1-84Q, or wild-type ATXN1 2Q in HeLa cells. In order to enrich for ATXN1 transfected cells, we performed fluorescence activated cell sorting (FACS) for the green fluorophore two days after transfection. In subsequent Western blots, lysates from GFP positive, mutant ATXN1 84Q expressing cells had significant decrease in the levels of acetylated histone H3 compared to untransfected cells (Fig. 1b). These data suggest that mutant ATXN1 can rapidly decrease histone acetylation in cells. Similar results were also observed in the neuronal PC12 cell line, further bolstering our findings (Supplementary Fig. 1). We noticed that wild type ATXN1 2Q expressing cells displayed a trend toward decreased levels of acetylated histone H3. This trend was not statistically significant in HeLa cells; but was in fact significant in PC12 cells. This suggests that wild type ATXN1 can also induce histone deacetylation particularly in neuronal cells and is consistent with the mild toxicity in animal models through a likely gain of normal function (Fernandez-Funez et al., 2000; Cvetanovic et al., 2011).
Synergistic repression by ATXN1 and LANP in the context of histone acetyl transferase-dependent transcription
One of the co-repressors that interacts with mutant ATXN1 is LANP, a protein belonging to the INHAT family of proteins that inhibit histone acetyl transferase activity by virtue of a histone masking mechanism (INHAT is an acronym of inhibitor of HAT activity) (Seo et al., 2001, 2002). There is data to suggest that LANP is recruited to specific chromatin sites by binding to transcription factors (Cvetanovic et al., 2007). With LANP recruitment to chromatin, its acidic tail domain binds basic histones preventing histone acetylation and thus LANP effectively shuts off gene expression in a targeted manner (Seo et al., 2001, 2002). In principle, there are two mutually non- exclusive ways by which expanded ATXN1 might affect LANP function. First, mutant ATXN1 could rob LANP from transcription factors and chromatin binding sites, where LANP might normally bind in the non-diseased state. The second possibility is that LANP is recruited by mutant ataxin-1 into a pathological complex with an exaggerated normal function. In a previous study, we have found that mutant ataxin-1 can reduce LANP's binding to an LANP-dependent transcriptional repressor, lending support to the former possibility (Cvetanovic et al., 2007). Here we decided to test for the latter.
Since LANP is an inhibitor of histone acetyl-transferases (HATs), we decided to test whether ATXN1 recruits LANP to inhibit HAT-transcriptional activity. For these experiments, we turned to a CREB binding protein (CBP) transcriptional assay. CBP is an important histone acetyl transferase in neurons; moreover LANP has been shown to significantly inhibit its activity (Seo et al., 2001, 2002).
CBP activity has also been shown to be down-regulated by other polyglutamine disorders, suggesting that this inhibition could be an underlying mechanism for shared pathology (McCampbell and Fischbeck, 2001; Nucifora et al., 2001; Steffan et al., 2000). In addition ATXN1 has been shown in the past to associate with CBP (Stenoien et al., 2002). Using a CBP-GAL4 construct in a GAL4-responsive luciferase assay, we confirmed that LANP does indeed inhibit CBP activity in a dose dependent manner as reported earlier (Seo et al., 2001) (Fig. 2a). Of note, we found that both wild type (2Q) and mutant (84Q) ATXN1 inhibit CBP activity (Fig. 2a; no statistical difference on CBP repression between wild type and mutant). These results suggest that one of the normal functions of ATXN1 might well be to cause transcriptional repression by inhibiting CBP. Importantly, using doses of ATXN1 and LANP that alone do not cause any inhibition of CBP activity, we found functional synergism between mutant ATXN1 84Q and LANP (Fig. 2b). In these experiments, wild-type ATXN1 2Q did not show significant synergism (p>0.05). These results suggest that it is the mutant ATXN1 that has a greater propensity to recruit LANP compared to wild-type ATXN1. These results are also consistent with ATXN1–LANP interaction studies using the yeast two-hybrid system, where expanded ATXN1 has approximately a ten-fold increased binding compared to wild-type ATXN1 (Matilla et al., 1997).
Fig. 2.
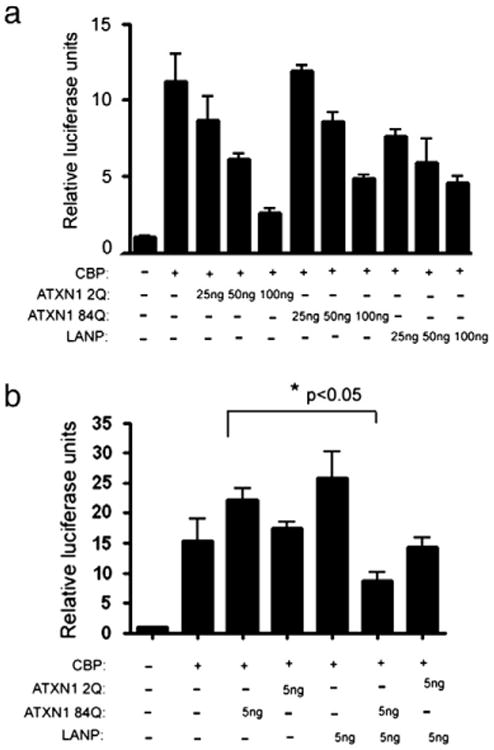
Synergistic transcriptional repression by ATXN1 and LANP. a) N2A cells were co-transfected with the CBP-Gal4 (100 ng/well) and increasing amounts (25, 50 and 100 ng) of LANP and ATXN1 2Q or 84Q-GFP to cause progressive repression of CBP activity when compared to cells transfected with CBP-Gal4 and empty vector. ANOVA p<0.05 comparing 50 or 100 ng of each construct (LANP, ATXN1 2Q or ATXN1 84Q) to CBP control. No statistical difference (p>0.05) was present when comparing repression caused by equal DNA amounts of ATXN1 2Q and ATXN 1 84Q. b) N2A cells were co-transfected with CBP-Gal4 and low levels (5 ng) of LANP and ATXN1 (2Q or 84Q) that by themselves do not cause any inhibition. When co-transfected LANP and ATXN1 84Q synergistically inhibit CBP activity (asterisk signifies p<0.05; Analysis of variance (ANOVA) with post-hoc t test comparing ATXN1 84Q to ATXN1 84Q and LANP). ATXN1 2Q did not show statistical synergism (p>0.05). The data in a. and b. is representative of three (a) or eight (b) independent experiments, Error bars=SE.
Depletion of LANP rescues toxicity engendered by mutant ATXN1 in vitro
As shown above, LANP is recruited by ATXN1 to cause potent transcriptional repression. In addition, we also have evidence to suggest that ATXN1 can take LANP away from LANP's normal targets (Cvetanovic et al., 2007). It is still unclear what the overall role of the interaction of LANP and ATXN1 is in the contextof SCA1 pathogenesis. To address this question, we turned to depleting the levels of LANP in SCA1 cell and mouse models.
Cell-death is a late feature in SCA1 pathology; moreover, it is not a major feature of ATXN1 induced toxicity in vitro. We have therefore developed a cellular assay of ATXN1 toxicity focusing on diminished neurite outgrowth and maintenance, a characteristic feature of SCA1 pathology in human patients and mouse models (Burright et al., 1995; Watase et al., 2002; Yamada et al., 2008). For these experiments we used PC12 cells, that extend neurites upon exposure to Nerve Growth Factor (NGF) (Greene and Tischler, 1976). As shown in Fig. 3a, mutant ATXN1 inhibits neurite extension in PC12 cells (19.67% of mutant ATXN1 (84Q) transfected PC12 cells extend neurites; compared to 44.26% observed in cells transfected with GFP alone; ANOVA p<0.05). We should mention that overexpression of even non-polyglutamine expanded ATXN1 (2Q) decreases neurite outgrowth though to a smaller extent (28.39%; ANOVA p<0.05). The deleterious effects observed with wild type ATXN1 on neurite outgrowth, albeit less than that seen with mutant ATXN1, are once again consistent with the observation that overexpression of wild-type ATXN1 can cause toxicity in animal models through a likely gain of normal function such as an ability to potentiate transcriptional repression as shown here with our CBP-luciferase assays (Fernandez-Funez et al., 2000; Cvetanovic et al., 2011).
Fig. 3.
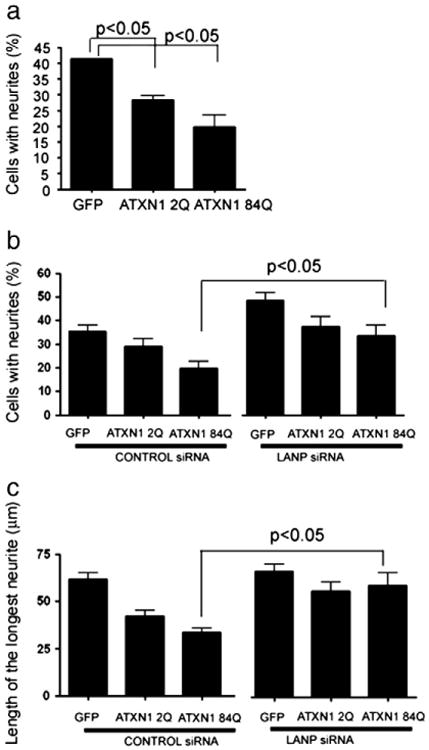
LANP depletion ameliorates the anti-neuritic effects of mutant ataxin-1 in PC12 cells. a) PC12 cells transfected with GFP (control) or GFP-tagged wild type (2Q) or mutant (84Q) ATXN1 were treated with NGF for 72 h. Cells displaying neurites were counted. Data is average of three independent experiments with error bars representing SE. Data was analyzed using ANOVA with Bonferroni post test, p<0.05 comparing GFP transfected control to ATXN1 2Q or ATXN1 84Q. b) and c) PC12 cells were transfected with one of the following constructs expressing: GFP (control), GFP-tagged wild type (2Q) or mutant (84Q) ATXN1 along with control and LANP siRNA as shown. Cells were treated with NGF for 72 h and cells displaying processes longer than one cell body length were counted (b). Length of the longest neurite in GFP positive cells was measured and plotted (c). Data is average of four (b) or three (c) independent experiments, with error bars representing SE. Data was analyzed using ANOVA with Bonferroni post test, for both b and c. p<0.05 comparing control siRNA ATXN1 84Q to LANP siRNA ATXN1 84Q; p>0.05 comparing control siRNA ATXN1 2Q to LANP siRNA ATXN1 2Q.
Regardless, we reasoned that if ATXN1 causes neuritic derangements through transcriptional repression then relieving this repression might prove beneficial. Intriguingly, we have previously shown that depleting LANP promotes neuritic outgrowth by a transcriptional mechanism (Kular et al., 2009) adding further credence to this notion. We therefore tested the effect of depleting LANP using siRNA on this ATXN1 induced neuritic phenotype. As described earlier (Kular et al., 2009), we found that depleting LANP alone (using short interfering RNA; siRNA) has a significant pro-neuritic effect (35.42% with control siRNA compared to 48.57% with LANP targeting siRNA; ANOVA p<0.05). Importantly, we noticed that depleting LANP relieves mutant ATXN1-induced cytopathology: 33.59% of mutant ATXN1 (84Q) transfected PC12 cells with depleted LANP extend neurites compared to 19.59% of mutant ATXN1 (84Q) transfected PC12 cells with normal levels of LANP (ANOVA p<0.05) (Fig. 3b). Moreover, the length of the neurites was also measurably increased upon LANP depletion: 33.55 μm for mutant ATXN1 (84Q) with normal levels of LANP and 59.1 μm for mutant ATXN1 (84Q) with depleted LANP (ANOVA p<0.05) (Fig. 3c). These results suggest that depleting LANP can rescue the neuritic toxicity engendered by mutant ATXN1. Incidentally, the degree of improvement in the case of cells transfected with ATXN1 2Q upon LANP siRNA did not result in statistical improvement, presumably because the deleterious effects caused by ATXN1 2Q were not as robust as that caused by expanded ATXN1. Moreover, depleting LANP rescues the decrease in histone acetylation induced by ATXN1 suggesting that this reversal is via its effects on histone acetylation (Supplementary Fig. 1).
LANP depletion ameliorates neuritic pathology in SCA1 mice
Since depleting LANP has ameliorative effects in vitro, we were eager to test whether depleting LANP in SCA1 mice might also prove beneficial, especially given that neuritic derangements are an early feature of pathology. We mated the LANP null line with the SCA1 knock-in line to generate SCA1 progeny with or without LANP. Given that genetic background can add a confounding factor to phenotypic analyses, we first backcrossed each of these mouse lines over ten generations to generate both SCA1 and LANP null mice in a pure C57/Bl6 background. [Note that LANP null mice are indistinguishable from the wild type mice based on behavioral analyses, including rotarod testing (Opal et al., 2004); however just as our in vitro studies suggest, neurons from LANP null mice show enhanced neurite outgrowth in culture, and an increase in histone acetylation detectable at specific promoters of LANP target genes (Kular et al., 2009), that can be discerned even in semi-quantitative Western blots of whole cerebellar lysates (Supplementary Fig. 2).]
Our initial observations focused around testing the ataxic behavior of SCA1 mice employing rotarod analysis. As expected, SCA1 mice have a diminished ability to stay on the accelerating rotarod (Watase et al., 2002). We did not observe significant improvement in the motor deficit in SCA1 mice that also lack LANP (Fig. 4a). In view of our observation that depleting LANP improved neurite outgrowth in the context of mutant ATXN1 expression, we decided to study the neuritic aspects of SCA1 pathology.
Fig. 4.
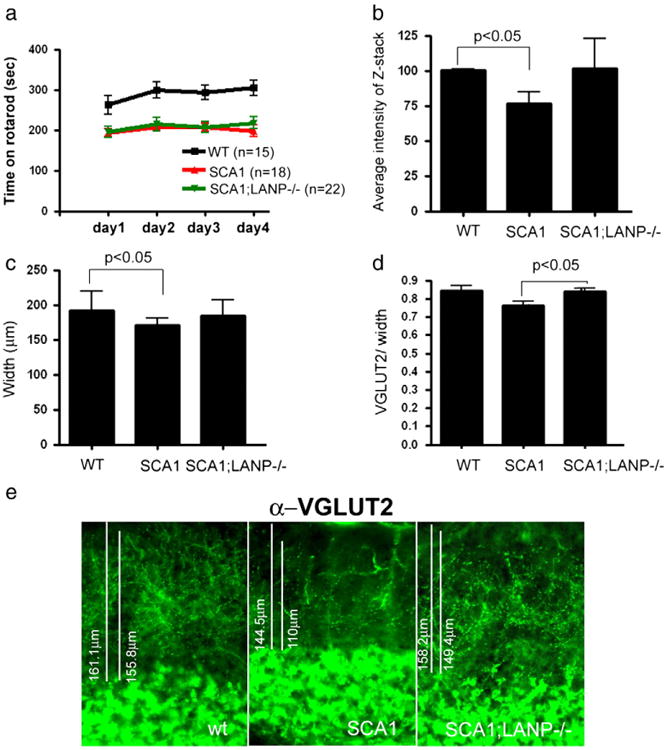
LANP deletion ameliorates pathology but not behavioral deficits in SCA1. a) Rotarod performance of 13 week old mice of indicated genotypes. Analyzed by two way ANOVA with Bonferroni post test p<0.05 comparing WT to SCA1 or WT to SCA1;LANP−/−, and p>0.05 comparing SCA1 to SCA1;LANP−/−. b-e) Cerebella of 1 year old wild type, SCA1, and SCA1;LANP−/− mice were stained with Purkinje cell specific marker calbindin or VGLUT2, labeling presynaptic terminals of climbing fibers. b) Average intensity of z-stacks was measured and normalized to the intensity of wild-type littermates (n=5). Paired t-test values were p=0.0185 and p=0.09 comparing SCA1 mice to wild-type and SCA1; LANP−/− mice respectively. c) An average width from at least six mice of each genotype is shown with error bars showing SE. Statistical analysis was performed using paired t-test with p=0.0367 and p=0.1211 comparing SCA1 mice to wild-type and SCA1;LANP−/− mice respectively. d) Quantification of VGLUT2 staining demonstrating retraction of dendritic arbor in SCA1 mice and rescue of this effect in SCA1;LANP−/− mice. n=3 mice of each genotype. Statistical analysis was performed using paired t-test with p<0.05 comparing SCA1 mice to wild-type or SCA1 mice to SCA1;LANP−/− mice. e) Representative images of VGLUT2 staining.
The most obvious neuritic pathology in SCA1 mice is the thinning of the molecular layer of the cerebellum. This layer is largely constituted of the dendrites of Purkinje cells, which bear the brunt of pathology. To visualize the molecular layer, we performed calbindin staining—a protein enriched in Purkinje cells, and often used as a Purkinje cell marker (Watase et al., 2002). In addition, we performed staining for the vesicular glutamate transporter 2 protein (VGLUT2), an important component of the presynaptic terminals formed by the climbing fibers on the primary dendrites of Purkinje neurons (Fremeau et al., 2001). In SCA1 mice, there is a highly reproducible decrease in the intensity of calbindin staining along with a reduction in the width of the molecular layer. In addition, there is a decrease in the number of climbing fibers terminals as measured by VGLUT2 staining (Duvick et al., 2010). Genetically depleting LANP increased the intensity of calbindin staining and the width of the molecular layer compared to SCA1 mice; however, this improvement failed to reach statistical significance (Figs. 4b and c; with a representative image shown in Supplementary Fig. 3). On the other hand, there was a statistically significant improvement in the VGLUT2 staining upon LANP depletion in SCA1 mice, suggesting an improvement in the neuritic pathology (Figs. 4d and e).
Discussion
SCA1 pathogenesis is characterized by early transcriptional derangements that predate the behavioral features. In this report, we show that some of the changes in gene expression could stem from decreased histone acetylation in SCA1 mice. We further demonstrate that LANP, an ATXN1 interacting co-repressor, synergizes with ATXN1 to cause potent repression of histone acetyl transferase CBP activity. To elucidate the role of histone acetylation in disease progression, we have targeted the HAT inhibitor LANP in the context of SCA1.
Although, we do not find a significant improvement at the level of behavior as assayed by the rotarod test, we do find that depleting LANP improves neuritic pathology in both cell-based and mouse models. Our findings implicating LANP in neuritic derangements in SCA1 add to the importance of transcriptional modulators in the pathology of polyglutamine disorders. Some of these transcriptional modulators are activators: such as CREB Binding Protein (CBP) (in virtually all polyQ disorders), Tip60 (in SCA1) (Serra et al., 2006), and the STAGA complex (in SCA7) (Helmlinger et al., 2006; Wood et al., 2000); while others are corepressors, such as: LANP (in SCA1), the NCoR/SMRT/HDAC3 complex (in SCA1, HD, SBMA and SCA3) (Boutell et al., 1999; Evert et al., 2006; Steffan et al., 2000; Tsai et al., 2004; Dotzlaw et al., 2002; Liao et al., 2003) and a Sin 3A, HDAC2 complex (in DRPLA) (Wood et al., 2000). Our studies with LANP in vivo suggest that specific corepressors might mediate only some aspects of the particular polyglutamine phenotype— for instance neuritic pathology in the case of LANP—leaving others unchanged. We also did not detect any rescue of histone acetylation deficits in SCA1 mice upon LANP depletion (i.e. comparing the histone acetylation in SCA1 mice to SCA1;LANP−/− mice; data not shown). This is likely because complexes other than LANP (such as NCoR/SMRT/HDAC3 and Tip60) also contribute to the alterations in histone acetylation in SCA1 knock-in mice; and the contribution of each individual complex might be difficult to discern in the context of whole cerebellar lysates. We should also point out that besides its role as a corepressor, LANP could also influence neuritic outgrowth directly via its effects on the cytoskeleton (Opal et al., 2003; Ulitzur et al., 1997). This role may be an additional reason why the neuritic pathology in SCA1 is most sensitive to LANP depletion. We foresee that a comprehensive dissection of the role of histone acetylation in SCA1 will require genetic depletion experiments of other corepressors, or pharmacological targeting of histone acetylation using drugs such as histone deacetylase inhibitors (Ferrante et al., 2003; Hockly et al., 2003; Steffan et al., 2001; Taylor et al., 2003).
Supplementary Material
Acknowledgments
We thank members of the Opal lab for their intellectual input. P.O. wishes to thank Huda Zoghbi for discussions in the early stages of this project. This work was funded by the U.S. National Institutes of Health grants K02 NS051340; R21 NS060080, and R01 NS062051; with additional funding from the National Organization for Rare Disorders, the National Ataxia Foundation, and the Brain Research Foundation (P.O.). M.C. and R.K. received funding from the U.S. National Institutes of Health training grant T32.
Footnotes
Available online on ScienceDirect (www.sciencedirect.com).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2012.07.024.
References
- Boutell JM, et al. Aberrant interactions of transcriptional repressor proteins with the Huntington's disease gene product, huntingtin. Hum Mol Genet. 1999;8:1647–1655. doi: 10.1093/hmg/8.9.1647. [DOI] [PubMed] [Google Scholar]
- Burright EN, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Cvetanovic M, et al. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat Med. 2011;17:1445–1447. doi: 10.1038/nm.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetanovic M, et al. The role of LANP and ataxin 1 in E4F-mediated transcriptional repression. EMBO Rep. 2007;8:671–677. doi: 10.1038/sj.embor.7400983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotzlaw H, et al. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol. 2002;16:661–673. doi: 10.1210/mend.16.4.0798. [DOI] [PubMed] [Google Scholar]
- Duvick L, et al. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evert BO, et al. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J Neurosci. 2006;26:11474–11486. doi: 10.1523/JNEUROSCI.2053-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31(2):247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, et al. The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc Natl Acad Sci U S A. 2008;105:1291–1296. doi: 10.1073/pnas.0711257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement IA, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Kular RK, et al. Neuronal differentiation is regulated by leucine-rich acidic nuclear protein (LANP), a member of the inhibitor of histone acetyltransferase complex. J Biol Chem. 2009;284:7783–7792. doi: 10.1074/jbc.M806150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kular RK, et al. Cpd-1 null mice display a subtle neurological phenotype. PLoS One. 2010;5(9):e12649 pii. doi: 10.1371/journal.pone.0012649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YC, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Liao G, et al. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003;278:5052–5061. doi: 10.1074/jbc.M206374200. [DOI] [PubMed] [Google Scholar]
- Lin X, et al. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- Matilla A, et al. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature. 1997;389:974–978. doi: 10.1038/40159. [DOI] [PubMed] [Google Scholar]
- Matilla A, Radrizzani M. The Anp32 family of proteins containing leucine-rich repeats. Cerebellum. 2005;4:7–18. doi: 10.1080/14734220410019020. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Fischbeck KH. Polyglutamine and CBP: fatal attraction? Nat Med. 2001;7:528–530. doi: 10.1038/87842. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Opal P, et al. Generation and characterization of LANP/pp 32 null mice. Mol Cell Biol. 2004;24:3140–3149. doi: 10.1128/MCB.24.8.3140-3149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opal P, et al. Mapmodulin/leucine-rich acidic nuclear protein binds the light chain of microtubule-associated protein 1B and modulates neuritogenesis. J Biol Chem. 2003;278:34691–34699. doi: 10.1074/jbc.M302785200. [DOI] [PubMed] [Google Scholar]
- Opal P, Zoghbi HY. Diseases of the nervous system. Dis Nerv Syst. 2002;II:1880–1895. [Google Scholar]
- Orr H, et al. Expansion of an unstable trinucleotide (CAG) repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Seo SB, et al. Regulation of histone acetylation and transcription by nuclear protein pp 32, a subunit of the INHAT complex. J Biol Chem. 2002;277:14005–14010. doi: 10.1074/jbc.M112455200. [DOI] [PubMed] [Google Scholar]
- Seo SB, et al. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104:119–130. doi: 10.1016/s0092-8674(01)00196-9. [DOI] [PubMed] [Google Scholar]
- Serra HG, et al. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- Serra HG, et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Steffan JS, et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, et al. Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat Cell Biol. 2002;4:806–810. doi: 10.1038/ncb859. [DOI] [PubMed] [Google Scholar]
- Taylor JP, et al. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003;17:1463–1468. doi: 10.1101/gad.1087503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CC, et al. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci U S A. 2004;101:4047–4052. doi: 10.1073/pnas.0400615101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H, et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Ulitzur N, et al. Mapmodulin: a possible modulator of the interaction of microtubule- associated proteins with microtubules. Proc Natl Acad Sci U S A. 1997;94:5084–5089. doi: 10.1073/pnas.94.10.5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34:905–919. doi: 10.1016/s0896-6273(02)00733-x. [DOI] [PubMed] [Google Scholar]
- Wells RD, Warren ST, editors. Genetic Instabilities and Hereditary Neurological Diseases. Academic Press; San Diego: 1998. [Google Scholar]
- Wood JD, et al. Atrophin-1, the dentato-rubral and pallido-luysian atrophy gene product, interacts with ETO/MTG8 in the nuclear matrix and represses transcription. J Cell Biol. 2000;150:939–948. doi: 10.1083/jcb.150.5.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, et al. CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:71–86. doi: 10.1007/s00401-007-0287-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.