

Abstract
The autosomal dominant form of Caffey disease is a largely self-limiting infantile bone disorder characterized by acute inflammation of soft tissues and localized thickening of the underlying bone cortex. It is caused by a recurrent arginine-to-cysteine substitution (R836C) in the α1(I) chain of type of I collagen. However, the functional link between this mutation and the underlying pathogenetic mechanisms still remains elusive. First, it remains to be established as to how a point-mutation in type I collagen leads to a cascade of inflammatory events and spatio-temporally limited hyperostotic bone lesions, and second, the contribution of the structural and inflammatory components to the different organ-specific manifestations in Caffey disease. In this review we attempt to shed light on these questions based on the current understanding of other mutations in type I collagen, their role in perturbing collagen biogenesis, and consequent effects on cell-cell and cell-matrix interactions.
Keywords: Caffey disease, Collagen, Extracellular matrix, COX-2, PGE2, TGFβ
CAFFEY DISEASE OVERVIEW
Caffey disease or infantile cortical hyperostosis (OMIM 114000) is a rare disease affecting various skeletal elements and contiguous connective tissue. First reported by Roske in 1930, the congenital and regressive aspects of the disease were described by Toni in 1943 [1, 2]. In 1945, based on radiographic observations in a large kindred with several affected members, Caffey and Silverman formally defined it as an infantile inherited disease, with a mean age at onset of 9 weeks [3]. Bennett et al., and later Lecolier et al. also described a lethal prenatal form of Caffey disease with distinct bone lesions that are recognized radiographically on average by 27 weeks of gestation [4, 5]. The infantile form of the disease is usually self-limiting, with spontaneous regression by two years; however, several well documented cases of recurrence of cortical hyperostosis during adolescence have also been reported [6, 7].
The infantile form of the disease is characterized by fever, soft tissue swelling and hyperirritability. Radiographic evaluation is used to establish the diagnosis. The disease is variably penetrant involving a single or multiple bones. Periosteal new bone formation leads to cortical thickening (hyperostosis) of the affected bone(s) and swelling of the overlying soft tissue (Fig. 1) [8]. The bone lesions are often asymmetric with a characteristic involvement of the mandible, clavicle, scapula, skull, ilium and long bones. The mandible is most frequently involved (70–90% of the cases) and is often used as a defining diagnostic criterion [9, 10]. In half the cases the clavicle is affected, whereas the ribs and the scapulae are affected in 20–30% of the cases. In the long bones, primary bone lesions are localized to the diaphysis resulting in grossly deformed spindle shaped bones. The epiphysis and metaphysis are typically not involved; however cortical hyperostosis frequently extends to these areas. Episodes of hyperostosis may also lead to the formation of bony bridges between adjacent bones [8, 11].
Figure 1. Radiographic features of Caffey disease.
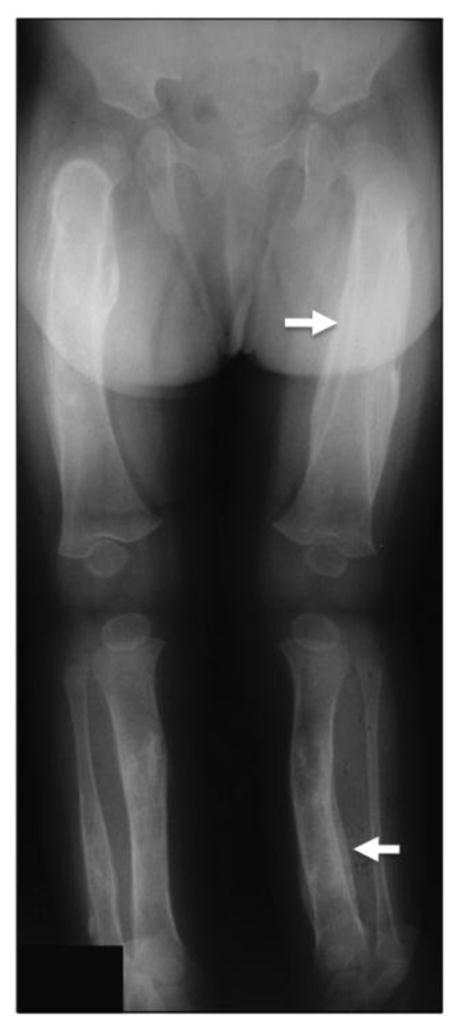
Representative radiograph of an affected individual showing periosteal thickening of the femur and the tibia (white arrow). Adapted from Gensure et al., 2005 [20].
Laboratory findings in affected patients include elevated erythrocyte sedimentation rates and alkaline phosphatase levels along with increased C-reactive protein and immunoglobulin levels suggesting a concurrent inflammatory distress [12–15]. Eversole et al. were the first to provide a detailed histological analysis of the affected bones with evidence for local inflammation along with subperiosteal new lamellar bone formation. Foci of acute inflammation were confined to the periosteum early on but progressively disrupted the adjacent soft tissue. As the disease resolves the periosteum reestablishes as a fibrous layer under which new subperiosteal bone is formed. Subsequently, the inflammation disappears and excess peripheral bone is eliminated through remodeling [16].
The role of prostaglandin E (PGE1 and 2) received strong support when Ueda et al. reported induction of cortical hyperostosis in infants with ductus-dependent cyanotic heart diseases receiving prophylactic PGE1 administration to prevent duct closure [17]. This PGE-induced side effect has considerable prevalence with radiologic changes evident in 62% of all infants receiving PGE1 infusion for more than 60 days [18]. Heyman et al. subsequently reported elevated PGE levels in five patients with cortical hyperostosis, whose symptoms resolved upon abrogation of PGE synthesis via indomethacin administration or other anti-inflammatory drugs, such as glucocorticoids [19]. However, the mechanistic context of PGE elevation and consequent immunoregulatory effects on disease onset and progression remain unresolved.
The pathophysiology of Caffey disease presents a unique clinical conundrum, as it is a spatio-temporally limited state of acute bone and soft tissue inflammation that is transmitted as an autosomal dominant trait with incomplete penetrance. Moreover, incidence of non-familial cases attributed to PGE therapy further complicates matters and indicates that there could be several distinct subtypes of the disease. In an attempt to identify the genetic defect responsible for the infantile form of Caffey disease, Gensure et al. undertook genetic linkage studies in one large kindred with the autosomal dominant form of the disease. These studies revealed that affected individuals (including patients with severe prenatal cortical hyperostosis) and obligate carriers are heterozygous for a missense mutation (3040C→T) in the COL1A1 gene. This mutation results in an Arg to Cys (R836C/p.R1014C) substitution within the helical domain of α1(I) chain of type of I collagen (COL1A1) [12, 20]. Furthermore, Gensure et al. identified the same mutation in several unrelated kindred and revealed previously unknown features of the disease in adults, namely hyperextensible skin and joint hypermobility. Subsequently, other groups identified the R836C mutation in Thai, Korean, Indian and Australian kindreds with the classical familial form of the disease and in sporadic cases of the infantile and prenatal forms of disease. Taken together these studies validated the pathogenetic role of this particular amino acid substitution in Caffey disease [21–24].
Ultrastructural analyses of mutant dermal collagen fibrils revealed variability in diameter and aberrant disulfide bonded α1(I) homodimers[20]. These structural defects in type I collagen may explain the skin and joint manifestations in adults, however their role in pathogenesis of the self-limiting bone lesions during infancy remains unresolved.
TYPE I COLLAGEN MUTATIONS AND PATHOPHYSIOLOGY
The structural building blocks of connective tissues, including the skeleton, involve collagens, proteoglycans, non-collagenous proteins along with enzymes capable of matrix assembly and degradation. The relative differences in molecular composition define the structural and functional topology of the matrix at various anatomical sites. Collagens are the most abundant extracellular matrix (ECM) components in the skeleton, with ~90% of the bone matrix consisting of type I collagen fibrils [25]. Apart from providing multidimensional strength, they serve as the primary substrate for mineralization. In addition, type I collagen fibrils within bone participate in the formation of supramolecular assemblies in conjunction with small leucine-rich proteoglycans and other non-collagenous proteins to produce an architecturally precise ECM that facilitates cell adhesion, migration and function [26]. Therefore, mutations in genes encoding different ECM components often result in similar phenotypes. A corollary is that in cases where the molecules are composed of multiple domains with different functional properties, different mutations in the same gene can result in different clinical manifestations. Cases in point are the various collagenopathies including osteogenesis imperfecta (OI), Ehlers-Danlos syndrome (EDS) and Caffey disease (Table.1).
Table 1.
Type I Collagen Disorders Genotype Phenotype Relationships.
| Type I Collagen Disorder | OMIM | Inheritance | Clinical Features | Gene Defect | Molecular Genetics |
|---|---|---|---|---|---|
| Osteogenesis Imperfecta, Type I | 166200 | Autosomal Dominant | Mild and non-deforming: Fractures with little or no limb deformity, blue sclerae, normal stature, dentinogenesis imperfecta, hearing loss. | null COL1A1 | Frameshifts or premature termination codon in COL1A1. |
| Osteogenesis Imperfecta, Type II | 166210 | Autosomal Dominant | Perinatal lethal: In utero fractures and bone deformity, blue sclerae, undermineralized skull, platyspondyly | COL1A1 or COL1A2 | Glycine substitutions in COL1A1 or COL1A2. |
| Osteogenesis Imperfecta, Type III | 259420 | Autosomal Dominant | Severely deforming: High fracture incidence with moderate bone deformity, blue/grey sclerae, very short stature, dentinogenesis imperfecta | COL1A1 or COL1A2 | Glycine substitutions in COL1A1 or COL1A2. |
| Osteogenesis Imperfecta, Type IV | 166220 | Autosomal Dominant | Moderately deforming: Fractures with mild limb deformity, normal sclerae, variable short stature, dentinogenesis imperfecta, hearing loss. | COL1A1 or COL1A2 | Glycine substitutions in COL1A1 or COL1A2. |
| Ehlers-Danlos Syndrome, Type I | 130000 | Autosomal Dominant | Marked skin involvement - fragile, bruisable and hyperextensible skin, joint laxity and subluxation. | COL1A1 | Arginine to cysteine substitution (R134C) in COL1A1 |
| Ehlers-Danlos Syndrome, Type VIIA | 130060 | Autosomal Dominant | Joint laxity and subluxations, congenital hip dislocations, minimal skin involvement | COL1A1 | Splice- junction mutations of exon 6 in COL1A1 |
| Ehlers-Danlos Syndrome VIIB | 130060 | Autosomal Dominant | Joint laxity and subluxations, congenital hip dislocations, minimal skin involvement | COL1A2 | Exon 6 deletions in COL1A2 |
| Ehlers-Danlos Syndrome, Cardiac Valvular | 225320 | Autosomal Recessive | Valvular insufficiency, joint laxity and subluxations, hyperextensible skin | null COL1A2 | Premature termination codon in COL1A2 |
| Caffey Disease | 114000 | Autosomal Dominant | Joint laxity and subluxation, hyperextensible skin with normal appearance, cortical hyperostosis | COL1A1 | Arginine to cysteine substitution (R836C) in COL1A1 |
The triple helical domain within the collagen chains enables self-assembly into highly organized collagen fibrils with high tensile strength that are then integrated into the bone matrix. Formation of type I collagen fibers involves C-propeptide-mediated intracellular assembly of two α1(I) and one α2(I) pro-collagen chains followed by extracellular cleavage of the N- and C-termini of procollagen monomer by specific proteinases resulting in the higher order assembly into fibrils and fibers [26] (Fig. 2). Tandem Gly-X-Y sequences within the triple helical domain are critical for this self-assembly and triple helical conformation. Consequently, Gly substitutions result in structural or quantitative defects of type I collagen leading to bone fragility and fractures characteristic of OI types II-IV. Rare mutations affecting type I procollagen processing sites or involving X/Y residues result in distinct variants of OI [27]. Mutations resulting in delayed N-propeptide processing cause a phenotype that overlaps OI and EDS, whereas mutations in C-propeptide cleavage site result in mild OI with increases in bone mineralization (29). Substitutions of X or Y residues cause clinically similar disorders, including mild OI, classical EDS, and Caffey disease, but only Caffey disease is characterized by increased cortical bone formation [20, 28, 29]. The overlapping clinical manifestations of these diseases are thought to result from delayed propeptide processing or impaired ligand interactions with COL1 fibrils, however precise mechanism(s) leading to bone fragility or increased cortical bone formation in these cases remains unclear.
Figure 2. Schematic representation of type I collagen fibril.
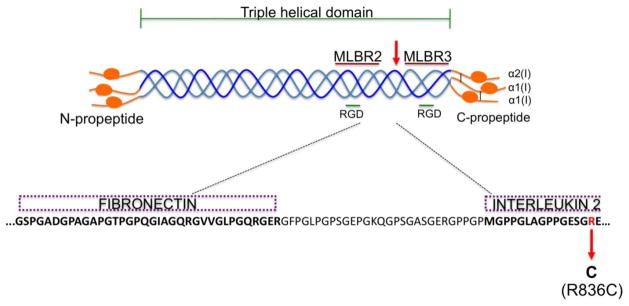
The R836C mutation (red arrow) is localized to a gap region between the major ligand binding regions 2 and 3 (MLBR2 and MLBR3, maroon lines) and flanked by integrin-binding RGD motifs (green line) within the C-terminus. The R836C mutation is located within the interleukin-2 binding site and juxtaposed to a fibronectin binding site (dashed purple boxes).
The mature triple helical collagen molecules contain multiple sites for interactions with other structural and signaling components leading to the formation of the heterotypic fibril that is then incorporated into a higher order structure within the extracellular matrix [30, 31]. In silico analyses by the San Antonio group of putative ligand binding sites along with the distribution of known type I collagen mutations resulted in a domain model that mapped various cell, signaling and matrix interactions to the type I collagen fibril. These studies identified three ligand-binding hotspots defined as major ligand binding regions (MLBR 1-3). MLBR 2 and 3 (helix positions 680-830 and 920-1012) within type I collagen fibril have been shown to be crucial for intermolecular crosslinking and self-assembly, binding to integrins α1β1/α2β1, and interactions with an array of non-collageneous proteins (fibronectin, cartilage oligomeric matrix protein, discoidin domain receptors, decorin and heparin) (Table 2). Clustering of lethal collagen mutations to MLBR 2 and 3 underscores the importance of these sites for maintaining matrix homeostasis. The Arg836 residue that causes Caffey disease when mutated, maps to a gap region between MLBR 2 and 3 (Fig. 2). Arg-to-Cys substitutions within the triple helical domain of type I collagen are thought to influence both structure and function of type I collagen fibrils. In fact, presence of cysteine residues results in aberrant disulfide bonded α1(I) dimers leading to abnormal fibril diameter often noticed in ultrastructural analyses of patient-derived dermis [20, 29]. Importantly, local unwinding surrounding the R-to-C substitutions is thought to interrupt ligand interactions [29]. Broadly, these mutations (i.e. R134C, R836C, and R888C) are characterized by hyperextensible skin and joint hypermobility. However, each mutation can also be recognized by specific phenotype during early childhood. For example, periosteal new bone formation and cortical hyperostosis associated with soft tissue inflammation are only observed in Caffey disease. Furthermore, these phenotypes are not replicated by other R-to-C substitutions, suggesting that the R836C mutation may disrupt specific type I collagen-ligand interaction(s), resulting in impaired resident cell function. Several lines of evidence support such a hypothesis: 1) R836C is localized within the region that was shown to interact with interleukin-2 (IL-2), and is in close vicinity of the site known to interact with fibronectin (FN) (Fig. 2) 2. Neonates have a hyporesponsive immune system with blunted inflammatory cytokine production[32]. Thus, suggesting inflammation in Caffey disease to be a local matrix-driven process. 3. Upregulation of COX2/PGE axis and transforming growth factor (TGFβ) signaling have been shown to cause cortical hyperostosis in humans [17, 33]. Taken together, these observations argue for a pathogenesis driven not necessarily by gross structural or quantitative defects in type I collagen fibrils, but by a subtle alteration in ECM composition that may affect cell signaling as a result of impaired collagen interactions.
Table 2.
Molecules interacting with type I collagen fibril and putative binding sites.
| Interacting Molecule | Putative Interaction Site on Type I Collagen Fibril [31, 52] |
|---|---|
| Amyloid precursor protein | 822–1040 |
| Cartilage oligomeric matrix protein | 582–629; 1035–1040 |
| Type V collagen | 1023–1036 |
| Decorin | 159–199; 345–387; 934–964 |
| Dermatan sulfate proteoglycan | 601–611; 622–632; 852–862; 856–866; 887–897 |
| Fibronectin | 757–790 |
| Heparin sulfate proteoglycan | 86–90 |
| Interleukin-2 | 124–402; 822–1015 |
| Keratin sulfate proteoglycan | 451–461; 565–575 |
| Matrix metalloproteinase 1, 2, 13 | 766–786 |
| Phosphophoryn | 680–750 |
| Secreted protein acidic and rich in cysteine | 100–200; 650–800 |
| Thrombospondin | 101–200 & 651–800 |
| von Willebrand factor | 396–404 |
Consistent with such a hypothesis, Somasundaram et al., demonstrated binding of IL-2 to native type 1 collagen and a carboxy-terminal peptide spanning Arg836 [34]. Although first isolated as a T-cell derived growth factor, IL-2 is expressed in the skin and gut of neonatal mice suggesting a role that is distinct from adult immunological responses to an antigen [35]. IL-2 has been shown to have dual and contrasting roles leading to induction and termination of inflammatory immune responses, by regulating T-cell proliferation and CD4+ T-helper 1 (TH1) and TH2 effector T-cell differentiation and inhibiting development of inflammatory TH17 cells respectively [36]. Moreover, CD4+ T-helper cells predominantly mediate neonatal adaptive immunity [37]. Thus, in the context of Caffey disease, it is conceivable that pre-existing immunological memory along with CD4+ T-helper mediated immunological response could possibly result in the acute inflammatory phase that regresses as the immune system matures. Given the central role of IL-2 in immunity and inflammation, it would be crucial to understand how the local bioavailability of this key cytokine is altered in Caffey disease and to evaluate its effects on resident cell function.
Cellular interactions with abnormal matrix lead to aberrant tissue development that in turn influences signaling between resident cells ultimately resulting in impaired tissue homeostasis. In the context of OI, aberrant signaling between osteoblasts and osteoclasts is thought to lead to enhanced bone remodeling exacerbating the bone fragility caused by the primary collagen defect [27]. Moreover, inter-chain cross-links and collagen fibril maturity have been shown to have instructive roles in osteoblast differentiation [38]. Binding of α2β1 integrins on the preosteoblast surface to type I collagen within the bone matrix has been shown to activate Runx2, a key regulator of osteoblast differentiation that drives expression of downstream genes such as osteocalcin, bone sialoprotein, alkaline phosphatase, and parathyroid hormone/parathyroid hormone-related protein receptor [39]. Furthermore, interaction of type I collagen with α2β1 integrin has been shown to enable osteoblast differentiation through the down-regulation of TGFβ receptor expression in osteoblastic cells. The down- regulation of TGFβ receptor complexes enables these cells to evade the inhibitory effects of TGFβ signaling on terminal osteoblast differentiation [40]. Moreover, FN has been shown to orchestrate type I collagen assembly and matrix sequestration of latent TGFβ in conjunction with latent transforming growth factor binding protein-1 [41]. Impaired FN matrix results in promiscuous TGFβ activation in both in vitro and in vivo experimental models [42, 43]. Consistent with the crucial role of TGFβ signaling in bone formation, cortical hyperostosis in Camurati-Engelmann disease results from constitutively active TGFβ signaling due to mutations within the latency-associated peptide of TGFβ1 [33, 44].
Taken together, these observations in rare human diseases are highly significant for the understanding of normal osteoblastogenesis and how collagen mutations lead to distinct bone phenotypes. For example, some of the type I collagen mutations identified in dominant OI are associated with impaired osteoblast maturation and increased osteoclast recruitment, whereas mutant collagen fibrils in Caffey disease lead to enhanced lamellar bone formation along the periosteal surface. Understanding the role of the two integrin-binding RGD domains (helix residues 567-569 and 915-917) juxtaposed to Arg 836, along with fine mapping of the protein-protein interacting domains within this region may provide important clues to further our understanding of type I collagen in the bone formation process (Fig. 2).
Spatiotemporal and quantitative coupling between bone forming osteoblasts and bone resorbing osteoclasts is crucial for the maintenance of adult bone mass. However, during embryogenesis and early postnatal development these activities remain uncoupled to facilitate rapid bone matrix synthesis and growth. This is called bone modeling, a process that helps in fine tuning the spatial distribution of accumulating tissue to evolving mechanical demands [45]. The endoplasmic reticulum (ER) plays a critical role in the synthesis, folding and processing of type I collagen, the predominant protein synthesized by metabolically active osteoblasts during this phase [26, 46]. To enable the increased metabolic demands of the cell, the ER undergoes active membrane expansion thus greatly increasing its surface area. However, expression of mutant proteins results in increased levels of unfolded or misfolded proteins within the ER lumen leading to a state of chronic ER stress. This triggers the activation of the unfolded protein response (UPR), a quality control mechanism designed to maintain the protein-folding equilibrium within the ER. Type I procollagen chains harboring mutations in the C-terminal propeptide or triple helical domain (G349C in COL1A1 and G904C in COL2A1) have been shown to trigger the UPR [47]. Importantly, ER stress has been shown to induce of COX-2 (cyclooxygenase-2) expression, a key enzyme necessary for the synthesis of PGE [48]. Furthermore, numerous murine studies have demonstrated the bone anabolic effects of PGE2 resulting from increased proliferation of periosteal osteoblasts [49–51].
Based on radiographic findings, Gensure et al., suggested that periosteal detachment from underlying bone could be the key pathological process driving hyperostosis in Caffey disease [20]. In such a scenario, the periosteal injury might elicit an inflammatory response that triggers a local positive feedback loop involving COX-2/PGE signaling driving new bone formation by periosteal osteoblasts. It would therefore be important to investigate signaling events downstream of COX-2/PGE axis and their potential contribution to Caffey disease pathogenesis (Fig. 3).
Figure 3. Hypothetical model representing pathogenetic mechanisms underlying Caffey Disease.

The R836C mutant proα1(I) chains may induce ER stress triggering an unfolded protein response leading to an upregulation of the COX2/PGE axis. Alternatively, the R836C mutant type I collagen fibrils may disrupt ligand interactions within the ECM resulting in signaling events that either directly or indirectly lead to elevated COX2/PGE signaling and altered resident cell function.
CONCLUSION
Characterization of the various collagenopathies has yielded new insights into the pathophysiology of connective tissue disorders. These findings have revised the notion that the molecular basis underlying the spectrum of clinical phenotypes is solely due to structural defects of the ECM. The emerging paradigm postulates metabolic dysfunction of the matrix and the consequent effects on resident cell function as concomitant causative drivers of disease progression. In light of these findings, a reevaluation of the pathogenetic mechanisms underlying Caffey disease may provide novel insights into bone biology and potentially offer therapeutic approaches for the clinical management of both the prenatal and infantile forms of the disease. Further investigations into the functional roles of the type I collagen fibril could also provide clues as to how the R836C mutation leads to periosteal new bone formation that is spatially and temporally limited. Apart from unraveling novel matrix-derived signals that drive bone anabolic responses, this understanding of the interplay between type I collagen integrity and bone formation may be relevant to the management of common disorders such as osteoporosis and conditions characterized by ectopic calcifications in skin, kidney, tendons and the cardiovascular tissues.
HIGHLIGHTS.
Caffey disease is caused by an Arg to Cys substitution (R836C) in the α1(I) chain of type I collagen.
The structural change caused by the R836C mutation may trigger collagen-dependent metabolic dysfunction of the matrix.
Abnormal COX2/PGE signaling is likely to play role in periosteal new bone formation in Caffey disease.
Altered interplay between collagen matrix and bone formation in Caffey could provide novel insights into common disorders affecting bone.
Acknowledgments
We would like to thank Drs. Francesco Ramirez and Roland Baron for their insightful comments and suggestions.
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roske G. Eine eigenartige Knochenerkrankung im Säuglingsalter. Mschr Kinderheilk. 1930;47:385. [Google Scholar]
- 2.Toni G. Una nuova osteopatia infantile: La poliosteopatia deformante connatale regressiva. Policlin Infantil Radiot Fis Med. 1943;9:1. [Google Scholar]
- 3.Caffey J, Silverman WA. Infantile cortical hyperostosis: preliminary report on a new syndrome. American Journal of Roentgenology, Radium Therapy and Nuclear Medicine. 1945;54:1–16. [Google Scholar]
- 4.Bennett H, Nelson T. Prenatal cortical hyperostosis. The British journal of radiology. 1953;26(301):47–49. doi: 10.1259/0007-1285-26-301-47. [DOI] [PubMed] [Google Scholar]
- 5.Lecolier B, et al. Radiographic, haematological, and biochemical findings in a fetus with Caffey disease. Prenatal diagnosis. 1992;12(8):637–641. doi: 10.1002/pd.1970120803. [DOI] [PubMed] [Google Scholar]
- 6.Navarre P, Pehlivanov I, Morin B. Recurrence of infantile cortical hyperostosis: a case report and review of the literature. J Pediatr Orthop. 2013;33(2):e10–7. doi: 10.1097/BPO.0b013e318277d3a2. [DOI] [PubMed] [Google Scholar]
- 7.Borochowitz Z, et al. Familial Caffey’s disease and late recurrence in a child. Clin Genet. 1991;40(4):329–35. doi: 10.1111/j.1399-0004.1991.tb03104.x. [DOI] [PubMed] [Google Scholar]
- 8.Caffey J. Infantile cortical hyperostosis; a review of the clinical and radiographic features. Proceedings of the Royal Society of Medicine. 1957;50(5):347–354. doi: 10.1177/003591575705000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufmann HJ, Mahboubi S, Mandell GA. Case Report 39: Monosteal infantile cortical hyperostosis. Skeletal Radiology. 1977;2:109. [Google Scholar]
- 10.Juniper R. Caffey’s disease. The British journal of oral surgery. 1982;20(4):281–287. doi: 10.1016/s0007-117x(82)80024-3. [DOI] [PubMed] [Google Scholar]
- 11.Greenfield GB. Radiology of bone disease. 4. 1986. [Google Scholar]
- 12.Kamoun-Goldrat A, le Merrer M. Infantile cortical hyperostosis (Caffey disease): a review. Journal of oral and maxillofacial surgery. 2008;66(10):2145–2150. doi: 10.1016/j.joms.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Barba W, Freriks D. The familial occurrence of infantile cortical hyperostosis in utero. The Journal of pediatrics. 1953;42(2):141–150. doi: 10.1016/s0022-3476(53)80207-6. [DOI] [PubMed] [Google Scholar]
- 14.Finsterbush A, Rang M. Infantile cortical hyperostosis. Follow-up of 29 cases. Acta orthopaedica Scandinavica. 1975;46(5):727–736. doi: 10.3109/17453677508989258. [DOI] [PubMed] [Google Scholar]
- 15.Silverman FN. Caffey’s pediatric X-ray diagnosis. 1984. [Google Scholar]
- 16.Eversole S, Holman G, Robinson R. Hitherto undescribed characteristics of the pathology of infantile cortical hyperostosis (Caffey’s disease) Bulletin of the Johns Hopkins Hospital. 1957;101(2):80–99. [PubMed] [Google Scholar]
- 17.Ueda K, et al. Cortical hyperostosis following long-term administration of prostaglandin E1 in infants with cyanotic congenital heart disease. The Journal of pediatrics. 1980;97(5):834–836. doi: 10.1016/s0022-3476(80)80282-4. [DOI] [PubMed] [Google Scholar]
- 18.Woo K, Emery J, Peabody J. Cortical hyperostosis: a complication of prolonged prostaglandin infusion in infants awaiting cardiac transplantation. Pediatrics. 1994;93(3):417–420. [PubMed] [Google Scholar]
- 19.Heyman E, Laver J, Beer S. Prostaglandin synthetase inhibitor in Caffey disease. The Journal of pediatrics. 1982;101(2):314. doi: 10.1016/s0022-3476(82)80153-4. [DOI] [PubMed] [Google Scholar]
- 20.Gensure R, et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. The Journal of clinical investigation. 2005;115(5):1250–1257. doi: 10.1172/JCI22760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suphapeetiporn K, et al. Expanding the phenotypic spectrum of Caffey disease. Clin Genet. 2007;71(3):280–4. doi: 10.1111/j.1399-0004.2007.00768.x. [DOI] [PubMed] [Google Scholar]
- 22.Ranganath P, et al. COL1A1 mutation in an Indian child with Caffey disease. Indian J Pediatr. 2011;78(7):877–9. doi: 10.1007/s12098-010-0339-z. [DOI] [PubMed] [Google Scholar]
- 23.Cho TJ, et al. The c.3040C > T mutation in COL1A1 is recurrent in Korean patients with infantile cortical hyperostosis (Caffey disease) J Hum Genet. 2008;53(10):947–9. doi: 10.1007/s10038-008-0328-5. [DOI] [PubMed] [Google Scholar]
- 24.Kamoun-Goldrat A, et al. Prenatal cortical hyperostosis with COL1A1 gene mutation. Am J Med Genet A. 2008;146A(14):1820–4. doi: 10.1002/ajmg.a.32351. [DOI] [PubMed] [Google Scholar]
- 25.Young M. Bone matrix proteins: their function, regulation, and relationship to osteoporosis. Osteoporosis international. 2003;14(Suppl 3):42. doi: 10.1007/s00198-002-1342-7. [DOI] [PubMed] [Google Scholar]
- 26.Myllyharju J, Kivirikko K. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends in genetics : TIG. 2004;20(1):33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Forlino A, et al. New perspectives on osteogenesis imperfecta. Nature reviews Endocrinology. 2011;7(9):540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nuytinck L, et al. Substitution of glycine-661 by serine in the alpha1(I) and alpha2(I) chains of type I collagen results in different clinical and biochemical phenotypes. Hum Genet. 1996;97(3):324–9. doi: 10.1007/BF02185764. [DOI] [PubMed] [Google Scholar]
- 29.Malfait F, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28(4):387–95. doi: 10.1002/humu.20455. [DOI] [PubMed] [Google Scholar]
- 30.Marini J, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Human mutation. 2007;28(3):209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Lullo G, et al. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. The Journal of biological chemistry. 2002;277(6):4223–4231. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- 32.PrabhuDas M, et al. Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol. 2011;12(3):189–94. doi: 10.1038/ni0311-189. [DOI] [PubMed] [Google Scholar]
- 33.Kinoshita A, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26(1):19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 34.Somasundaram R, et al. Collagens serve as an extracellular store of bioactive interleukin 2. The Journal of biological chemistry. 2000;275(49):38170–38175. doi: 10.1074/jbc.M006616200. [DOI] [PubMed] [Google Scholar]
- 35.Yang-Snyder JA, Rothenberg EV. Spontaneous expression of interleukin-2 in vivo in specific tissues of young mice. Dev Immunol. 1998;5(4):223–45. doi: 10.1155/1998/12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoyer KK, et al. Interleukin-2 in the development and control of inflammatory disease. Immunol Rev. 2008;226:19–28. doi: 10.1111/j.1600-065X.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 37.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4(7):553–64. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 38.Turecek C, et al. Collagen cross-linking influences osteoblastic differentiation. Calcif Tissue Int. 2008;82(5):392–400. doi: 10.1007/s00223-008-9136-3. [DOI] [PubMed] [Google Scholar]
- 39.Xiao G, et al. Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. The Journal of biological chemistry. 1998;273(49):32988–32994. doi: 10.1074/jbc.273.49.32988. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi Y, Nakayama K, Matsumoto T. Differentiation and cell surface expression of transforming growth factor-beta receptors are regulated by interaction with matrix collagen in murine osteoblastic cells. J Biol Chem. 1996;271(7):3938–44. doi: 10.1074/jbc.271.7.3938. [DOI] [PubMed] [Google Scholar]
- 41.Dallas SL, et al. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem. 2005;280(19):18871–80. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- 42.Fontana L, et al. Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J. 2005;19(13):1798–808. doi: 10.1096/fj.05-4134com. [DOI] [PubMed] [Google Scholar]
- 43.Kawelke N, et al. Fibronectin protects from excessive liver fibrosis by modulating the availability of and responsiveness of stellate cells to active TGF-beta. PLoS One. 2011;6(11):e28181. doi: 10.1371/journal.pone.0028181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janssens K, et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26(3):273–5. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 45.Jee W, Frost H. Skeletal adaptations during growth. Triangle; the Sandoz journal of medical science. 1992;31(2/3):77–88. [PubMed] [Google Scholar]
- 46.Lamande S, Bateman J. Procollagen folding and assembly: the role of endoplasmic reticulum enzymes and molecular chaperones. Seminars in cell & developmental biology. 1999;10(5):455–464. doi: 10.1006/scdb.1999.0317. [DOI] [PubMed] [Google Scholar]
- 47.Boot-Handford R, Briggs M. The unfolded protein response and its relevance to connective tissue diseases. Cell and tissue research. 2010;339(1):197–211. doi: 10.1007/s00441-009-0877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hung JH, et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. The Journal of biological chemistry. 2004;279(45):46384–46392. doi: 10.1074/jbc.M403568200. [DOI] [PubMed] [Google Scholar]
- 49.Ueno K, et al. The effects of prostaglandin E2 in rapidly growing rats: depressed longitudinal and radial growth and increased metaphyseal hard tissue mass. Bone. 1985;6(2):79–86. doi: 10.1016/8756-3282(85)90311-4. [DOI] [PubMed] [Google Scholar]
- 50.Jee W, Ma Y. The in vivo anabolic actions of prostaglandins in bone. Bone. 1997;21(4):297–304. doi: 10.1016/s8756-3282(97)00147-6. [DOI] [PubMed] [Google Scholar]
- 51.Suponitzky I, Weinreb M. Differential effects of systemic prostaglandin E2 on bone mass in rat long bones and calvariae. The Journal of endocrinology. 1998;156(1):51–57. doi: 10.1677/joe.0.1560051. [DOI] [PubMed] [Google Scholar]
- 52.Sweeney SM, et al. Candidate Cell and Matrix Interaction Domains on the Collagen Fibril, the Predominant Protein of Vertebrates. Journal of Biological Chemistry. 2008:283. doi: 10.1074/jbc.M709319200. [DOI] [PMC free article] [PubMed] [Google Scholar]