

Abstract
Despite available antivirals and vaccines, influenza infections continue to be a major cause of mortality worldwide. Vaccination generally induces an effective, but strain-specific antibody response. As the virus continually evolves, new vaccines have to be administered almost annually when a novel strain becomes dominant. Furthermore, the sporadic emerging resistance to neuraminidase inhibitors among circulating strains suggests an urgent need for new therapeutic agents. Recently, several cross-reactive antibodies have been described, which neutralize an unprecedented spectrum of influenza viruses. These broadly neutralizing antibodies generally target conserved functional regions on the major influenza surface glycoprotein hemagglutinin (HA). The characterization of their neutralization breadth and epitopes on HA could stimulate the development of new antibody-based antivirals and broader influenza vaccines.
Infections with influenza virus have a major impact on human health and economy. The annual epidemics result in a substantial number of hospitalizations with an estimated 3 to 5 million cases of severe disease, and 300,000 to 500,000 deaths globally. Furthermore, during the 20th century, three major influenza pandemics have occurred with a total mortality of 50 –100 million people (Lambert and Fauci, 2010). Influenza types A and B are enveloped RNA viruses and belong to the Orthomyxoviridae family and can lead to respiratory or gastro-intestinal tract infections in mammalian or avian species. Both types are responsible for recurrent annual influenza epidemics, but only influenza A has so far lead to pandemics. Influenza A viruses circulates in a variety of animals including birds, humans, horses, pigs and sea mammals, while influenza B is restricted to humans and seals (Osterhaus et al., 2000; Webster et al., 1992). Influenza A and B viruses contain two surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), that are embedded in the viral membrane envelope. HA mediates binding to sialic acid receptors on host cells and subsequent fusion between the virus and host membranes, while NA is responsible for virus progeny release. There are 17 different subtypes of influenza A HA (H1–H17), which are divided into two markedly distinct antigenically phylogenetic groups, group 1 (H1, H2, H5, H6, H8, H9, H11–H13, H16 and H17) and group 2 (H3, H4, H7, H10, H14 and H15). Most subtypes are present in the avian host, but only H1, H2 and H3 are or have been resident in the human population. Influenza B is classified in two distinct phylogenetic lineages, the Yamagata and Victoria lineages (Yamashita et al., 1988). HA is synthesized as a single polypeptide and folds into a trimeric spike (HA0) that is cleaved by host proteases into HA1 and HA2 subunits. Each trimer comprises a membrane distal globular head composed of HA1, which contains the receptor-binding site, and a stem region, which houses the fusion machinery (Wilson et al., 1981) (Fig. 1). The receptor-binding site is located in a small depression on the head of the HA and mediates virus binding to host cell sialic-acid receptors. The stem region is primarily composed of HA2 and some HA1 residues and is mostly helical. Like the surface spikes of many other viruses, HA is highly glycosylated (Wiley et al., 1981; Wilson et al., 1981). Although some glycans may be required for correct protein folding (Roberts et al., 1993), most are used as a mean for the virus to circumvent the immune response. The glycans are synthesized by host enzymes and are observed by the immune system as “self-structures” and do not normally induce an adaptive immune response. Moreover, glycans can directly shield vulnerable epitopes on HA and thereby prevent immune recognition.
Fig. 1.
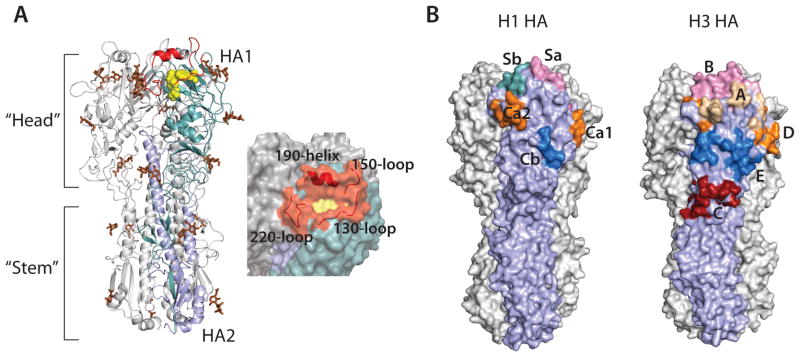
Crystal structure of HA. (A) Structure of the trimeric HA spike (PDB code; 4FNK) (Ekiert et al., 2012). One protomer is colored in cyan (HA1) and light blue (HA2). The receptor binding site is colored in yellow and the surrounding loops and helix in red. Glycans are colored brown (left). Surface representation of the receptor binding site and its surroundings (right). (B) The antigenic sites on HA. Antigenic sites Sa (pink), Sb (cyan), Ca1 and Ca2 (orange), and Cb (blue) on H1 HAs (left) (PDB code; 3LZG) (Xu et al., 2010). Antigenic sites A (wheat), B (pink), D (orange), E (blue) and C (red) on H3 HAs (right) (PDB code; 4FNK) (Ekiert et al., 2012).
Vaccination provides the best method for prevention and control of influenza and normally elicits a potent neutralizing antibody response. Most vaccines are trivalent and contain representative HAs from two influenza A strains and one influenza B strain. However, FDA recently approved quadrivalent influenza vaccines containing two influenza A strains and two influenza B strains. Current licensed vaccines include trivalent inactivated vaccines, live-attenuated vaccines and subunit vaccines. The trivalent inactivated vaccines contain killed influenza viruses and induce a protective serum antibody response, but a poor cell-mediated response, while the live attenuated vaccine contains weakened viruses and induce both a humoral and cellular immune response. These vaccines are grown in chicken eggs, which is relatively time consuming. The subunit vaccine contains purified baculovirus-expressed HA0 protein and, thus, circumvents the lengthy process of egg adaption of influenza virus (He et al., 2006). Most antibodies, which are generated upon vaccination or infection, are targeted towards the highly variable head of HA and are often strain specific. As a consequence, new formulations of the vaccine are generated almost annually when a new strain starts to dominate (Fiore et al., 2010). The strains to include in the upcoming vaccine are predicted by the WHO and the efficiency of the vaccine thus depends on the match between the vaccine strains and the circulating strains. Influenza undergoes continual antigenic drift where mutations are accumulated in HA due to an error prone influenza RNA polymerase, and the selective pressure from the host immune system. Additionally, co-infection of a single host with more than one virus strain can result in an antigenic shift, where re-assortment of genes from different viruses generates novel subtypes (Carrat and Flahault, 2007). As a result, cross-species transmission of newly adapted strains might occur, as in the case of the highly pathogenic avian H5N1 (Claas et al., 1998). If minor or no immunity exists in the bulk of the human population, as with the 1918 Spanish flu, new pandemics can arise. Thus, the emergence of new influenza strains and lack of herd immunity in the population remains a persistent threat to human health.
Given the sporadic occurrence of oseltamivir-resistant viruses, the characterization of zanamivir-resistant viruses and the lack of heterovariant vaccines, alternative treatment strategies for influenza are urgently needed (de Jong et al., 2005; Hurt et al., 2009; Monto et al., 2006). Immunotherapy with monoclonal antibodies represents a complementary strategy to current antivirals. The use of monoclonal antibodies for treatment of medical conditions, including viral diseases like hepatitis and respiratory syncytial virus, is well established (Sawyer, 2000).
Monoclonal antibody therapy could be employed alone for the treatment of influenza infections with strains that are resistant to current antivirals or in combination with antivirals in the case of terminal patients with severe influenza infections. Prophylactic administration of antibodies could be valuable in the case of a pandemic with a highly pathogenic virus like H5N1 especially for persons which are particular susceptible to illness like elderly and immunocompromised individuals, and people with a higher risk of getting infections as health-related and hospital workers.
Recently, exciting new influenza monoclonal antibodies have been identified that are capable of neutralizing a wide range of influenza viruses (Corti et al., 2011; Dreyfus et al., 2012; Ekiert et al., 2009; Ekiert et al., 2011; Ekiert et al., 2012; Krause et al., 2012; Lee et al., 2012; Sui et al., 2009; Tan et al., 2012; Throsby et al., 2008; Tsibane et al., 2012; Wang et al., 2010b; Yoshida et al., 2009). These broadly neutralizing antibodies show an unprecedented breadth of cross reactivity enabling them to neutralize many different strains within a subtype, group or even between groups and different types of influenza virus. The therapeutic and prophylactic efficiency of theses antibodies have been characterized in both mouse and ferret models and shows promising results. Furthermore, the molecular basis of influenza virus recognition has been elucidated for many of these antibodies through biochemical and structural studies and will be discussed in this review.
Antibody recognition of the HA head
The globular membrane distal head of HA is highly immunogenic and is the main target of antibodies generated during vaccination. However, the variability of HA leads to annual influenza epidemics. Five distinct antigenic sites have been characterized for the H1 subtype in the head and are designated Sa, Sb, Ca1, Ca2 and Cb, while those in the H3 subtype are called A through E (Caton et al., 1982; Wiley et al., 1981) (Fig. 1B). These sites are hypervariable and are the prime locations for escape from the host immune detection. In contrast, the receptor-binding site is relatively conserved as it is functionally constrained. The receptor-binding site is located in a small, shallow pocket at the top of the HA and is bordered by the 130-loop, the 150-loop, the 190-helix and the 220-loop. This site represents a possible target for broad-spectrum antibodies. However, the typical footprint of an antibody is rather large and would inevitably contact the less conserved areas outside the receptor-binding site. Notwithstanding, broadly neutralizing antibodies, which target the receptor-binding site and its surroundings, have recently been identified (Ekiert et al., 2012; Lee et al., 2012; Tsibane et al., 2012; Whittle et al., 2011). These antibodies all insert a CDR loop into the receptor-binding site to contact conserved residues.
Broad H1 neutralization by antibody CH65
In 2011, Whittle et al. (2011) described the identification and characterization of the human monoclonal antibody CH65, which in a microneutralization assay neutralizes a broad spectrum of H1 viruses spanning more than three decades. The antibody was isolated from a recipient of the 2007 trivalent vaccine by screening single sorted plasma cells. CH65 binds to the head of HA with an epitope covering the receptor binding site and antigenic sites Sa, Sb and Ca2 (Fig. 2A) and buries a total surface area of 748 Å2 on HA, using five out of six possible CDR loops. The most substantial contacts are made by a 19-residue HCDR3, which inserts into the receptor-binding site, and displays receptor mimicry by making similar interactions with many of the conserved residues involved in sialic-acid binding. However, the variability of the H1 strains in and around the receptor pocket dictate binding selectivity of CH65. Most often, insertion of a basic amino acid at position 133a (between residues 133 and 134), which is present in ~ 75 % H1, ~ 94 % H5, all H6 and all H10 viruses, will clash with CH65 and result in escape from neutralization (Whittle et al., 2011).
Fig. 2.

Structure of HA in complex with head binding antibodies. (A) Structure of Fab CH65 in complex with H1 HA (PDB code; 3SM5) (Whittle et al., 2011). HA is shown as surface representation with one protomer colored light blue. Residues in the epitope that are part of the receptor binding site colored in red. Residues in the epitope adjacent to the receptor-binding site are colored in blue. The Fab is shown in cartoon with the light chain (LC) in yellow and the heavy chain (HC) in green (top). CDR loops from the heavy chain (green) and the light chain (yellow) involved in the interaction with HA are shown. (B) Structure of Fab S139/1 in complex with H3 HA (PDB code; 4GMS) (Lee et al., 2012). Colored as in Fig. 2A. (C) Structure of Fab C05 in complex with H3 HA (PDB code 4FQR) (Ekiert et al., 2012).
Heterosubtypic neutralization by avidity
The mouse monoclonal antibody S139/1 was initially described in 2009 and followed by structural characterization in complex with HA in 2012 (Lee et al., 2012; Yoshida et al., 2009). The antibody shows hemaglutination inhibition (HI) against particular viruses from H1, H2, H3, and H13 subtypes with high HI titers to one H1 strain and several H3 strains, and moderate activity against H2 and H13 strains tested. Passive immunization of mice by intraperitoneal injection of S139/1 IgG (200 μg) one day before or after challenge with 10 times the 50% mouse infectious dose of H1N1 and H3N2 viruses significantly reduces viral lung titers. Neutralization escape mutants of H1, H2 and H3 were selected in the presence of S139/1, which acquired mutations in antigenic sites B (H3) and Sb (H1).
S139/1 binds the HA head using both its heavy and light chain and five of its six complementarity determining region (CDR) loops (Fig. 2B) (Lee et al., 2012). The epitope maps onto highly conserved residues in the receptor-binding site and contacts antigenic sites A, B and D (H3 designation). Like CH65, S139/1 competes directly with the receptor by inserting a CDR loop (in this case HDCR2) into the receptor pocket. Additional interactions include contacts to the antigenic site B made by LCDR1 and LCDR3 that binds to the 150-loop as well as HCDR3, which interacts with conserved residues in the 190-helix. Several changes in the epitope surrounding the receptor-binding site lead to resistance of neutralization by S139/1. As for CH65, an insertion at 133a abolishes binding due to steric hindrance. Furthermore, an insertion in the 150-loop observed in almost all H4, H6–H8, H10, H14 and H15 subtypes most likely contributes to the lack of neutralization of these viruses by S139/1.
Interestingly, although the S139/1 Fab fragment shows a wide range of binding affinities for different subtypes (2 μM -10 nM), all of these subtypes (H1, H2, H3, H13 and H16) are neutralized by S139/1 IgG in a plaque reduction assay. Thus, for all strains tested, low affinity binding with monovalent Fab is effectively converted into high avidity through IgG multivalency. The orientation of S139/1 in respect to HA likely prevents protomer cross-linking within a single HA trimer by IgG, suggesting that the IgG binding cross-links adjacent trimers on the virus surface. Thus, the bivalent nature of IgG broadens the neutralization potential significantly by making S139/1 more permissive towards minor changes in the epitope that would otherwise reduce monovalent Fab binding (Lee et al., 2012).
Neutralization by a single antibody loop
Recently, a monoclonal antibody capable of binding HA using a single CDR loop was characterized. Antibody C05 neutralizes H1–H3 and H9 viruses in vitro as shown by HI or microneutralization using MDCK cells. Prophylactic peritoneal injection of 1 mg/kg and 10 mg/kg C05 prior to lethal challenge with H1N1 and H3N2 viruses, respectively, protected 100 % of mice from death. Administration of 15 mg/kg IgG at 1, 2 or 3 days after lethal challenge of mice with H1N1 or H3N2 viruses provided full protection (Ekiert et al., 2012). C05 was isolated using a vary large phage display library derived from the bone marrow of a person that was identified from an advertisement on Craigslist placed by Sea Lane for an individual previously infected with certain strains of influenza virus. The antibody contains a 24-residue HCDR3 and a five-residue somatic insertion in LCDR1. The crystal structure of C05 in complex with HA revealed that the antibody binds to HA primarily with its HCDR3, which is inserted into the receptor-binding site (Fig. 2C). Only minor additional interactions are made from HCDR1 to the 190-loop in antigenic site B. As a result, the buried surface area on HA (550 Å2) upon complex formation is substantially smaller that the footprint of a typical antibody to a protein. C05 inhibits receptor binding by steric hindrance, but does not directly mimic the receptor as in the case of CH65. Similar to S139/1, C05 employs avidity by cross-linking HA spikes on the surface of influenza virus. Common insertions in the loops surrounding the receptor-binding site affect the C05 interaction. Like CH65 and S139/1, the 133a insertion abolishes C05 binding due to steric clashes. Likewise, insertions around residue 158 and in the 220-loop explain why C05 does not neutralize H4, H6, H7, H10, H14 and H16 viruses (Ekiert et al., 2012).
The studies on S139/1 and C05 show that heterosubtypic neutralizing antibodies that primarily target the receptor-binding site can be generated. The relative dense clustering of HA spikes on the virus surface, unlike HIV, can facilitate multivalent binding by IgG (Harris et al., 2006; Zhu et al., 2006). The use of avidity allows S139/1 and C05 to be more permissive to minor changes in the epitope, gaining the ability to bind highly divergent HA strains. Although the receptor-binding pocket contains residues that are universally conserved among all influenza A strains sequenced to date, influenza seems to have adapted especially well to protect this vulnerable site. Thus, subtype differences in the loops and helices surrounding the receptor-binding site generally makes the head binding antibodies more restricted in specificity than the stem-targeted antibodies.
Stem reactive broadly neutralizing antibodies
Neutralizing influenza antibodies were initially believed only to be generated against the immunodominant head of HA. However, in 1993, the first indication emerged that neutralizing antibodies could also target the highly conserved stem. The mouse antibody C179 (Okuno et al., 1993) was shown to neutralize viruses from the H1 and H2 subtypes. C179 does not inhibit hemagglutination and was, therefore, suggested to target the helical stem region of HA. Recently, several stem-reactive antibodies have been identified, which have reinvigorated the influenza field (Corti et al., 2011; Dreyfus et al., 2012; Ekiert et al., 2009; Ekiert et al., 2011; Kashyap et al., 2010; Sui et al., 2009; Tan et al., 2012; Wang et al., 2010b). In contrast to head binding antibodies, the stem-directed antibodies appear to neutralize influenza virus by preventing fusion of the host and virus membranes in a process that is trigged in the low-pH environment of late endosomes (Fig. 3A and 3B). In each protomer, the B-loop, which connects helix A and C in the pre-fusion structure, now adopts a helical conformation at low pH and brings helix A to the top of an extended α-helix that forms a helical coil-coil in the trimer. The movement of helix A relocates and exposes the fusion peptide towards the target membrane. Additionally, the lower part of helix C refolds into a loop allowing helix D to turn toward helix C (Bullough et al., 1994).
Fig. 3.


Structures of HA in the pre-fusion and post-fusion conformation, and HA in complex with stem reactive Fab fragments. (A) Pre-fusion conformation of HA with helices A (red), C (yellow), and D (blue) and B loop (orange) in one protomer colored distinctly (PDB code; 4FNK) (Ekiert et al., 2012). (B) Post-fusion conformation of HA (PDB code; 1HTM) (Bullough et al., 1994). Colored as in (A). Note that most of HA1 is not present in the structure and the B loop folds into helix B. (C) Structure of Fab CR6261 in complex with H5 HA (PDB code; 3GBM) (Ekiert et al., 2009). HA is colored as in (A) shown as surface representation and in the same orientation (left). The epitope recognized by CR6261 is shown on the right. CDR loops from the heavy chain (green) and the light chain (yellow) involved in the interaction with HA are shown. The position of the glycan at Asn38 in group 2 viruses is indicated by #. Trp21 is shown by *. The side chain of the Phe residue interacting with Trp21 is shown (D) Structure of Fab CR8020 in complex with H3 HA (PDB code; 3SDY) (Ekiert et al., 2011). The glycan at Asn21 in most group 2 viruses is shown by # and Tyr34 as *, both in red. (E) Structure of Fab CR9114 in complex with H5 HA (PDB code; 4FQI) (Dreyfus et al., 2012). Colored as in (A). (F) Structure of Fab FI6v3 in complex with H3 HA (PDB code; 3ZTJ) (Corti et al., 2011). The glycan at Asn38 is shown in brown.
Most stem-directed antibodies appear to use a common mechanism of neutralization by binding the highly conserved HA stem region that is essential for fusion. These antibodies can lock HA in a pre-fusion conformation and, therefore, prevent the major conformational changes associated with pH-activation of the fusion machinery, which is indispensible for the viral entry into the cell.
Group 1 neutralizing antibodies
In 2009, two broadly neutralizing antibodies, CR6261 and F10, against group 1 viruses were independently functionally and structurally characterized (Ekiert et al., 2009; Sui et al., 2009). CR6261 and F10 are both derived from the VH1-69 germline and were identified by panning immobilized HA using phage display libraries generated from recently vaccinated donors. They share a very similar breadth of neutralization, bind to most group 1 viruses, and protect against H5N1 and H1N1 viruses. F10 neutralizes H5N1, H1N1, H2N2, H6N2, H8N4 and H9N2 viruses in a microneutralization assay. Intraperitoneal injection of 10 mg/kg F10 IgG one hour before lethal challenge with H1N1 or H5N1 viruses protected 80–100% of mice from death. Therapeutic administration of 15 mg/kg of F10 IgG 24 hours following lethal challenge with H5 viruses protected 80–100% of mice. Viral replication in the lungs was substantially suppressed when F10 was given 48 or 72 hours following inoculation with virus. No neutralization escape variants were generated in the presence of F10 following 3 passages (Sui et al., 2009).
CR6261 showed heterosuptypic neutralization activity against H1, H2, H5, H6, H8 and H9 viruses in a microneutralization assay (MDCK cells). Prophylaxis by intraperitoneal injection of CR6261 (5 mg/kg) protected all mice from lethal challenge with H5N1 virus. Administration of 2mg/kg CR6261 fully protected mice from lethal challenge with H1N1 virus. Therapeutic injection of CR6261 (15 mg/kg) to mice one day following infection with lethal doses of H1N1 or H5N1 viruses provided full protection. In vitro neutralization escape variants of H5N1 were were difficult to generate in the presence of CR6261; only after 10 passages were viruses selected with an H111L mutation (Throsby et al., 2008).
Both antibodies recognize a similar highly conserved epitope in the stem region of HA. Binding by CR6261 is mediated by the heavy chain using HCDR1-3 and framework region 3 (Fig. 3C). The epitope mainly consists of helix A in HA2 and a few residues in HA1. The primary interactions are made by HCDR1, which make important contacts to helix A, and HCDR2, which interact with a hydrophobic pocket in HA1 next to helix A. The base of HCDR3 makes a few contacts to the lower part of helix A, while framework region 3 makes minor interactions with the upper region of helix A. Both antibodies do not cross-react with group 2 H3, H7 and H10 primarily due to presence of a highly conserved glycan on Asn38, which is part of the epitope contacted by the heavy chain, as well as some other group specific residues. Additionally, HCDR2 interacts with Trp21 in HA1, which has a different conformation in group 2 viruses that cannot be accommodated by the HCDR2 loop (Ekiert et al., 2009).
Group 2 neutralizing antibodies
Following the characterization of CR6261 and F10, a complementary monoclonal antibody CR8020 was identified that targets group 2 influenza viruses (Ekiert et al., 2011). CR8020 has neutralizing activity against H3, H7 and H10 viruses in a microneutralization assay and binds H3, H4, H7, H10, H14 and H15 HAs. Administration of CR8020 (3 mg/kg) one day before lethal challenge with H3N2 or H7N7 viruses protected all mice from death. No signs of respiratory distress were observed and all mice showed an increased body weight following the study. Therapeutic injection of CR8020 (15 mg/kg) 2 days after infection with H3N2 virus prevented death in all animals, whereas treatment 3 days after infection with H7N7 virus prevented mortality in all animals. Like CR6261 and F10, CR8020 interacts with the stem of HA, but binds at the base of the stem substantially closer to the viral membrane than CR6261 and uses both heavy and light chains (Fig. 3D). The epitope is highly conserved across group 2 viruses and includes residues in the C-terminal region of the fusion peptide, which is recognized by HCDR1 and HCDR3. By directly contacting residues in the fusion peptide, CR8020 prevents its release in the endosome and inhibits the conformational changes in HA that are necessary for fusion. Additionally, CR8020 binds to HA0 and inhibits maturation to HA1 and HA2, sterically preventing cleavage by host proteases (Ekiert et al., 2011). Two neutralization escape mutants (D19N or G33E) located in the stem region were selected for an H3N2 virus in the presence of CR8020, but only after 4 passages.
Only two residues are conserved between the epitopes of CR6261 and CR8020, and CR8020 does not neutralize group 1 viruses. More specifically, a conserved glycan at Asn21 in group 1 viruses prevents binding as a result of steric clash with HCDR1. Likewise, a Tyr residue at position 34 conserved in most group 1 viruses clashes with HCDR3.
Universal influenza A antibodies
For antibody-based influenza treatments, a potent and broad-spectrum antibody capable of neutralizing all influenza A strains represent the ultimate goal. Remarkably, two antibodies FI6v3 and CR9114 neutralize all group 1 and group 2 viruses tested (Corti et al., 2011; Dreyfus et al., 2012).
In vitro CR9114 binds HAs from H1–H5, H7 and H9, H10, H12, H13, H15 and H16 viruses and neutralizes H1–H12 and H14 viruses in a microneutralization assay. Administration of 1.7 mg/kg and 5 mg/kg CR9114 one day prior to challenge with lethal doses of H1N1 and H3N2 viruses, respectively, protected all animals from mortality and resulted in a significant reduction in weight loss compared to control animals (Dreyfus et al., 2012). CR9114 is VH1-69 derived, and uses HCDR1-3 and framework 3 for binding. Its epitope is highly similar to the epitopes of CR6261 and F10 (Fig. 3E) (Dreyfus et al., 2012). However, minor important differences are present in the way that HA is recognized, allowing CR9114 to bind both group 1 and group 2 viruses. In CR6261, the tip of HCDR2 (Phe54) interacts with Trp21 in HA2, which is orientated slightly differently in group 1 and 2 viruses and thereby prevents cross-group reactivity. In contrast, the flexibility of HCDR2 in CR9114 and the different orientation of Phe54 allows it to accommodate the minor differences in the conformation of Trp21. Furthermore, the glycan at HA1 Asn38 in group 2 HAs, which interfere with CR6261 and F10 from binding group 2 viruses, is physically displaced by CR9114 (Dreyfus et al., 2012).
FI6v3 binds H1–H10 HAs in ELISA and stains cells tranfected with genes of H4, and H11–H16 HAs. In addition, FI6v3 neutralizes H1, H3, H5 and H7 pseudoviruses and viruses. Prophylatic administration of FI6v2 (4 mg/kg) protected all mice from lethal challenge with H1N1 and therapeutic administration of FI6v3 (15 mg/mg) 1 or 2 days after lethal infection with H1N1 was fully protective. FI6v3 also significantly reduced mice lung virus titers when administered up to 1 day after infection with a lethal dose of H1N1 and reduced weight loss in mice challenged with an H3 virus. Like CR9114, FI6v3 recognizes an epitope, which is similar to the epitopes of CR6261 and F10 (Corti et al., 2011). However, the angle of approach in respect to the HA trimer and the nature of the interaction is substantially different (Fig. 3F). FI6v3 is rotated approximately 90° compared to CR9114 and uses both heavy and light chain in binding to HA. FI6v3 is VH3-30 encoded and has a long HCDR3 (22 residues) that alone mediates the binding to the hydrophobic groove between HA1 and helix A. A Phe residue at the tip of the flexible HCDR3 loop interacts with Trp21 in both group 1 and 2 viruses. Like CR9114, FI6v3 is able to displace the conserved glycan at Asn38 to avoiding steric clash with group 2 HAs. LCDR1 makes contacts with helix A at the opposite end of the hydrophobic groove, but also interacts with the fusion peptide in the neighboring HA monomer. Thus, crosslinking of monomers in the HA trimer may additionally contribute to inhibition of membrane fusion (Corti et al., 2011). Remarkably, the CR9114 epitope is conserved between influenza A and B. Electron microscopy reconstructions show that CR9114 similarly binds and recognizes the stem of influenza B HA. Moreover, prophylaxis with 5 mg/kg and 15 mg/kg of CR9114 protects against lethal challenge in mice with influenza B viruses B/Malaysia/2506/2004 and B/Florida/4/2006, respectively (Dreyfus et al., 2012). In this regard, CR9114 represents the only universal influenza antibody described to date.
Broadly neutralizing influenza B antibodies
The number of people infected with influenza B virus varies dramatically between each influenza season and can in some years contribute substantially to the annual epidemic. Although influenza B is restricted to humans and seals and has not lead to a pandemic, it is estimated that influenza B accounted for ~ 15 % of all influenza related deaths in the US between 1990 and 1998 (Ambrose and Levin, 2012; Thompson et al., 2004). Influenza B is not divided into subtypes and groups, but into two distinct phylogenetic lineages: the Yamagata lineage and the Victoria linage. The two lineages have co-circulated globally since 1985 where the Victoria lineage emerged (Rota et al., 1990; Yamashita et al., 1988). Both contribute to the annual epidemics with one lineage dominating each season. Influenza B undergoes antigenic variation due to antigenic drift. However, the mutation rate of influenza B HA is approximately 4 times less than that of influenza A HA (Nobusawa and Sato, 2006).
Recently, two influenza B antibodies, CR8033 and CR8071 were identified that neutralize viruses from both influenza B lineages (Dreyfus et al., 2012). Prophylaxis with 0.6 mg/kg and 0.2 mg/kg of CR8033 protected all mice from death upon lethal challenge with B/Florida/4/2006 (Yamagata) and B/Malaysia/2506/2004 (Victoria), respectively. Administration of 0.6 mg/kg and 1.7 mg/kg of CR8071 prior to lethal challenge with B/Florida/4/2006 and B/Malaysia/2506/2004 fully protected from mortality. For B/Florida/4/2006, no neutralization escape variants were generated with CR8033 after 20 passages but a P161Q escape variant was generated with B/Malaysia/2506/2004 virus following 15 passages. Using CR8071 and B/Florida/4/2006, 15 passages generated K38E and Y40H escape mutations while no escape variants were observed after 20 passages of B/Malaysia/2506/2004 virus (Dreyfus et al., 2012).
Structural analysis by x-ray crystallography and electron microscopy shows that CR8033 and CR8071 recognize two distinct conserved epitopes on influenza B HA. CR8033 binds to the top of HA, while CR8071 recognizes the vestigial esterase domain in the base of the head. In contrast to the influenza A antibodies described, CR8071 and CR8033 appear to uniquely neutralize virus by preventing virus progeny release (Dreyfus et al., 2012). Virus progeny bud from infected cells, which requires receptor cleavage by NA on the cell surface. Additionally, the newly synthesized HA and NA may contain sialic acid residues that need to be cleaved to facilitate virus spread. The neuraminidase inhibitor zanamivir prevents progeny virus budding by inhibiting receptor cleavage that leads to aggregation of infected cells (Gubareva et al., 2000). Similarly, when cells infected with influenza B are incubated with CR8033 or CR8071, cell clumping occur, which may be due to HA trimer cross-linking during virus budding. In this regard, these antibodies neutralize influenza B viruses by a mechanism different from the previously described head or stem binding influenza A antibodies. Whether neutralization of influenza A by egress inhibition represents an important but previously unrecognized mechanism of neutralization has still to be determined.
Perspectives and concluding remarks
Drug resistance between circulating H1N1 and H3N2 strains indicates an urgent need for new antivirals agents (Bright et al., 2005; Kiso et al., 2004). The recent identification of broadly neutralizing human antibodies has sparked new interest in the use of antibody therapy in treatment of influenza. Their broad and heterosubtypic reactivity is clearly beneficial and suggests that these antibodies could be used as novel antivirals against current and future circulating viruses. Additionally, the information gained from the structural characterizations may be used as guides for rational design of therapeutic molecules. Indeed, the structure of CR6261 in complex with HA stimulated the generation of computationally designed scaffolds that binds to a very similar epitope as CR6261 (Fleishman et al., 2011; Whitehead et al., 2012). These scaffolds interact with a range of group 1 HAs and inhibit the conformational change in HA associated with membrane fusion. Likewise, the use of a single extended loop by C05 to target the receptor-binding site and the resent description of three antibodies, that all targets a conserved pocket in the receptor binding site using an aromatic residue, indicates that it may be possible to design small molecules or peptides, which mimic the antibody interactions and thereby compete with receptor binding (Ekiert et al., 2012; Xu et al., 2013).
Most importantly, the aforementioned discoveries may lead the way for rational structure-based vaccine design. Although effective strain-specific vaccines exist against influenza infection, new dominant strains continually arise such that new vaccines have to be produced regularly. Furthermore, a long period of time is required for vaccine manufacturing which minimizes the possibility for efficiently controlling a potential pandemic by vaccination, as herd immunity cannot be generated quickly enough. The most broadly neutralizing antibody identified so far CR9114 that neutralizes influenza A and B uses essentially the same conserved epitope as the influenza A group 1 specific CR6261. However, minor differences in recognition means that CR9114 has acquired the ability to also cross-react with group 2 influenza A viruses. For vaccine design and elicitation of broadly neutralizing antibodies, it is interesting that CR9114, CR6261 and F10 all use the VH1-69 germline segment. Each antibody is relatively conventional with a normal degree of affinity maturation, which indicates that it might be possible to induce CR6261- or even CR9114-like antibodies using the appropriate strategy. Indeed, broadly neutralizing stem-directed antibodies have recently been generated by priming with a DNA vaccine followed by boosting with a seasonal vaccine (Wei et al., 2010; Wei et al., 2012). The VH1-69 gene codes for hydrophobic residues on the tip of HCDR2 (Ile53 and Phe54), that in CR6261 and CR9114 are directed into a hydrophobic pocket next to helix A on HA. Moreover, the germline of CR6261 can bind HA when the antibody is expressed as membrane-bound IgM and triggers B-cell receptor-associated tyrosine kinase signaling. For HA binding, Ile53 and Phe54 on HCDR2 are indispensible and may provide the essential binding energy in the initial encounter with antigen, which then stimulates subsequent maturation to a higher affinity interaction (Lingwood et al., 2012). Thus, by using an immunogen designed to specifically bind the CR6261 germline, one might be able to focus the B-cell response towards CR6261-like antibodies. To date, a few “headless” HA immunogens have been designed to direct the antibody response towards the more conserved areas in the stem. However, for reasons not fully understood, these immunogens have not yet generated a very cross-reactive neutralizing antibody response (Bommakanti et al., 2010; Sagawa et al., 1996; Steel et al., 2010; Wang et al., 2010a). Several fundamental questions vital for vaccine design still remain unclear and need to be investigated, including how to induce a focused, broad and long lasting immune response. More research is required to evaluate how to present specific antigens, how to administer the antigens, and how to predict human immune responses based on animal studies before a universal influenza vaccine can be formulated. However, the recent identification of several potent, broadly neutralizing antibodies to influenza virus has shown that, although the virus is especially well adapted to evade the host immune system, conserved vulnerable sites on the virus can be targeted by antibodies. Collectively, this knowledge should be of use in the development of future antivirals and in structure-based vaccine design.
Acknowledgments
We thank P.S. Lee and R. L. Stanfield for analysis and helpful discussions. Influenza work in the Wilson lab has been supported in part by National Institutes of Health (NIH) grant P01AI058113 (I.A.W.), the National Institute of Allergy and Infectious Diseases, NIH, Department of Health and Human Services, USA, under contract HHSN272200900060C (to Crucell) and the Skaggs Institute, TSRI. N.S.L. has a Saper Aude Postdoc fellowship from the Danish Council for Independent Research, Natural Sciences. This is manuscript #23061 from The Scripps Research Institute.
Footnotes
The authors have no financial or personal relationships that could be viewed as a potential conflict of interest.
References
- Ambrose CS, Levin MJ. The rationale for quadrivalent influenza vaccines. Hum Vaccin Immunother. 2012;8:81–88. doi: 10.4161/hv.8.1.17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommakanti G, Citron MP, Hepler RW, Callahan C, Heidecker GJ, Najar TA, Lu X, Joyce JG, Shiver JW, Casimiro DR, ter Meulen J, Liang X, Varadarajan R. Design of an HA2-based Escherichia coli expressed influenza immunogen that protects mice from pathogenic challenge. Proc Natl Acad Sci U S A. 2010;107:13701–13706. doi: 10.1073/pnas.1007465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Carrat F, Flahault A. Influenza vaccine: the challenge of antigenic drift. Vaccine. 2007;25:6852–6862. doi: 10.1016/j.vaccine.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype) Cell. 1982;31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, Krauss S, Shortridge KF, Webster RG. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet. 1998;351:472–477. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- de Jong MD, Tran TT, Truong HK, Vo MH, Smith GJ, Nguyen VC, Bach VC, Phan TQ, Do QH, Guan Y, Peiris JS, Tran TH, Farrar J. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, van der Vlugt R, Lamrani M, Korse HJ, Geelen E, Sahin O, Sieuwerts M, Brakenhoff JP, Vogels R, Li OT, Poon LL, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RH. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science. 2011;333:843–850. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O’Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature. 2012;489:526–532. doi: 10.1038/nature11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA, Iskander JK, Wortley PM, Shay DK, Bresee JS, Cox NJ. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep. 2010;59:1–62. [PubMed] [Google Scholar]
- Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubareva LV, Kaiser L, Hayden FG. Influenza virus neuraminidase inhibitors. Lancet. 2000;355:827–835. doi: 10.1016/S0140-6736(99)11433-8. [DOI] [PubMed] [Google Scholar]
- Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XS, Holmes TH, Zhang C, Mahmood K, Kemble GW, Lewis DB, Dekker CL, Greenberg HB, Arvin AM. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J Virol. 2006;80:11756–11766. doi: 10.1128/JVI.01460-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt AC, Holien JK, Parker M, Kelso A, Barr IG. Zanamivir-resistant influenza viruses with a novel neuraminidase mutation. J Virol. 2009;83:10366–10373. doi: 10.1128/JVI.01200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap AK, Steel J, Rubrum A, Estelles A, Briante R, Ilyushina NA, Xu L, Swale RE, Faynboym AM, Foreman PK, Horowitz M, Horowitz L, Webby R, Palese P, Lerner RA, Bhatt RR. Protection from the 2009 H1N1 pandemic influenza by an antibody from combinatorial survivor-based libraries. PLoS Pathog. 2010;6:e1000990. doi: 10.1371/journal.ppat.1000990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, Kimura K, Hayden FG, Sugaya N, Kawaoka Y. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759–765. doi: 10.1016/S0140-6736(04)16934-1. [DOI] [PubMed] [Google Scholar]
- Krause JC, Tsibane T, Tumpey TM, Huffman CJ, Albrecht R, Blum DL, Ramos I, Fernandez-Sesma A, Edwards KM, Garcia-Sastre A, Basler CF, Crowe JE., Jr Human monoclonal antibodies to pandemic 1957 H2N2 and pandemic 1968 H3N2 influenza viruses. J Virol. 2012;86:6334–6340. doi: 10.1128/JVI.07158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med. 2010;363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- Lee PS, Yoshida R, Ekiert DC, Sakai N, Suzuki Y, Takada A, Wilson IA. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc Natl Acad Sci U S A. 2012;109:17040–17045. doi: 10.1073/pnas.1212371109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, McTamney PM, Yassine HM, Whittle JR, Guo X, Boyington JC, Wei CJ, Nabel GJ. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 2012;489:566–570. doi: 10.1038/nature11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto AS, McKimm-Breschkin JL, Macken C, Hampson AW, Hay A, Klimov A, Tashiro M, Webster RG, Aymard M, Hayden FG, Zambon M. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob Agents Chemother. 2006;50:2395–2402. doi: 10.1128/AAC.01339-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobusawa E, Sato K. Comparison of the mutation rates of human influenza A and B viruses. J Virol. 2006;80:3675–3678. doi: 10.1128/JVI.80.7.3675-3678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y, Isegawa Y, Sasao F, Ueda S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol. 1993;67:2552–2558. doi: 10.1128/jvi.67.5.2552-2558.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterhaus AD, Rimmelzwaan GF, Martina BE, Bestebroer TM, Fouchier RA. Influenza B virus in seals. Science. 2000;288:1051–1053. doi: 10.1126/science.288.5468.1051. [DOI] [PubMed] [Google Scholar]
- Roberts PC, Garten W, Klenk HD. Role of conserved glycosylation sites in maturation and transport of influenza A virus hemagglutinin. J Virol. 1993;67:3048–3060. doi: 10.1128/jvi.67.6.3048-3060.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology. 1990;175:59–68. doi: 10.1016/0042-6822(90)90186-u. [DOI] [PubMed] [Google Scholar]
- Sagawa H, Ohshima A, Kato I, Okuno Y, Isegawa Y. The immunological activity of a deletion mutant of influenza virus haemagglutinin lacking the globular region. J Gen Virol. 1996;77 (Pt 7):1483–1487. doi: 10.1099/0022-1317-77-7-1483. [DOI] [PubMed] [Google Scholar]
- Sawyer LA. Antibodies for the prevention and treatment of viral diseases. Antiviral Res. 2000;47:57–77. doi: 10.1016/s0166-3542(00)00111-x. [DOI] [PubMed] [Google Scholar]
- Steel J, Lowen AC, Wang TT, Yondola M, Gao Q, Haye K, Garcia-Sastre A, Palese P. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. MBio. 2010;1 doi: 10.1128/mBio.00018-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, Wan H, Murakami A, Yammanuru A, Han T, Cox NJ, Bankston LA, Donis RO, Liddington RC, Marasco WA. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P. A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J Virol. 2012;86:6179–6188. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. Influenza-associated hospitalizations in the United States. JAMA. 2004;292:1333–1340. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- Throsby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, Guan Y, Cinatl J, ter Meulen J, Lasters I, Carsetti R, Peiris M, de Kruif J, Goudsmit J. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsibane T, Ekiert DC, Krause JC, Martinez O, Crowe JE, Jr, Wilson IA, Basler CF. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog. 2012;8:e1003067. doi: 10.1371/journal.ppat.1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TT, Tan GS, Hai R, Pica N, Ngai L, Ekiert DC, Wilson IA, Garcia-Sastre A, Moran TM, Palese P. Vaccination with a synthetic peptide from the influenza virus hemagglutinin provides protection against distinct viral subtypes. Proc Natl Acad Sci U S A. 2010a;107:18979–18984. doi: 10.1073/pnas.1013387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TT, Tan GS, Hai R, Pica N, Petersen E, Moran TM, Palese P. Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog. 2010b;6:e1000796. doi: 10.1371/journal.ppat.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CJ, Boyington JC, McTamney PM, Kong WP, Pearce MB, Xu L, Andersen H, Rao S, Tumpey TM, Yang ZY, Nabel GJ. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science. 2010;329:1060–1064. doi: 10.1126/science.1192517. [DOI] [PubMed] [Google Scholar]
- Wei CJ, Yassine HM, McTamney PM, Gall JG, Whittle JR, Boyington JC, Nabel GJ. Elicitation of broadly neutralizing influenza antibodies in animals with previous influenza exposure. Sci Transl Med. 2012;4:147ra114. doi: 10.1126/scitranslmed.3004273. [DOI] [PubMed] [Google Scholar]
- Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, Baker D. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci U S A. 2011;108:14216–14221. doi: 10.1073/pnas.1111497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Xu R, Krause JC, McBride R, Paulson JC, Crowe JE, Jr, Wilson IA. A recurring motif for antibody recognition of the receptor-binding site of influenza hemagglutinin. Nat Struct Mol Biol. 2013;20:363–370. doi: 10.1038/nsmb.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Krystal M, Fitch WM, Palese P. Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology. 1988;163:112–122. doi: 10.1016/0042-6822(88)90238-3. [DOI] [PubMed] [Google Scholar]
- Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog. 2009;5:e1000350. doi: 10.1371/journal.ppat.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]