

Abstract
We report the discovery and crystal structure of a human mycoplasma protein, Protein M, which binds with high affinity to antibodies, predominantly through attachment to the variable region of the κ and λ light chains. Protein M broadly blocks antibody-antigen union and its mechanism of inhibition is of considerable interest because, as a diversity system, the binding mode of each antibody is different. Protein M thus appears to function by a mechanism that is independent of the sequences of members of the extensive antibody repertoire. By anchoring to conserved regions of the antibody light chains, Protein M is in a position to extend its large C-terminal domain over the antibody combining site and block entrance to macromolecular antigens.
Clonal B-cell proliferation, as well as lymphomas and myelomas, can result from chronic infections with organisms such as Escherichia coli (E. coli) and Helicobacter pylori (H. pylori) (1–6). The main feature seems to be the approximation of two systems each capable of sustained replication where the replicating microbe induces proliferation and selection of members of the replicating B-cell repertoire. In this regard, we investigated mycoplasma infection because it has the features of chronicity (7) and, as an obligatory parasite, is largely confined to the surface of cells (8, 9). In the course of our experiments, we discovered that some human mycoplasmas produce a protein that binds to immunoglobulins (Igs) with high affinity. This protein, which we refer to as Protein M, has a structure differs from all others in the Protein Data Bank (PDB).
Clonal immune cell proliferation can occur in the context of sustained chronic infections. Since we were interested in such responses, we investigated whether monoclonal antibodies produced as a result of multiple myeloma react with mycoplasma antigens. We tested the ability of plasma from 20 multiple myeloma patients to bind to total cellular extracts from multiple mycoplasma species, including human pathogens or commensal organisms and others that infect non-human vertebrates (10). Remarkably, these experiments showed that the antibodies in the plasma all reacted strongly with molecules present in human, but not non-human, pathogenic mycoplasmas (Fig. 1A, fig. S1A). The main reactivity was with a protein with an apparent molecular weight of ~ 50 kDa in Mycoplasma genitalium (M. genitalium) and with several proteins with apparent molecular weights of 40–65 kDa in Mycoplasma penetrans (Fig. 1A). We focused our attention on the protein from M. genitalium because it appeared to be more homogeneous as determined by gel electrophoresis (Fig. 1A). The Ig reactivity with the M. genitalium protein was similar for all patients' plasma tested (fig. S1A). To substantiate that the clonal multiple myeloma Ig is the component responsible for binding to the M. genitalium protein, as opposed to a highly reactive protein that co-purifies with it, the Fab' (fragment antigen-binding) of the primary monoclonal antibody in the plasma of multiple myeloma patient 13PL (13PL Fab') was highly purified by chromatography followed by crystallization, and its reactivity was studied using dissolved crystals as a source of the antibody (Fig. 1, B to D). The 13PL Fab' from the dissolved crystals bound to the same antigen in mycoplasmas as antibodies isolated from whole sera (Fig. 1C). To confirm that the crystals contained an antibody, the x-ray structure was determined at 1.2 Å resolution and only an Fab' was present (Fig. 1D) (10). To test whether a similar reactivity could be found in blood from non-myeloma normal donors (fig. S1B), samples from random donors were studied. The sera from these normal donors also surprisingly reacted with the same mycoplasma proteins as the clonal myeloma Igs, indicating that the ability of human Igs to react with this mycoplasma protein was not confined to those produced in multiple myeloma. We therefore termed the M. genitalium protein that reacts with Igs Protein M.
Fig. 1. Immunoglobulins selectively bind to proteins in human mycoplasma.
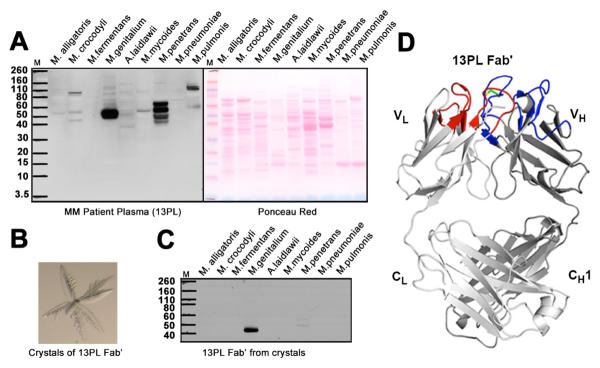
(A) (Left panel) Western blot analysis of the reactivity of plasma from multiple myeloma patient 13PL with cell extracts from Mycoplasma alligatoris, Mycoplasma crocodyli, Mycoplasma fermentans, M. genitalium, Acholeplasma Mycoplasma laidlawii, Mycoplasma mycoides, Mycoplasma penetrans, Mycoplasma pneumoniae and Mycoplasma pulmonis. All mycoplasma cells were grown in appropriate media. Cells were lysed according to manufacturer's protocol using lysis buffer from Sigma Aldrich. Nucleic acids were degraded by treatment with DNAase and RNAase. A protease inhibitor cocktail (Roche) was added to prevent proteolytic degradation. The extracts from the same number of cells were separated on SDS-PAGE gels and transferred to nitrocellulose membranes for Western blot analysis. (Right panel) Ponceau red-stained protein bands of the cell extracts. (B) Crystals of 13PL Fab' from a multiple myeloma patient's monoclonal immunoglobulin. (C) Western blot analysis of the reactivity of 13PL Fab' from the plasma of a multiple myeloma patient with the same cell extracts in A. The 13PL Fab' was purified by crystallization. The extracts were separated on SDS-PAGE gels as described in (A). (D) Crystal structure of 13PL Fab' shown in ribbon diagram with the light and heavy chains colored in light and dark gray, respectively. The loops corresponding to CDRs L1, L2, and L3 are colored blue, whereas CDRs H1, H2, and H3 are colored red. The relatively rare disulfide in human CDR3's is colored green.
At this point, the possibilities were that Protein M was an antigen to which most people make an antibody, or was a protein that binds to Ig domains or other features that are present in most antibodies. To study these possibilities, we first isolated Protein M using an affinity column constructed from antibody 13PL. The affinity purified Protein M was separated on SDS-PAGE gels followed by Western blot analysis using a different myeloma antibody to confirm the presence of the binding protein. The band on the SDS-PAGE gel corresponding to Protein M was excised and proteomics analysis by mass spectrometry was carried out (fig. S2A) (10). These studies showed that Protein M was M. genitalium protein MG281, which is an uncharacterized membrane protein (UniProtKB accession no. P47523) (fig. S2B) of 556 amino-acids with a predicted trans-membrane domain (residues 16 to 36) (fig. S2C). Furthermore, homologs of Protein M are present in other mycoplasma strains such as Mycoplasma pneumonia, Mycoplasma iowae and Mycoplasma gallisepticum (UniProtKB data base). Antibodies did not bind to mycoplasma extracts from a Protein M-null M. genitalium mutant, again suggesting that Protein M might be the molecule to which antibodies bind (Fig. S3A). To establish that Protein M alone is sufficient for antibody binding, a His-tagged Protein M lacking the membrane-spanning region (recombinant Protein M, residues 37–556) was cloned, expressed in E. coli, and purified by affinity chromatography and size-exclusion chromatography (10). Western blot analysis of purified Protein M showed that it reacted strongly with the monoclonal Ig from a multiple myeloma patient (Fig. S3B). Protein M also bound to all isotypes of human IgGs (fig. S4A) as well as mouse, rat, rabbit, goat, and bovine IgGs (fig. S4B). To further elucidate the minimum sequence responsible for antibody binding, the Protein M and 13PL IgG complex (mixed in a 1:1.1 molar ratio) was incubated with trypsin for 5 hours. SDS-PAGE gel analysis showed that a truncated protein remained intact after 5 hours as compared to uncomplexed Protein M that was totally digested into smaller fragments. The trypsin-digested Protein M (Protein M TD) was found by mass spectroscopy to contain residues 74 to 482 (fig. S5). A His-tagged Protein M TD consisting of residues 74 to 468 (recombinant Protein M TD) was then cloned, expressed in E. coli, and purified by affinity chromatography and size-exclusion chromatography. Protein M and Protein M TD showed similar binding affinities to a panel of Igs or Fabs with Kd values in the nM range (fig. S6), as determined using Biolayer Interferometry (11).
Ig binding to Protein M was confined to the Fab domain of the antibody molecule as shown by Biolayer Interferometry (fig. S6). Since a variety of antibodies with different complementarity determining regions (CDRs) all bind to Protein M, specific interaction with the combining site of the antibody molecule appeared to be excluded. To understand the molecular basis for this interaction, crystal structures of recombinant Protein M TD in complex with two antibody Fabs (PGT135 against HIV-1 gp120 with a κ light chain (Kd = 3.7 nM) (12) and Fab CR9114 against influenza hemagglutinin with a λ light chain (Kd = 1.9 nM) (13)) were determined to 1.65 and 2.50 Å resolution, respectively (table S1, Fig. 2). Although the 13PL Fab'–Protein M TD complex could not be crystallized, we were able to obtain the structure by electron microscopy, which showed a similar mode of binding (fig. S7) (10). The Protein M structure is very different from any other known Ig binding proteins or, indeed, any other structures in the Protein Data Bank (www.pdb.org) (14). Protein M TD comprises a large domain (residues 78–440) that includes a leucine-rich repeat (LRR)-like subdomain, and a smaller domain (residues 441–468) (Fig. 2A). Protein M TD binds predominantly to the variable light (VL) domains of both PGT135 Fab (Fig. 2B) and CR9114 Fab (Fig. 2C), but makes some very limited interactions with the other three Fab domains. The Fab-Protein M TD interactions bury total solvent accessible surface areas of 3590 Å2 and 2870 Å2 for PGT135 Fab (Fig. 2D, figs. S8A and S9A) and CR9114 Fab (Fig. 2E, figs. S8B and S9B), respectively, mainly from the VL domains of the Fabs (15). The common interacting positions, which are about two-thirds occupied by hydrophilic residues in both antibodies, are located on one edge of VL (Fig. 2F, tables S2 and S3). Ten conserved hydrogen bonds and one salt bridge are made from Protein M TD to each Fab VL, including 6 hydrogen bonds to the main chain of VL residues 15, 16, 18, 54 and 77, with the rest to the side chains of VL Arg61, Gln79 and Glu81 (tables S2 and S3), almost all of which are highly conserved among human antibodies with both κ and λ light chains (table S4). Other residues at the common paratope positions, which make van der Waals contacts and non-conserved H-bonds or salt bridges, are less conserved except for VL Gln37 and Pro59 (table S5) as well as the completely conserved CL Ser168 and VH1 Ser168. However, some of the non-polar interactions may be conserved even with different amino acids. The N- and C-terminal fragments (residues 37–74 and residues 469–556), which were truncated in Protein M TD as compared to Protein M, are likely disordered as the 3D reconstructions of a Fab in complex with Protein M and Protein M TD using negative-stain electron microscopy are nearly identical (figs. S10 and S11).
Fig. 2. Crystal structures of recombinant protein M TD in complex with PGT135 Fab and with CR9114 Fab.

(A) Overall structure of Protein M TD from its complex with PGT135 Fab in ribbon representation. Protein M TD appears to have two main domains: a larger domain I in green (residues 78 to 440), which includes a leucine-rich repeat-like structure in purple (LRR; residues 240 to 330) and a smaller domain II in cyan (residues 441 to 468). (B) Overall structure of Protein M TD in complex with PGT135 Fab. Protein M TD is colored in green, and PGT135 Fab is colored in light gray for the light chain and dark gray for the heavy chain. Protein M TD predominantly binds VL of PGT135 Fab, but also interacts to a lesser extent with VH, CL and CH1. (C) Overall structure of Protein M TD in complex with CR9114 Fab. The coloring scheme is similar to that of (B). Residues 455–468 in domain II of Protein M were flexible and not modeled. Protein M TD predominantly binds VL of CR9114 Fab. (D) Molecular surface representation of PGT135 Fab with the paratope of Protein M TD in red, Fab light chain in light gray and Fab heavy chain in dark gray. (E) Molecular surface representation of CR9144 Fab with the paratope of Protein M TD and colored as in (D). (F) The common paratope residue locations (in red) of Protein M TD for PGT135 Fab and CR9114 Fab are shown on PGT135 Fab, including VL residues 14–18 of FR1; 37, 45, 47 and 49 of FR2; 53–56 of CDR2; 57–61, 76, 77, 79 and 81 of FR3; residues 107 and 109 connecting VL and CL; as well as residues 168 and 170 of CL and residue 168 of CH1.
To determine the scope of Protein M binding to antibodies with different light chain allotypes, the binding affinities (Kds) of protein M TD to 24 different light chains in a variety of formats were determined. Because the heavy chains could alter potentially the light-chain conformations, we studied the same germline light chain paired with three different heavy chains. The particular heavy chains made little difference and the Kds varied between 2.1 and 4.8 nM for four VH/VL combinations (fig. S12, A to C). Similarly, when the same heavy chain was paired with four different light chains, Protein M binding ranged from 1.8 to 2.3 nM (fig. S12, D to G). The effect of allotypic variation was evaluated using five κ chain allotypes (κ1, κ2, κ3, κ4 and κ6) and three λ chain allotypes (λ2, λ5, and λ11). Allotypic variation had little effect and the Kds for the allotypic variants ranged between 1.0 and 4.8 nM (fig. S12 and table S6). The preservation of binding affinity in the presence of allotypic variation is to be expected because the critical Protein M contacts are largely conserved among allotypes (tables S4 and S5). To determine whether antigen specificity impacted Protein M, we determined the Kd's for Protein M TD binding to a panel of eight affinity-matured monoclonal antibodies against the same HIV-1 gp120 antigen but with different epitopes and found that the Kd's only varied between 0.7–3.8 nM (fig. S12, table S6).
Finally, we assessed the percentage of polyclonal human Igs from the plasma of normal blood donors that was capable of binding to Protein M. After two passages thru a column containing Protein M TD immobilized on Ni-NTA matrix at a flow rate of 1 ml/min, greater than 90% of all the Igs were removed, in agreement with our data that showed Protein M binds to all human monoclonal antibodies that we have tested to date.
These structural studies suggested that Protein M should preclude the ability of the antibody to bind to its antigen because it displaces or distorts the CDRs and/or may use its C-terminal domain to sterically block entrance to the antibody combining site (fig. S13). We tested the ability of recombinant Protein M and Protein M TD to block antigen-antibody union for six different antigen-antibody pairs, including two polyclonal auto-antibodies. The monoclonal antibodies used were generated against human influenza virus (13), HIV-1 (16), human Ebola (17) and mouse Ebola (18); polyclonal antibodies were purified from Goodpasture's disease patient serum (19) and lupus mouse serum (20). Blocking of the binding of serum polyclonal antibodies to antigens by Protein M is important because such sera represents a collection of antibodies rather than a single monoclonal species. Prior incubation of the antibodies with Protein M or Protein M TD (in a 1:8 molar ratio) strongly inhibited antibody binding to its cognate antigen (Fig. 3), but the order of addition is critical. Once antigen-antibody union has occurred for high affinity antigens, Protein M does not disrupt the antibody-antigen complex (fig. S14).
Fig 3. Protein-M blocks antigen-antibody union.

(A) Binding of CR9114 IgG, a human broadly neutralizing antibody against influenza virus to one of its antigens, H5 hemagglutinin (influenza virus strain A/Viet Nam/1203/2004 (H5N1)) was evaluated using ELISA after precomplexing with recombinant Protein M (red) and Protein M TD (blue). Binding of the IgG to the HA in the absence of protein M was used as control (purple). The extent of binding was analyzed by a colorimetric assay. The curves were obtained by a nonlinear regression analysis where the data were fit to a four-parameter logistic equation based on a simple binding model. Error bars represent standard deviation of duplicate measurements. (B) Binding of PGT135 IgG, a human broadly neutralizing antibody against its antigen HIV-1 gp120 (JR-FL gp120 core construct) was evaluated (purple) after precomplexing PGT135 IgG with Protein M (red) and Protein M TD (blue) (at a 1:8 molar ratio). ELISA assay was performed as in (A). (C) Binding of KZ52 IgG, a human broadly neutralizing antibody against its Ebola antigen glycoprotein (purple) was evaluated as in B. ELISA assay was performed as in (A). (D) Binding of 13C6 IgG, a mouse broadly neutralizing antibody against its Ebola glycoprotein antigen (purple) was evaluated as in B. (E) Binding of anti-COL4A3 human polyclonal serum from a patient with Goodpasture's disease to its antigen COL4A3 (purple) was evaluated as in B. (F) Binding of anti-DNA polyclonal serum a mouse with lupus to its antigen chromatin (purple) was evaluated as in B.
It is important to consider this discovery of a heretofore unknown high affinity Ig binding protein in human M. genitalium in the context of other known Ig binding proteins, such as Protein G, Protein A, and Protein L (21–23), which have been invaluable reagents and tools in the antibody field. The Protein M structure is very different from these other Ig binding proteins and is also very different from any other known protein structures. Unlike Protein G, Protein A, and Protein L that all contain multiple, small, Ig binding domains, Protein M has a large domain of 360 residues, which binds principally to antibody VL domains, as well as a leucine-rich repeat (LRR) like motif (24) that faces away from the antibody molecule and may have an as yet uncharacterized function. Importantly, Protein M also contains a 115-residue C-terminal domain that likely protrudes over the antibody combining site. To our knowledge, compared to other known Ig binding proteins, the Protein M TD-antibody Fab buried surface area is the largest (25).
Protein M binds to antibodies with either κ or λ light chains using conserved hydrogen bonds and salt bridges from backbone atoms and conserved side chains, and some conserved van der Waals interactions, as well as other non-conserved interactions. These conserved interactions provide a structural basis for the broad reactivity with Fvs, Fabs or Igs. In contrast, the primary binding site for Protein G and Protein A is the antibody Fc domain, although secondary lower affinity binding sites include the CH1 domain of IgG for Protein G (26) or VH of the human VH3 gene family for Protein A (22). Protein L binds only to the VL of most human κ light chains, except for the VκII subgroup (23) (Fig. 4). Thus, this new broad-scope, high affinity antibody binding protein, which binds both κ and λ chains, is likely to find a myriad of applications in immunochemistry. In addition to its general use, Protein M may be particularly important for large-scale purification of therapeutic antibodies. Finally, while Protein M may be important for the host-parasite relationship, further studies are necessary to elucidate the consequences of its expression on the parasite surface.
Fig. 4. Structural comparison of the antibody binding sites of Protein M and other immunoglobulin binding proteins.

All antibody Fabs or Fcs are shown in ribbon representation with light chain in light gray and one heavy chain in dark. (A) Protein M TD is depicted in green ribbon representation in its complex with PGT135 Fab. Protein M mainly binds to the VL domain. (B) One of the Ig-binding domains of Protein L (in orange) bound to VL of an antibody Fab (PDB code 1HEZ). Noticeably, although the Protein L domain can binds to two Fabs with similar paratope sites (23), only one binding site is shown here. (C) One of the Ig-binding domains of Protein A (in skyblue) bound to VH (weaker secondary site) of an antibody Fab (PDB code 1DEE). (D) One of the Ig-binding domains of Protein G (in pink) bound to CH1 (weaker secondary site) of an antibody Fab (PDB code 1IGC). (E) One of the Ig-binding domains of Protein A (in skyblue) bound to the antibody Fc region (primary site) near the CH2 and CH3 domain interface (PDB code 1FC2). (F) One of the Ig-binding domains of Protein G (in pink) also bound to antibody Fc region (primary site) near CH2 and CH3 domain interface (PDB code 1FCC). Although Protein M and Protein L predominantly bind the VL domain, their binding sites are very different with only one common residue (position 18 of VL). These Ig-binding proteins appear to have different ranges of affinities for antibodies. Generally for human antibodies, Protein M binds strongly to all the types, Protein G binds strongly to the antibody Fc region and weakly to the CH1 domain, Protein A binds strongly to antibody Fc region (except to the IgG3 subtype) and weakly to VH of VH3 gene family, and Protein L binds strongly to VL of κ light-chains, except the VκII subgroup.
Supplementary Material
Acknowledgements
We thank Prof. Benjamin Cravatt for mass spectral studies, Megan Lee and Cameron Locke for assistance in Western blot and ELISA studies, Wenli Yu for protein purification and Henry Tien of the Robotics Core at the Joint Center for Structural Genomics (jcsg.org) for automated crystal screening. X-ray diffraction data sets were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) beamlines 11–1 and 12–2. The b12 IgG and PGT135 IgG were kindly provided by Dr. Brian Moldt in the laboratory of Prof. Dennis Burton, and the other anti-HIV antibody Fabs were provided by Dr. Jean-Philippe Julien and Yuanzi Hua. The Lupus mouse serum and mouse chromatin were kindly provided by Prof. Roberto Baccala. The plasmapheresis fluid from a Goodpasture patient and human recombinant COL4A3 antigen were provided by Dr. Vadim Pedchenko. The KZ52 IgG, 13C6 IgG and Ebola GP were kindly provided by Prof. Erica O. Saphire. The electron microscopy studies were supported by startup funds from The Scripps Research Institute (to A.B.W.) and conducted at the National Resource for Automated Molecular Microscopy at the Scripps Research Institute, which is supported by NIH through the National Center for Research Resources' P41 program (RR017573). We also thank NIH RO1 AI042266 (I.A.W.) for support. Coordinates and structure factors will be deposited in the Protein Data Bank under accession codes AAAA for Protein M TD/PGT135 Fab complex and BBBB for Protein M TD/CR9114 Fab complex and released immediately upon publication. Image reconstruction will be deposited at the EMDataBank and released immediately upon publication. This is publication 25063 from The Scripps Research Institute.
References and Notes
- 1.Grover RK, et al. The costimulatory immunogen LPS induces the B-cell clones that infiltrate transplanted human kidneys. Proc. Natl. Acad. Sci. U.S.A. 2012;109:6036–6041. doi: 10.1073/pnas.1202214109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertoni F, Zucca E. Delving deeper into MALT lymphoma biology. J. Clin. Invest. 2006;116:22–26. doi: 10.1172/JCI27476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111:2962–2972. doi: 10.1182/blood-2007-10-078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minguet S, et al. Enhanced B-cell activation mediated by TLR4 and BCR crosstalk. Eur. J. Immunol. 2008;38:2475–2487. doi: 10.1002/eji.200738094. [DOI] [PubMed] [Google Scholar]
- 5.Vianello A, et al. Vesicoureteral reflux after kidney transplantation: clinical significance in the medium to long-term. Clin. Nephrol. 1997;47:356–361. [PubMed] [Google Scholar]
- 6.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor-Robinson D, Jensen JS. Mycoplasma genitalium: from Chrysalis to multicolored butterfly. Clin. Microbiol. Rev. 2011;24:498–514. doi: 10.1128/CMR.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rottem S. Interaction of Mycoplasmas with host cells. Physiol. Rev. 2003;83:417–432. doi: 10.1152/physrev.00030.2002. [DOI] [PubMed] [Google Scholar]
- 9.Citti C, Nouvel LX, Baranowski E. Phase and antigenic variation in Mycoplasmas. Future Microbiol. 2010;5:1073–1085. doi: 10.2217/fmb.10.71. [DOI] [PubMed] [Google Scholar]
- 10.Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin. Proc. 2001;76:476–487. doi: 10.4065/76.5.476. [DOI] [PubMed] [Google Scholar]
- 11.ForteBio - A Division of Pall Life Sciences All rights reserved. 2013. [Google Scholar]
- 12.Kong L, et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat. Struct. Mol. Biol. 2013;20:796–803. doi: 10.1038/nsmb.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreyfus C, et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.A structural similarity search performed using the Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/start) indicated that Protein M TD is not significantly similar to other structures, except for the leucine-rich repeat-like motif.
- 15.The Fab-Protein M TD interactions bury total solvent accessible surface areas of 2490 Å2 for PGT135 VL domain (1280 Å2 from Protein M TD and 1210 Å2 from PGT135) and 2380 Å2 for CR9114 VL domain (1230 Å2 from Protein M TD and 1150 Å2 from CR9114), respectively.
- 16.Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JE, et al. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson JA, et al. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–1666. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- 19.Gunwar S, et al. Glomerular basement membrane. Identification of dimeric subunits of the noncollagenous domain (hexamer) of collagen IV and the Goodpasture antigen. J. Biol. Chem. 1991;266:15318–15324. [PubMed] [Google Scholar]
- 20.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv. Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 21.Gronenborn AM, et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 22.Graille M, et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5399–5404. doi: 10.1073/pnas.97.10.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graille M, et al. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure. 2001;9:679–687. doi: 10.1016/s0969-2126(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 24.Doxey AC, McConkey BJ. Prediction of molecular mimicry candidates in human pathogenic bacteria. Virulence. 2013;4:453–466. doi: 10.4161/viru.25180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 26.Derrick JP, Wigley DB. The third IgG-binding domain from streptococcal protein G. An analysis by X-ray crystallography of the structure alone and in a complex with Fab. J. Mol. Biol. 1994;243:906–918. doi: 10.1006/jmbi.1994.1691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.