

Abstract
The PTPN11 (protein-tyrosine phosphatase, non-receptor type 11) gene encodes SHP2, a cytoplasmic PTP that is essential for vertebrate development. Mutations in PTPN11 are associated with Noonan and LEOPARD syndrome. Human patients with these autosomal dominant disorders display various symptoms, including short stature, craniofacial defects and heart abnormalities. We have used the zebrafish as a model to investigate the role of Shp2 in embryonic development. The zebrafish genome encodes two ptpn11 genes, ptpn11a and ptpn11b. Here, we report that ptpn11a is expressed constitutively and ptpn11b expression is strongly upregulated during development. In addition, the products of both ptpn11 genes, Shp2a and Shp2b, are functional. Target-selected inactivation of ptpn11a and ptpn11b revealed that double homozygous mutants are embryonic lethal at 5–6 days post fertilization (dpf). Ptpn11a-/-ptpn11b-/- embryos showed pleiotropic defects from 4 dpf onwards, including reduced body axis extension and craniofacial defects, which was accompanied by low levels of phosphorylated Erk at 5 dpf. Interestingly, defects in homozygous ptpn11a-/- mutants overlapped with defects in the double mutants albeit they were milder, whereas ptpn11b-/- single mutants did not show detectable developmental defects and were viable and fertile. Ptpn11a-/-ptpn11b-/- mutants were rescued by expression of exogenous ptpn11a and ptpn11b alike, indicating functional redundance of Shp2a and Shp2b. The ptpn11 mutants provide a good basis for further unravelling of the function of Shp2 in vertebrate development.
Introduction
The PTPN11 (protein-tyrosine phosphatase, non-receptor type 11) gene encodes SHP2, a ubiquitously expressed cytoplasmic protein-tyrosine phosphatase (PTP) with two Src homology 2 (SH2) domains that is essential for normal development [1]–[3]. SHP2 is a positive effector of extracellular regulated kinase (ERK)/MAPK signal transduction, downstream of most receptor tyrosine kinases (RTKs) [2] [3]. Moreover, Shp2 is involved in other signal transduction processes as well, including Jak-STAT signaling [3] and PI3K-AKT (also known as PI3K-PKB) signaling [2].
Missense mutations in the PTPN11 gene are associated with the dominantly inherited genetic disorders, Noonan syndrome (NS) (OMIM 163950) and LEOPARD syndrome (LS) (OMIM 151100) [4]–[9]. These syndromes share several overlapping features, including facial dysmorphism, cardiovascular defects, hearing loss, growth retardation and scoliosis [7], [10]. NS variants of SHP2 have increased PTP activity [2]. By contrast, most LS variants of SHP2 display reduced catalytic activity [11]–[14]. NS-associated mutations in Shp2 promote Erk/MAPK signaling [2]. Yet, the role of LS Shp2-variants in Erk/MAPK signaling is less clear, in that both activation and inhibition of Erk/MAPK signaling have been observed in LS [11], [14]–[18]. Nevertheless, signaling by NS- and LS-variants of SHP2 is distinct, because LS-SHP2, but not NS-SHP2, affects AKT (PKB) signaling [12], [19]. The mechanism underlying how NS- and LS-variants of SHP2 induce similar defects in development remains to be determined.
Shp2 has an essential role in early development in the mouse [20]. Homozygous Ptpn11-/- embryos die pre-implantation due to defective Erk activation and trophoblast stem cell death [20], precluding the possibility to study Shp2 function in differentiated cell types in adult animals. This problem was partially overcome with the use of conditional Shp2 knockout mice [21]–[23]. For example, mice with a Ptpn11 deletion in the cardiomyocytes show that Shp2 is required for the suppression of dilated cardiomyopathy [22]. Conditional deletion of Shp2 in neural progenitor cells results in early postnatal lethality, impaired corticogenesis, and reduced proliferation of progenitor cells in the ventricular zone [21]. In an aged mouse model, hepatocyte-specific deletion of Ptpn11 promotes inflammatory signalling and hepatic inflammation/necrosis, resulting in regenerative hyperplasia and spontaneous development of tumors [24]. Furthermore, a study in Xenopus shows that dominant-negative Shp2 mutants failed to complete gastrulation, resulting in severe posterior truncations [25]. In zebrafish, Shp2 knockdown embryos show a reduction of the body axis extension at 10 hours post fertilization (hpf) that is consistent with gastrulation defects. In addition, at later stages, the embryos are shorter and develop a hammerhead phenotype [26], [27]. These data show that Shp2 is a crucial player in the development of many different organisms.
In our study we used zebrafish as a model to further investigate the role of Shp2. The zebrafish has proven to be an excellent experimental model to study gene function in vivo and to analyze vertebrate embryogenesis as development occurs externally and the embryos are transparent [28]. We identified two zebrafish ptpn11 genes (ptpn11a and ptpn11b), encoding Shp2a and Shp2b, respectively. We demonstrate that ptpn11a was constitutively expressed and ptpn11b expression increased during early embryogenesis. Ptpn11a and ptpn11b both encoded functional proteins with similar catalytic activity. We used target selected gene inactivation (TSGI) to identify stop mutations in the N-terminal SH2 domain of each of the two ptpn11 genes. Our study showed that ptpn11b-/- zebrafish were viable and fertile and showed no abnormal phenotype during development. However, single homozygous mutants for ptpn11a and double homozygous mutant embryos exhibited abnormalities such as craniofacial defects and reduced length, and these mutants died at 5 days post fertilization (dpf). Moreover, we found that Shp2 deletion suppressed Erk activation in double mutant embryos, but not in embryos lacking only Shp2a or Shp2b. Together, our observations demonstrate that Shp2 is essential for zebrafish development, and that ptpn11a has a more pronounced role as the result of its higher expression level at early stages.
Materials and Methods
Ethics statement
All procedures involving experimental animals described here were approved by the local animal experiments committee (KNAW-DEC protocol HL05.1501) and performed according to local guidelines and policies in compliance with national and European law.
Zebrafish care and generation of ptpn11 mutant lines
Zebrafish maintenance, breeding and staging were performed following published protocols [29]. ENU mutagenesis was performed on TL males [30] and exons 1–4 of the zebrafish ptpn11a and ptpn11b genes were sequenced using DNA from F1 generation zebrafish. Each mutation was confirmed by resequencing. Genomic DNA was extracted from adult fish fin clip or fixed embryos. The genotyping assay for the ptpn11 mutations was performed by nested PCR with primer sets: ptpn11a, 5′-GCGCTGTCACACACATTAAGA, 5′-TCACAGCCAATAAAGAGAAGC; nested ptpn11a 5′-CGACCTGTATGGTGGAGAGAA, 5′-TCCCAAATTGTCATGTAAGG; ptpn11b, 5′- TTCAACAACATCCTCCTAACTG, 5′-AAACAAACCACAGCTCTTCC and nested ptpn11b 5′-GTCTGTCATCCCTCATTTCC, 5′-GCAGGATTTATTCTGTCCAC, followed by sequencing to detect the mutations.
RNA isolation, cDNA synthesis and quantitative PCR
For the reverse transcription PCR (RT-PCR) experiment, thirty zebrafish embryos at various stages of development (4 hpf, 10 hpf, 1 dpf, 2 dpf, 3 dpf and 5 dpf) were collected and homogenized in TRIzol Reagent (Invitrogen; Carlsbad, CA). The homogenate was centrifuged (12000 g, 4°C, 10 min.). 0.2 ml of Chloroform were added to the supernatant, vortexed for 15 sec., incubated for 3 min. at room temperature (RT) and then centrifuged (12000 g, 4°C, 15 min.). 0.5 ml isopropanol were added to the upper phase, incubated for 10 min. at RT and centrifuged (12000 g, 4°C, 15 min.). The pellet was washed with 75% EtOH and after vortexing, centrifuged (10000 g, 4°C, 5 min.). The pellet was then dried for 5 min. and dissolved in 100 µl MQ. After, the solution was incubated for 10 min. at 37°C, precipitated in 250 µl 100% EtOH and 10 µl 3 M NaAc and stored at −80°C.
For RT-PCR with M-MLV-RT (Promega) 1 µg of total RNA was used. PCR of cDNA was performed using the standard protocol of GoTaq (Promega) using oligonucleotides listed as follow; ptpn11a forward: 5′- ATGTGCCCAAGACTATCCAGATG, ptpn11a reverse: 5′- CCCACGTTCTCATAGACTCGAGA, ptpn11b forward: 5′- ACTGTGACATTGACATCCCAAAAA, ptpn11b reverse: TACTGCTGGTGGAGCCCTTTGGAT. The same primers were used for sequencing of the DNA fragments.
Real-time RT-PCR was performed using the IQ SYBR Green Supermix (Biorad). Real time PCR MyIQ software (Bio-rad) was used to determine the amplification cycle in which product accumulation was above the threshold cycle values (Ct). PCR was carried out after incubation at 50°C for 2 min and pre-denaturing at 95°C for 3 min, followed by 40 cycles at 95°C for 30 sec and 62°C for 1 min. The relative quantification was given by the CT values, determined by triplicate reactions for all of the samples for ptpn11a, ptpn11b and β-actin. Real time PCR Ct values were analyzed using the 2−ΔCT method [31].
For the quantification of ptpn11a and ptpn11b mRNA levels, the housekeeping gene actin was used as internal standard. ΔCT-values were calculated as follows: CT(β-actin)-CT(ptpn11) and were normalized to the starting point T0 (4 hpf).
In situ hybridization
In situ hybridizations were done essentially as described [32]. Digoxigenin-UTP-labeled riboprobes for ptpn11a and ptpn11b were synthesized from PCR products. The PCR products were amplified with primers containing the T7 promoter and then sequenced. Primers were designed as follows: ptpn11a forward, 5′- ATTTAGGTGACACTATAGGGAGCTACATTGCCACACAA, ptpn11a reverse: 5′- TAATACGACTCACTATAGGGTTTTTCATCTCTCGGTTTAGTCA. For ptpn11b, primers that attached to the 3′UTR region of the gene were designed: ptpn11b forward: 5′- ATTTAGGTGACACTATAGGGAACAAAACGAAGGAGAGG, ptpn11b reverse: 5′- TAATACGACTCACTATAGGGGCCTGCCTCATTTTTAGCTG. Embryos were cleared in methanol and mounted in Benzyl Benzoate/Benzyl Alcohol (2∶1) before pictures were taken.
Alcian blue staining
Morphological phenotypes were assessed at 5 dpf. Embryos were anesthesized at 5 dpf with MS-222 (Sigma), fixed in 4% paraformaldehyde (PFA) and the cartilage was stained with alcian blue. Briefly, fixed embryos were washed for 10 min in 50% ETOH in water. Next, the embryos were stained overnight at 4C in staining solution (0.04% alcian blue, 50 mM MgCl2, 70% EtOH). Embryos were washed in 0.2% of Triton X-100 in water. Pictures of lateral and ventral views were taken. The width of the ceratohyal angle was determined using Image J software and the ratio was determined as a direct measure for craniofacial defects. Averages were determined and a student t-test was done to determine whether the differences between the diverse conditions were statistically significant.
Immunoblotting
Zebrafish embryos were raised in standard conditions. At 5 dpf, 5 embryos were pooled and lysed in buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM sodium orthovanadate, 1% Nonidet P-40, 0.1% sodium deoxycholate, protease inhibitor mixture (Complete Mini, Roche Diagnostics) and vanadate, and 40 µl lysisbuffer. Lysates were spun down and 4× sample buffer was added to supernatant; Samples were run on SDS-PAGE gel (10%) and transferred to PVDF membrane. After transfer the membrane was stained with Coomassie Blue stain to verify equal loading of the lysates. Subsequently the PVDF membrane was blocked with 5% BSA and then incubated with the corresponding antibodies targeting mouse monoclonal antibody anti-pERK, rabbit monoclonal antibody anti-ERK, rabbit monoclonal antibody pAKT and rabbit monoclonal antibody anti-AKT. All the primary antibodies were diluted 1∶1000 in blocking solution and they were purchased from Cell-Signaling. The secondary antibodies (mouse and rabbit) conjugated to horseradish peroxidase (HRP), were purchased from BD bioscience Pharmaceutical and they were diluted 1∶10,000 in TBST. The membranes were subjected to detection by enhanced chemiluminescence (Thermo Scientific kit).
Phosphatase assay
We generated zebrafish ptpn11a and ptpn11b cDNA constructs and cloned them in a pGEX-4t vector, allowing production and purification of recombinant Shp2 proteins, fused to six histidine residues in their C-termini. Purified GST-fusion proteins were directly incubated in PTP assay buffer (20 mM MES buffer pH 6.0, 1 mM EDTA, 150 mM NaCl, 1 mM dithiotreitol, and 10 mM p-nitrophenylphosphate) for 45 min. at 30 °C. The reactions were quenched with 0.4 M NaOH, and optical density was measured with a spectrophotometer at 415 nm (wavelength).
Statistical analysis
Statistical significance was assessed by two-tailed Student's t-test in all experiments. Significance is represented in the graphs. Results are considered significant when p<0.05 and are expressed as mean ± standard error of the mean. * indicates a p value of <0.05, ** indicates a p value of <0.01 and *** indicates a p value of <0.001.
Results
Characterization of zebrafish ptpn11 genes
The zebrafish genome encodes two ptpn11 genes: ptpn11a and ptpn11b [33]. The homology between human Shp2 and zebrafish Shp2a and Shp2b, encoded by ptpn11a and ptpn11b genes, is 91% and 64% respectively (Fig. S1). Due to the genome duplication that occurred in teleosts it is not uncommon for zebrafish to have two homologous copies of a gene that is present as a single copy in other vertebrates [34], [35]. To analyze stage-specific expression of ptpn11a and ptpn11b mRNA in wild-type (WT) embryos, we used a reverse transcription PCR (RT-PCR) approach. Expression levels were analysed at 4 hpf, 10 hpf, 1 dpf, 2 dpf, 3 dpf and 5 dpf. Ptpn11a and ptpn11b mRNA are both expressed from 4 hpf to 5 dpf (Fig. 1A). Ptpn11a mRNA was expressed at similar levels at all stages. By contrast, ptpn11b mRNA was hardly detectable at early stages and ptpn11b expression increased during development. To confirm this result, ptpn11a and ptpn11b expression levels were examined at 10 hpf, 1 dpf, 2 dpf, 3 dpf and 5 dpf by quantitative PCR. Ptpn11a expression was constantly maintained in all the stages whereas ptpn11b gene was expressed at low levels at early stages and became strongly upregulated over time (Fig. 1B). In situ hybridization experiments showed that the two zebrafish ptpn11 genes were broadly expressed throughout embryonic development (Fig. 1C). Both ptpn11 genes were maternally expressed and later in development the expression was restricted to the more anterior region of the developing embryo. Overall, these results show that both ptpn11 genes were expressed throughout developing zebrafish embryos. Ptpn11a appears to be expressed at a constant level, whereas ptpn11b expression is low initially and is strongly upregulated over time.
Figure 1. Expression of zebrafish ptpn11a and ptpn11b mRNA during embryogenesis.

(a) RNA was isolated from zebrafish embryos at 4 hpf, 10 hpf, 1 dpf, 2 dpf, 3 dpf and 5 dpf. Reverse transcription PCR was performed to detect the indicated genes; reverse transcriptase was omitted from the reaction as a control (-RT). (b) Quantitative PCR for ptpn11a and ptpn11b gene expression was performed from a pool of wild-type embryos (n = 30) collected at 10 hpf, 1 dpf, 2 dpf, 3 dpf and 5 dpf. The values were normalized using Actin as a control and relative expression is depicted here (value at 5 dpf set to 1.0). Error bars indicate standard error of the mean. (c) Whole-mount in situ hybridization using ptpn11a and ptpn11b specific probes of 4 hpf, 10 hpf, 30 hpf, 2 dpf, 3 dpf and 5 dpf embryos. Lateral and dorsal views are given as indicated.
Zebrafish Shp2a and Shp2b displayed phosphatase activity
Previously, we assessed that Shp2a is an active phosphatase and that mutant Shp2a-D61G with a mutation that was identified in human NS patients shows an increased phosphatase activity compared to wild type Shp2a [26]. To investigate if the ptpn11b gene product was functional, we expressed Shp2b in bacteria in parallel with Shp2a and investigated phosphatase activity. Both Shp2a and Shp2b displayed phosphatase activity and dephosphorylated p-nitrophenylphosphate (pNPP) to a similar extent. Moreover, Shp2b-D61G showed an increase in activity compared to WT-Shp2b, resembling the Shp2a-D61G mutant (Fig. 2). These results demonstrate that both Shp2 proteins encoded by the zebrafish genome were functional PTPs in vitro.
Figure 2. Shp2a and Shp2b proteins displayed phosphatase activity.
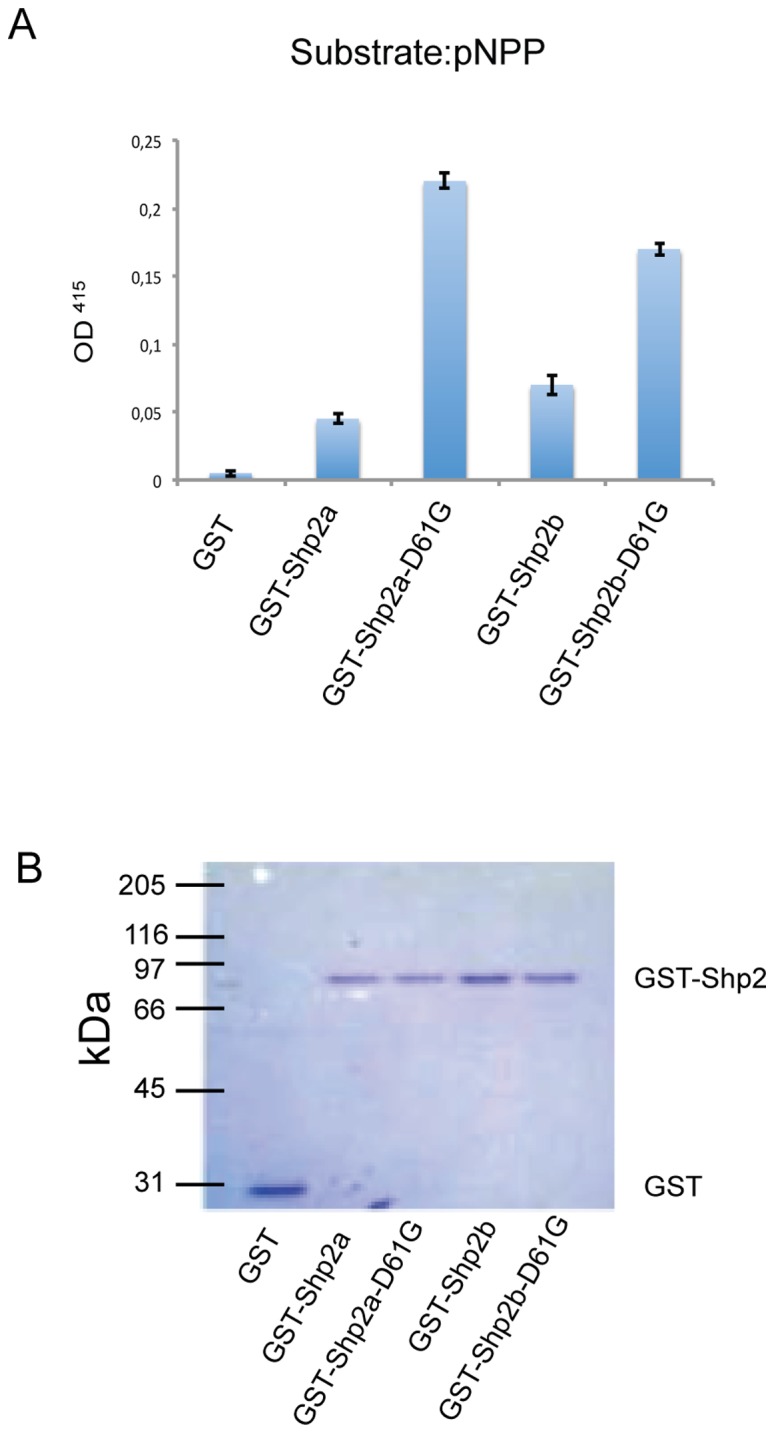
(a) PTP activity of pGEX, WT-Shp2a, Shp2a-D61G, WT-Shp2b and Shp2b-D61G was assayed using p-nitrophenylphosphate (pNPP) and quantified spectrophotometrically. Approximately 1 µg purified fusion protein was used in the assay. Experiments were done in quadruplicate; averages are depicted and error bars indicate standard error of the mean. (b) Equivalent amounts of fusion protein that were used in the activity assays in (a) were run on an SDS-PAGE gel and stained with Coomassie blue.
Generation of ptpn11 knock-out zebrafish
We identified germline mutations in the zebrafish ptpn11 genes, using the target-selected gene inactivation (TSGI) strategy, developed at the Hubrecht Institute [30]. Mutant alleles ptpn11ahu3459 and ptpn11bhu5920 contained nonsense mutations in exon 3 of ptpn11a and ptpn11b, respectively (Fig. 3). We denoted the homozygous mutants as ptpn11a-/- and ptpn11b-/- because the premature stops are well upstream of the phosphatase catalytic site and hence, no functional protein is produced. Ptpn11a+/- and ptpn11b+/- fish were viable and fertile and these fish were incrossed to generate homozygous fish. Genotypes were obtained at mendelian ratios until 5 dpf. Ptpn11a-/- fish did not reach adulthood. By contrast, homozygous ptpn11b-/- fish were viable and fertile. Next, fifth generation ptpn11a+/- and ptpn11b+/- fish were incrossed and double heterozygous ptpn11a+/-ptpn11b+/- fish were selected. Incrosses were used to investigate zebrafish development in double homozygous ptpn11a-/-ptpn11b-/- embryos. Phenotypically, all genotypes were indistinguishable from wild type embryos up to 3 dpf (data not shown). However, at 5dpf, ptpn11a-/- and double mutant embryos displayed significant phenotypic changes such as reduced body length, malformed head, absence of swim bladder, heart edema and eye edema (Fig. 4A-D). Furthermore, these embryos were unable to move, even after stimulation. Ptpn11b-/- embryos were indistinguishable from wild type embryos at 5 dpf and did not display morphological defects (Fig. 4A,C; Table 1). Ptpn11a-/- embryos and ptpn11a-/-ptpn11b-/- embryos were embryonic lethal. Whereas the developmental defects of ptpn11a-/- and ptpn11a-/-ptpn11b-/- embryos were overlapping (Fig. 4B,D), the phenotype of double mutant embryos was more severe than that of ptpn11a-/- embryos.
Figure 3. Nonsense mutations in zebrafish ptpn11a and ptpn11b.
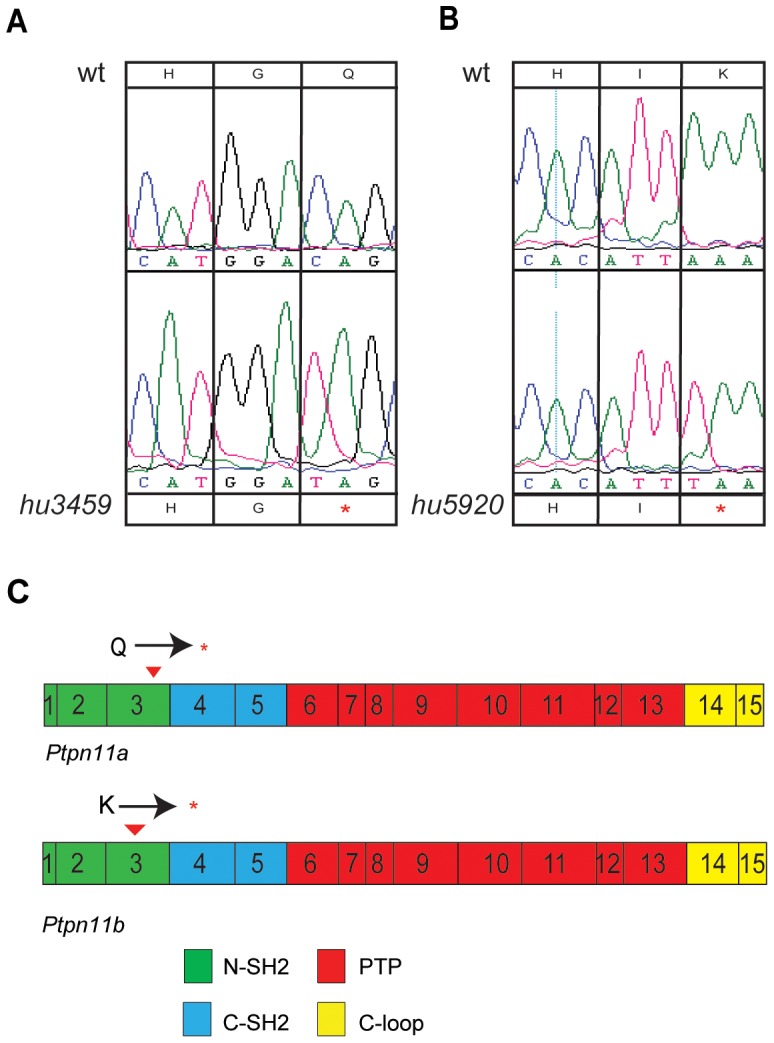
Sequence analysis of homozygous ptpn11a and ptpn11b mutants. (a) A C to T mutation in ptpn11ahu3459 resulted in a glutamine to a STOP mutation. (b) A T to A mutation in ptpn11bhu5920 changed lysine to a STOP mutation. (c) Schematic representation of the exon organization and structural domains of ptpn11a and ptpn11b. Nonsense mutations upstream of the catalytic domain are represented by red arrows.
Figure 4. Loss of Shp2 causes severe morphological defects at 5 dpf.
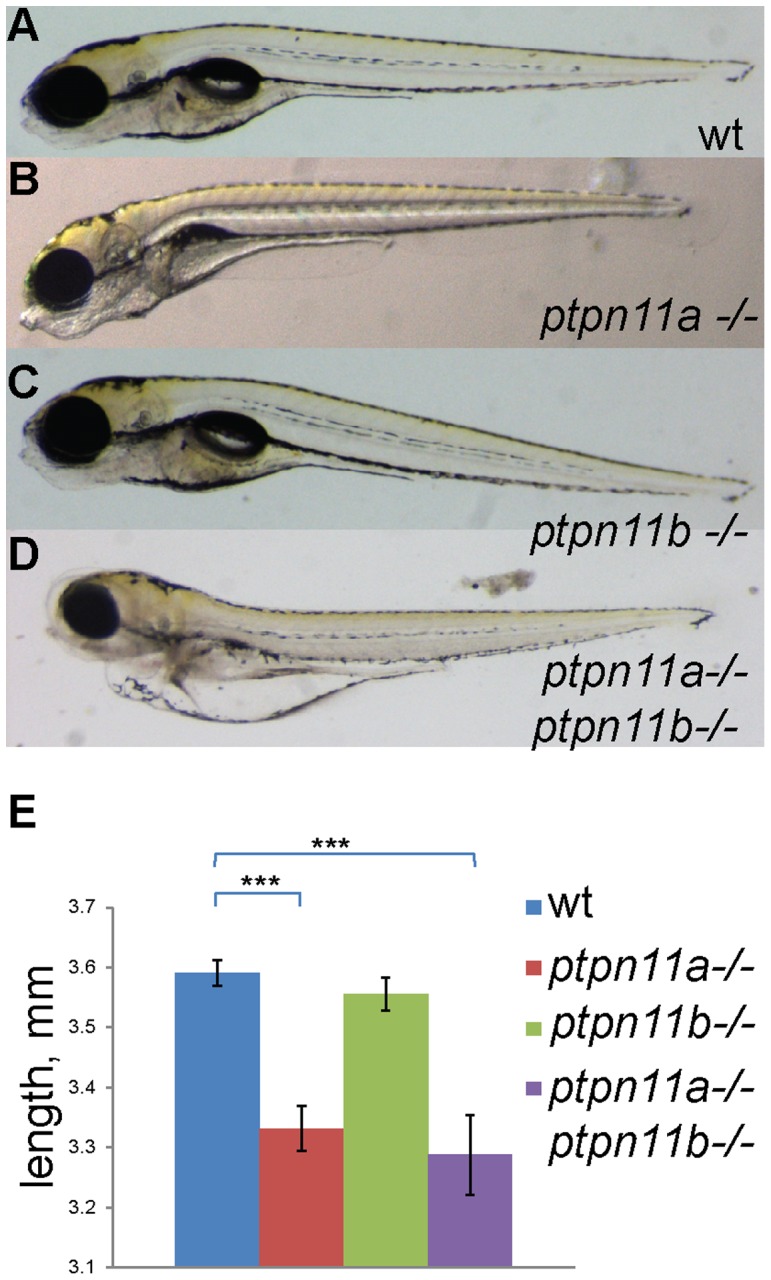
Morphology of 5(a) Representative wild-type, (b) ptpn11a-/-, (c) ptpn11b-/- and (d) double homozygous mutant are depicted. (e) The length was measured from nose to tip of tail at the same magnification. Bars show average length of the studied genotypes (n = 10-20). Statistics were determined using a student's t-test; * indicates a p value <0.05.
Table 1. Characteristic features of ptpn11a-/-, ptpn11b -/- and double mutant embryos.
| Characteristics | ptpn11a-/- | ptpn11b-/- | ptpn11a-/-ptpn11b-/- |
| Head | Reduced in some cases | Normal | Enlarged by edema. Prominent jaw |
| Heart | Normal | Normal | Severe edema |
| Swim bladder | Absent | Normal | Absent |
| Length | Shorter | Normal | Shorter |
| Ventral region | Reduced, skinny appearance | Normal | Grossly edematous. Reduced organs. |
Ptpn11a-/- and double mutants appeared shorter than wild type embryos and we measured the body length of wildtype and mutant embryos at 5 dpf. Ptpn11a-/- (3.33 mm±0.03, n = 20) and ptpn11a-/-ptpn11b-/- (3.28 mm±0.05, n = 10) embryos were significantly shorter (p<0,001, Student's t-test) than wild type embryos (3.59 mm±0.02, n = 21) (Fig. 4E). The length of wildtype embryos and ptpn11b-/- embryos (3.55 mm±0.02, n = 15) did not differ significantly. Collectively, our results show that ptpn11a, but not ptpn11b, is essential for normal development. Yet, ptpn11b appears to be functional, because the double mutant ptpn11a-/-ptpn11b-/- displayed more severe defects than ptpn11a-/- by itself.
Craniofacial defects in Ptpn11a-/- and double mutant Shp2 embryos
Patients affected by NS and LS often suffer from craniofacial defects, including positional anomalies of the eyes and ears and a short, webbed neck. Craniofacial defects were apparent in double mutant ptpn11a-/-ptpn11b-/- embryos at 5 dpf. Alcian blue staining of the cartilage revealed craniofacial defects that were quantified by assessment of the angle of the ceratohyal, a direct measure for the width of the head (Fig. 5A). There was a significant difference between wildtype (60.72°±1.51, n = 24) and double mutant embryos (95.14°±6.12, n = 10) (p<0.001, Student's t-test). Moreover, also ptpn11a-/- embryos (79.56°±5.29, n = 15, p<0.01) had a significantly wider ceratohyal compared to wildtype embryos, although the defect was milder. Ptpn11b-/- embryos (62.84°±5.73, n = 11) did not show a significant difference compared to wildtype embryos (Fig. 5I). Taken together, these results show that particularly ptpn11a is indispensable for craniofacial development.
Figure 5. Ptpn11a-/- and double mutant embryos exhibit craniofacial defects at 5 dpf.
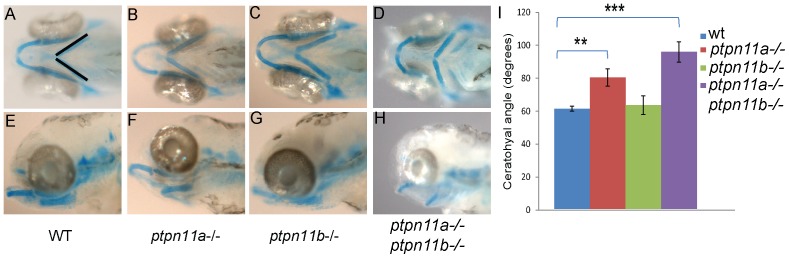
Alcian blue staining was done to visualize the cartilage. (a,e) wild-type, (b,f) ptpn11a-/-, (c,g) ptpn11b-/- and (d,h) double homozygous mutant. (i) Average width of the ceratohyal angle (indicated in panel a) was determined for each genotype (n = 10–24). Statistics were determined using a student's t-test; ** indicates a p value of <0.01, *** indicates a p value <0.001.
Double mutant Shp2 phenotype was rescued upon ptpn11a and ptpn11b mRNA injections
To investigate if the double mutant phenotype could be suppressed by introduction of the full-length ptpn11a and ptpn11b mRNA, we performed rescue experiments. Clutches of embryos from ptpn11a+/-ptpn11b-/- zebrafish were split and injected with ptpn11a or ptpn11b mRNA, or were not injected. The embryos were sorted based on their morphology at 5 dpf and genotyped by sequencing. The overall morphology of double mutant embryos is nicely distinguishable from wildtype embryos and 40/199 - close to the expected 25% - of the embryos are shorter and particularly the trunk and tail region is wider (Fig. 6A). Microinjection of ptpn11a mRNA in offspring of ptpn11a+/-ptpn11b-/- mutants at the one-cell stage largely suppressed the morphological phenotype in double mutant embryos at 5 dpf (Fig.6B, Table 2). Interestingly, injection of ptpn11b mRNA also led to rescue of the morphological defects (Fig.6C, Table 2), indicating that ptpn11a and ptpn11b have redundant biochemical functions. The difference in the function of ptpn11a and ptpn11b in development may be caused by the observed difference in expression level at early stages of development. To test this hypothesis directly, we investigated whether ptpn11b RNA was able to rescue the ptpn11a-/-ptpn11b+/+ phenotype. Ptpn11b mRNA injections at 1 cell stage indeed largely suppressed the ptpn11a-/- embryo phenotype (Fig.6D,E, Table 2). These results show that ptpn11a and ptpn11b mRNA injections largely rescued the developmental defects in the double mutant embryos at 5 dpf and that injections of ptpn11b alone suppressed the ptpn11a-/- phenotype.
Figure 6. Rescue of mutant phenotype by exogenous ptpn11a and ptpn11b.
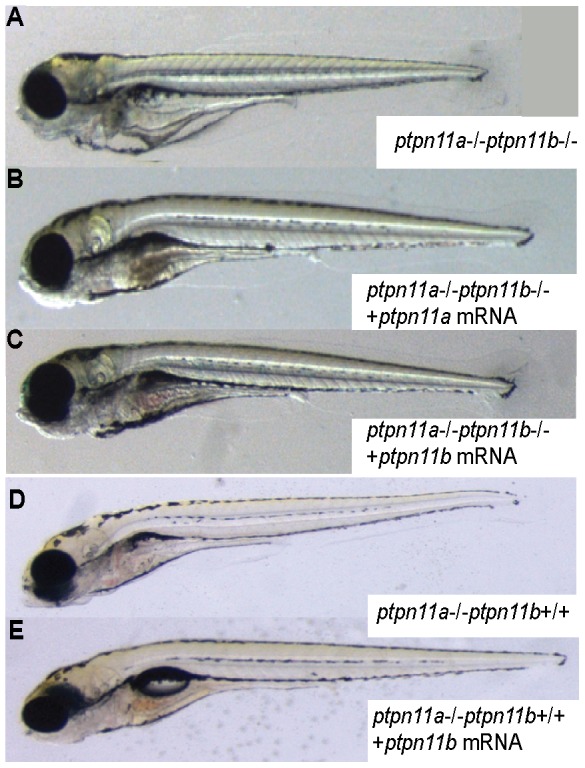
Morphology of 5(a) ptpn11a-/-ptpn11b-/- double mutant, (b) double mutant expressing exogenous ptpn11a, (c) double mutant expressing exogenous ptpn11b, (d) ptpn11a-/- mutant, (e) ptpn11a-/- mutant expressing exogenous ptpn11b. Representative embryos are depicted here. See Table 2 for quantification.
Table 2. Rescue of ptpn11a mutant phenotype by injection of ptpn11a and ptpn11b.
| incross | injection (mRNA) | phenotype | ptpn11a-/-/total |
| ptpn11a+/-ptpn11b-/- | - | 40/199 | 40/199 |
| ptpn11a+/-ptpn11b-/- | ptpn11a | 1/98 | 23/98 |
| ptpn11a+/-ptpn11b-/- | ptpn11b | 5/65 | 12/65 |
| ptpn11a+/-ptpn11b+/+ | - | 25/102 | 25/102 |
| ptpn11a+/-ptpn11b+/+ | ptpn11b | 15/360 | 75/360 |
Clutches of embryos from ptpn11a+/-ptpn11b-/- or ptpn11a+/-ptpn11b+/+ zebrafish were split and injected with ptpn11a mRNA or ptpn11b mRNA. The embryos were scored based on their morphology at 5 dpf. Phenotype represents the number of embryos with mutant morphology/total number of embryos. Subsequently, the genotype was determined by sequencing and is depicted as number of ptpn11a-/- embryos/total number of embryos analyzed. Ptpn11b was homozygous (ptpn11b-/- or ptpn11b+/+) and was not determined in these embryos.
Impaired Erk/MAPK, but not Akt/PKB signalling in Shp2 double mutant embryos
Shp2 is required to promote the activation of the MAPK cascade in response to growth factors, cytokine receptors and integrins [3], [36]. Thus, we examined the functional consequences of loss of Shp2 on downstream signaling by investigating Erk phosphorylation in wildtype, ptpn11a-/-, ptpn11b-/- and double mutant embryos at 5 dpf. Immunoblotting showed that Erk activation was decreased in double mutant embryos compared to wild type, whereas Erk levels were not impaired in the single homozygous mutants (Fig. 7A). Shp2 has been reported to act upstream of Akt/PKB signalling [12], [18]. We investigated Akt signaling by immunoblotting for phosphorylated Akt in wild-type, ptpn11a-/-, ptpn11b-/- and double mutant embryos at 5 dpf. We found that Akt activation was not affected in both single ptpn11 homozygous mutants, nor in the double mutants (Fig. 7B). Together, these data indicate that Erk/MAPK signaling, but not Akt (PKB) signaling is affected in mutants lacking functional Shp2.
Figure 7. Impaired Erk/MAPK, but not Akt/PKB signalling in Shp2 double mutant embryos.
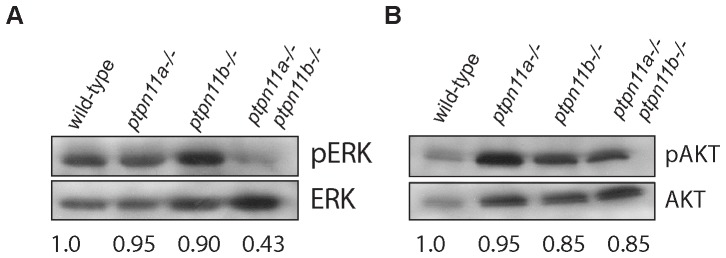
WT, ptpn11a-/-, ptpn11b-/- and ptpn11a-/- ptpn11b-/- embryos were lyzed and immunoblotted with phosho-specific antibody for ERK and AKT; membranes were then reprobed for total Erk and Akt expression to control for loading. The blots were quantified and the ratio of phosphorylated protein/total protein from three independent experiments is indicated below each lane.
Discussion
In this paper we characterized the two ptpn11 genes in zebrafish, ptpn11a and ptpn11b. The products of these genes, Shp2a and Shp2b, respectively, are both active and exhibit comparable phosphatase activities. Using a TSGI approach, we identified nonsense mutations in ptpn11a and ptpn11b, well upstream of the catalytic site that abolished Shp2 function. In agreement with previous studies in mice with targeted ptpn11 [20], [37], double mutant zebrafish embryos lacking all functional Shp2 were found to be embryonic lethal. Mouse embryos lacking functional Shp2 die around implantation [20], [37], whereas zebrafish embryos lacking functional Shp2 survive until 5-6 dpf. The difference in the developmental stage at which the lack of Shp2 is embryonic lethal between mice and zebrafish might be explained by the difference in maternal contribution of Shp2 in zebrafish eggs compared to mouse embryos.
Whereas double mutant zebrafish embryos are embryonic lethal, lack of ptpn11b in homozygous single mutant fish resulted in viable and fertile fish without developmental defects. Yet, ptpn11a-/- single mutant embryos did not survive, suggesting a distinct role of the two ptpn11 genes in zebrafish development. Biochemically, Shp2a and Shp2b have similar catalytic activities. However, expression analyses showed that ptpn11a was abundant in all developmental stages whereas ptpn11b was hardly detectable at early stages. Ptpn11b expression increased strongly over time. This may explain why the lack of ptpn11a affected development and lack of ptpn11b did not. Low levels of ptpn11b in ptpn11a-/- embryos at early stages cannot compensate for the lack of ptpn11a. Conversely, high levels of ptpn11a in ptpn11b-/- embryos can compensate for the lack of ptpn11b. Ptpn11a as well as ptpn11b rescued developmental defects in double mutant embryos, indicating that Shp2a and Shp2b have redundant functions. It is noteworthy that expression of exogenous ptpn11b rescued ptpn11a-/- embryos, demonstrating that the difference in developmental function of ptpn11a and ptpn11b is mainly caused by differences in expression levels during early development.
The developmental defects we observed in ptpn11a-/-ptpn11b-/- embryos became apparent relatively late during development. Only at 4-5 dpf the first morphological defects were visible. This is in contrast with earlier reports of morpholino (MO)-mediated knockdown of Shp2, where we and others observed developmental defects as early as gastrulation [26], [27]. The lack of gastrulation defects in ptpn11a-/-ptpn11b-/- embryos may be due to maternal contribution of ptpn11a in the double mutants.
The reduced body axis and craniofacial defects we observed in double mutants are comparable to the phenotypes of Shp2 knockdown embryos and of embryos expressing NS or LS variants of Shp2, that we and others reported earlier [26], [27]. It is evident that Shp2 has an important role in body axis extension and craniofacial development: short stature is one of the most recognized symptoms in individuals with NS and LS. During the first years of life the body length drops below the 10th percentile for height in NS [38] and below the 25th percentile in LS [39]. Knock-in mice expressing Shp2 variants of NS or LS show postnatal growth retardation [40], [41]. Moreover, human patients with NS or LS show visible craniofacial symptoms, including high forehead, hypertelorism, downslanting palpebral fissures, epicantal folds, ptosis, low-set and/or posteriorly rotated ears [5], [42], [43]. The ablation of Shp2 in neural crest cell leads to severe craniofacial anomalies in mice [44], [45]. Similarly, expression of morpholino Shp2 and Noonan or LEOPARD Shp2 embryos, induced craniofacial defects in zebrafish at 4dpf [26], [27]. Collectively, our results showed craniofacial defects and body axis extension defects in mutant zebrafish embryos, lacking functional Shp2, that are consistent with defects observed in mouse models lacking functional Shp2 in relevant cell types as well as with developmental defects in mouse models for NS or LS and symptoms in human individuals with NS or LS.
SHP2 positively regulates (ERK)/MAPK signal transduction [2] and downregulation of the (ERK)/MAPK pathway, due to the disruption of Shp2, leads to the formation of skeletal abnormalities and skull defects in mice [46], [47].We show that at 5dpf, pERK levels are downregulated in ptpn11a-/-ptpn11b-/- embryos, which is consistent with an essential role for Shp2 in Erk/MAPK signaling upstream of craniofacial development. However, even though in the ptpn11a-/- embryos pERK levels are comparable to wildtype, these embryos still showed craniofacial defects and reduced body length at 5dpf, albeit to a lesser extent than ptpn11a-/-ptpn11b-/- double mutant embryos. We cannot exclude the possibility that craniofacial development is independent of Erk/MAPK signaling. However, the ptpn11a-/- phenotype may be the consequence of reduced Erk/MAPK signaling at earlier stages, i.e. prior to 5 dpf. Ptpn11b is only expressed at low levels at these early developmental stages, which may not compensate for the loss of ptpn11a to sustain physiological levels of Shp2-dependent Erk/MAPK signaling at stages that are crucial for craniofacial development. At 5 dpf, ptpn11b expression is high and may compensate for the lack of Shp2a in ptpn11a-/- mutants with respect to Erk/MAPK signaling. To prove temporal dependence on Erk/MAPK signaling in ptpn11a-/- mutant embryos is not trivial as it requires assessment of Erk phosphorylation at early developmental stages, i.e. in embryos that do not yet display developmental defects. Collectively, our data show that functional Shp2 is required for Erk/MAPK signaling at 5 dpf and whether Erk/MAPK signaling is involved in the developmental defects remains to be determined definitively.
In conclusion, we characterized the two ptpn11 genes in zebrafish and found that the function of the two genes in development is distinct which is due to differences in their temporal expression patterns. The developmental defects that were observed in double mutant zebrafish embryos lacking functional Shp2 are consistent with developmental defects in mouse models lacking Shp2. The observed body axis extension and craniofacial defects are reminiscent of defects observed in mouse models for NS and LS, as well as the symptoms observed in human patients with NS and LS, underlining the importance of Shp2 for craniofacial development and body axis extension. The ptpn11 mutants that we have identified constitute a useful system to further unravel the function of Shp2 in development. Moreover, these mutants provide a good starting point for the development of genetic models for NS and LS in zebrafish that will complement other animal models for these syndromes.
Supporting Information
Comparison of human SHP2 and zebrafish Shp2a and Shp2b polypeptides. The alignment was done using ClustalW. Identical amino acids are highlighted in black. The consensus sequence is shown underlined, on top of the rows. Amino acids are numbered to the right of each row.
(PDF)
Funding Statement
This work was funded, in part, by a grant from the Research Council for Earth and Life Sciences (ALW 819.02.021) with financial aid from the Netherlands Organisation for Scientific Research (NWO). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dance M, Montagner A, Salles JP, Yart A, Raynal P (2008) The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 20: 453–459. [DOI] [PubMed] [Google Scholar]
- 2. Feng GS (1999) Shp-2 tyrosine phosphatase: signaling one cell or many. Exp Cell Res 253: 47–54. [DOI] [PubMed] [Google Scholar]
- 3. Neel BG, Gu H, Pao L (2003) The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28: 284–293. [DOI] [PubMed] [Google Scholar]
- 4. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, et al. (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29: 465–468. [DOI] [PubMed] [Google Scholar]
- 5. Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, et al. (2002) PTPN11 mutations in LEOPARD syndrome. J Med Genet 39: 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, et al. (2002) Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet 71: 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Quintana E, Rodriguez-Gonzalez F (2013) RASopathies: From Noonan to LEOPARD Syndrome. Rev Esp Cardiol. [DOI] [PubMed]
- 8. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, et al. (2006) Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311: 1287–1290. [DOI] [PubMed] [Google Scholar]
- 9.Zenker M (2009) Noonan Syndrome and Related Disorders. A matter of Deregulated Ras Signaling.; Schmid M, editor. Basel, Switzerland: Karger. 24 p. [Google Scholar]
- 10. Tartaglia M, Zampino G, Gelb BD (2010) Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol 1: 2–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanna N, Montagner A, Lee WH, Miteva M, Vidal M, et al. (2006) Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: consequences for PI3K binding on Gab1. FEBS Lett 580: 2477–2482. [DOI] [PubMed] [Google Scholar]
- 12. Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem 281: 6785–6792. [DOI] [PubMed] [Google Scholar]
- 13. Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, et al. (2006) Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 78: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu ZH, Xu J, Walls CD, Chen L, Zhang S, et al. (2013) Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J Biol Chem 288: 10472–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keilhack H, David FS, McGregor M, Cantley LC, Neel BG (2005) Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem 280: 30984–30993. [DOI] [PubMed] [Google Scholar]
- 16. Oishi K, Gaengel K, Krishnamoorthy S, Kamiya K, Kim IK, et al. (2006) Transgenic Drosophila models of Noonan syndrome causing PTPN11 gain-of-function mutations. Hum Mol Genet 15: 543–553. [DOI] [PubMed] [Google Scholar]
- 17. Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, et al. (2009) Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum Mol Genet 18: 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edouard T, Combier JP, Nedelec A, Bel-Vialar S, Metrich M, et al. (2010) Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3beta signaling. Mol Cell Biol 30: 2498–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Rocca Serra-Nedelec A, Edouard T, Treguer K, Tajan M, Araki T, et al. (2012) Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci U S A 109: 4257–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang W, Klaman LD, Chen B, Araki T, Harada H, et al. (2006) An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell 10: 317–327. [DOI] [PubMed] [Google Scholar]
- 21. Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, et al. (2007) Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol 27: 6706–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kontaridis MI, Yang W, Bence KK, Cullen D, Wang B, et al. (2008) Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal-regulated kinase/mitogen-activated protein kinase and RhoA signaling pathways. Circulation 117: 1423–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bard-Chapeau EA, Yuan J, Droin N, Long S, Zhang EE, et al. (2006) Concerted functions of Gab1 and Shp2 in liver regeneration and hepatoprotection. Mol Cell Biol 26: 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, et al. (2011) Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 19: 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang TL, Freeman RM Jr, O'Reilly AM, Neel BG, Sokol SY (1995) The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell 80: 473–483. [DOI] [PubMed] [Google Scholar]
- 26. Jopling C, van Geemen D, den Hertog J (2007) Shp2 knockdown and Noonan/LEOPARD mutant Shp2-induced gastrulation defects. PLoS Genet 3: e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stewart RA, Sanda T, Widlund HR, Zhu S, Swanson KD, et al. (2010) Phosphatase-dependent and -independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Dev Cell 18: 750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lieschke GJ, Currie PD (2007) Animal models of human disease: zebrafish swim into view. Nat Rev Genet 8: 353–367. [DOI] [PubMed] [Google Scholar]
- 29. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 30. Wienholds E, van Eeden F, Kosters M, Mudde J, Plasterk RH, et al. (2003) Efficient target-selected mutagenesis in zebrafish. Genome Res 13: 2700–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 32. Thisse C, Thisse B, Schilling TF, Postlethwait JH (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119: 1203–1215. [DOI] [PubMed] [Google Scholar]
- 33. van Eekelen M, Overvoorde J, van Rooijen C, den Hertog J (2010) Identification and expression of the family of classical protein-tyrosine phosphatases in zebrafish. PLoS One 5: e12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vandepoele K, De Vos W, Taylor JS, Meyer A, Van de Peer Y (2004) Major events in the genome evolution of vertebrates: paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc Natl Acad Sci U S A 101: 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, et al. (2004) Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431: 946–957. [DOI] [PubMed] [Google Scholar]
- 36. Nadler MJ, Chen B, Anderson JS, Wortis HH, Neel BG (1997) Protein-tyrosine phosphatase SHP-1 is dispensable for FcgammaRIIB-mediated inhibition of B cell antigen receptor activation. J Biol Chem 272: 20038–20043. [DOI] [PubMed] [Google Scholar]
- 37. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, et al. (1997) Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 16: 2352–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Noonan JA, Raaijmakers R, Hall BD (2003) Adult height in Noonan syndrome. Am J Med Genet A 123A: 68–71. [DOI] [PubMed] [Google Scholar]
- 39. Gorlin RJ, Anderson RC, Blaw M (1969) Multiple lentigenes syndrome. Am J Dis Child 117: 652–662. [DOI] [PubMed] [Google Scholar]
- 40. Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, et al. (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10: 849–857. [DOI] [PubMed] [Google Scholar]
- 41. Marin TM, Keith K, Davies B, Conner DA, Guha P, et al. (2011) Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest 121: 1026–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noonan JA (1968) Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am J Dis Child 116: 373–380. [DOI] [PubMed] [Google Scholar]
- 43. Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, et al. (2002) PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 70: 1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nakamura T, Gulick J, Colbert MC, Robbins J (2009) Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci U S A 106: 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakamura T, Gulick J, Pratt R, Robbins J (2009) Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc Natl Acad Sci U S A 106: 15436–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lapinski PE, Meyer MF, Feng GS, Kamiya N, King PD (2013) Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis Model Mech 6: 1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bauler TJ, Kamiya N, Lapinski PE, Langewisch E, Mishina Y, et al. (2011) Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis Model Mech 4: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of human SHP2 and zebrafish Shp2a and Shp2b polypeptides. The alignment was done using ClustalW. Identical amino acids are highlighted in black. The consensus sequence is shown underlined, on top of the rows. Amino acids are numbered to the right of each row.
(PDF)