

Abstract
Type 2 diabetes mellitus (T2DM) is a complex metabolic disease characterized by the loss of beta-cell secretory function and mass. The pathophysiology of beta-cell failure in T2DM involves a complex interaction between genetic susceptibilities and environmental risk factors. One environmental condition that is gaining greater appreciation as a risk factor for T2DM is the disruption of circadian rhythms (eg, shift-work and sleep loss). In recent years, circadian disruption has become increasingly prevalent in modern societies and consistently shown to augment T2DM susceptibility (partly mediated through its effects on pancreatic beta-cells). Since beta-cell failure is essential for development of T2DM, we will review current work from epidemiologic, clinical, and animal studies designed to gain insights into the molecular and physiological mechanisms underlying the predisposition to beta-cell failure associated with circadian disruption. Elucidating the role of circadian clocks in regulating beta-cell health will add to our understanding of T2DM pathophysiology and may contribute to the development of novel therapeutic and preventative approaches.
Keywords: Circadian rhythms, Circadian clocks, Circadian disruption, Hyperglycemia, Type 2 diabetes, Insulin secretion, Beta-cell mass, Oxidative stress, Beta-cell failure
Introduction
Accumulating evidence collected over the last few decades point to pancreatic beta-cell failure as one of the key pathophysiological events responsible for the development of Type 2 diabetes mellitus (T2DM) [1]. Thus, preservation of beta-cell health presents a critical barrier for the development of successful preventative and therapeutic strategies to combat the rise in diabetes prevalence. Beta-cell failure in patients with T2DM is multifaceted and manifests as (1) nearly absent insulin release in response to intravenous glucose challenge [2], (2) diminished insulin response following oral glucose or meal tolerance test [3], (3) reduced incretin-mediated amplification of insulin secretion [4], and (4) impaired insulin release in response to nonglucose secretagogues (ie, arginine) [5]. Given that insulin is preferentially released in discrete secretory pulses, patients with T2DM also exhibit a decline in the amplitude and orderliness of pulsatile insulin release [6]. Importantly, many of the characteristic features of beta-cell failure are seen “early” in the etiology of the disease (ie, impaired fasting glucose (IFG) and/or impaired glucose tolerance (IGT) stages [1], and even present in normoglycemic first degree relatives of patients with T2DM [7]. This suggests that beta-cell failure often precedes diabetes diagnosis and likely exhibits a strong genetic component. In support of this, recent genome-wide association studies (GWAS) have identified several T2DM-associated gene variants mechanistically involved in the regulation of insulin secretion [8].
The causes of beta-cell failure in T2DM are likely complex and multifactorial, but can be generally ascribed to an interplay between impaired beta-cell secretory function and the loss (or inappropriate formation) of beta-cell mass. The estimate of collective numbers of beta-cells in the pancreas is referred to as the beta-cell mass and the appropriate release of insulin by pancreatic beta-cells in response to secretory stimuli as the beta-cell secretory function (terms that will be used hereafter). The quantitative contribution of beta-cell dysfunction vs loss of beta-cell mass to T2DM onset and beta-cell failure remains an area of investigation [9].
A ~20 %–60 % deficit in beta-cell mass in T2DM has been documented by a number of independent groups utilizing postmortem autopsied pancreatic tissues for morphometric analysis [10–14]. In addition, beta-cell loss has been reported in pancreatic tissues collected from individuals with IFG and/or IGT, implying that the decline in beta-cell numbers is evident “early” in the evolution of diabetes, and deteriorates further with disease progression [10, 14]. Mechanisms underlying beta-cell loss in T2DM are likely multifaceted, but have been attributed in part to elevated beta-cell apoptosis reported in autopsied pancreatic tissues as well as protein lysates from T2DM islets [10, 14, 15]. Although the exact molecular pathways contributing to beta-cell apoptosis in T2DM remain to be elucidated, distinct mechanisms have been proposed including cytotoxicity due to prolonged exposure to high glucose levels (glucotoxicity) [16, 17], cytotoxicity due to high concentrations of free fatty acids (lipotoxicity) [18], and cytotoxicity due to intracellular formation of human islet amyloid polypeptide (h-IAPP) toxic oligomers (proteotoxicity) [19]. These mechanisms appear to converge to induce beta-cell apoptosis via activation of endoplasmic reticulum, oxidative, and/or inflammatory stress pathways [19–21].
Loss of beta-cell secretory function, akin to beta-cell loss, also precedes diabetes diagnosis and becomes more pronounced with disease progression [1]. Pancreatic beta-cells are uniquely designed to respond to changes in ambient glucose levels through complex coupling of glycolytic metabolism and insulin secretion [22]. Beta-cell glucose uptake is initiated via glucose transporter (GLUT)-2 (GLUT-1 in human beta-cells) facilitative glucose transporters, which are characterized by high Km for extracellular glucose and localized to the outer edges of the beta-cell plasma membrane. Upon cellular entry, glucose is phosphorylated by glucokinase (GK), a high Km (lower affinity) enzyme with kinetics that make it well suited to serve as a glucose sensor and permit changes in the intracellular glucose metabolism to reflect changes in the extracellular glucose concentrations [23]. Therefore, the extent of pyruvate production and its subsequent entry into the tricarboxylic acid (TCA) cycle is directly proportional to the postprandial rise in blood glucose [24]. In beta-cells, pyruvate almost exclusively enters the mitochondria to fuel TCA cycle reactions leading to generation of ATP [25]. The cellular rise in intracellular ATP/ADP ratio couples glucose oxidation with insulin exocytosis through closure of ATP-sensitive K+ (KATP) channels, resulting in membrane depolarization and Ca2+ influx, which triggers exocytosis of the readily releasable pool of insulin granules. In addition, insulin secretion can also be potentiated via various secondary messengers and metabolites such as cyclic AMP (cAMP), which trigger insulin exocytosis by activating protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC1) [26].
Mechanisms purported to mediate beta-cell secretory dysfunction in T2DM include (1) defects in beta-cell glucose transport and sensing, (2) defects in glucose oxidation and mitochondrial function, and (3) impairments in the insulin exocytosis machinery [27–31]. Furthermore, increased oxidative stress and generation of mitochondrial reactive oxygen species (ROS) in T2DM beta-cells contributes to secretory dysfunction by inactivating key beta-cell-specific genes and transcription factors purported to mediate insulin production, storage, and secretory apparatus (eg, Insulin (Ins), Pancreatic and duodenal homeobox 1 (Pdx1), Musculoaponeurotic fibrosarcoma A (MafA), and Homeobox protein (Nkx6.1)) [32].
Taken together, beta-cell failure is a prerequisite pathophysiological event in the development of T2DM. Diverse molecular and physiological mechanisms underlying beta-cell failure can be distilled to progressive decline in beta-cell mass and insulin secretory function, attributed in part to the activation of beta-cell endoplasmic reticulum, oxidative and inflammatory stress pathways. Having introduced the mechanisms underlying beta-cell failure in T2DM, we will next focus on the discussion of potential mechanisms that mediate increased susceptibility to beta-cell failure following circadian misalignment, an increasingly regarded risk factor for T2DM [33–35]. We will start our discussion with a review of the organization of the circadian system in mammals.
Organization of the Mammalian Circadian System
The circadian (Latin circa- around; diem- day) system is a fundamental property of nearly all living organisms. Circadian clocks are highly conserved endogenous transcriptional-translational time-keeping mechanisms that permit organisms ranging from bacteria to humans to synchronize behavioral, physiological, and molecular processes with the earth’s 24 hour light/dark cycle (LD]. These clocks can be set to local time or “entrained” by environmental stimuli called Zeitgebers (German “time giver” or “synchronizer”) most commonly light, temperature, food, etc; however, rhythms also persist in the absence of environmental cues [36]. This fundamental ability of clocks to “free-run” in constant conditions yet entrain to periodic external factors allows organisms to anticipate time of the day and generate daily rhythms in behavior (eg, sleep-wake cycles), physiology (eg, cortisol secretion), metabolism (eg, glycolytic metabolism), as well as regulate a multitude of gene/protein expression profiles.
In mammals, the circadian system is organized as a multilevel oscillator network. The pacemaker (or master clock) is located in the suprachiasmatic nucleus (SCN) of the hypothalamus, where it receives photic information from specialized ganglion cells in the retina, synchronizing the SCN clock to the solar day [37]. In fact, light is the most salient stimulus responsible for the entrainment of the SCN to daily changes in the LD cycle [38]. The SCN subsequently drives circadian rhythms throughout the body via a combination of neural, endocrine, and systemic outputs [39]. Molecular circadian oscillators are present in most mammalian cell types (including pancreatic beta-cells [40•]) and the SCN functions to synchronize the transcription of peripheral circadian clocks with changes in the LD cycle [41].
At the molecular level, the mammalian circadian clock is composed of several “clock genes” and proteins involved in rhythmic ~24 hour transcription-translation feedback loops [42]. In the positive limb of this feedback circuit, genes Circadian locomotor output cycles kaput (CLOCK) and Brain and muscle ARNT-like1 (Bmal1) encode basic helix-loop-helix Per-Arnt-Single-minded (bHLH-PAS) proteins that form CLOCK-BMAL1 activator complexes and initiate transcription by binding to specific DNA sequences (E-boxes) in the promoter regions of target genes [43]. One set of target genes include Period (Per1/2/3) and Cryptochrome (Cry1/2), which comprise the negative limb of the feedback loop. PER and CRY proteins form heterodimers and inhibit the transcriptional activation by CLOCK-BMAL1, allowing a new circadian cycle to repeat [43, 44]. In another coordinated feedback loop, CLOCK-Bmal1 heterodimers also initiate the rhythmic E-box mediated transcription of orphan nuclear receptor genes RevErbα/β and RORα/β [45]. REV-ERB and ROR proteins compete for retinoic acid-related orphan receptor response element (RORE) binding sites upstream of Bmal1 to inhibit or activate its transcription, respectively [45, 46]. Post-translational modifications of clock proteins such as phosphorylation, ubiquitination, and sumoylation greatly determine their stability and degradation, which plays a critical role in circadian cycle progression and setting the clock period. GWAS studies indicate that ~3 %–20 % of genes may be under circadian control and are purported to mediate an array of vital cellular processes and functions [47].
This complex multilevel circadian oscillator system undoubtedly provides an evolutionary advantage for human health and survival [35]; however, this system becomes disadvantageous when lifestyle factors impose time constrains that produce misalignment between internal circadian oscillators and the external environment (circadian disruption or misalignment). In recent decades, human population has become increasingly exposed to conditions associated with the disruption of normal circadian rhythms, which can be partly attributed to an increased reliance on shift-work, extended work schedules, advent of new media technologies with 24-hour news and entertainment, and technological advances in lighting leading to extensive light pollution [48]. As an example, 70 % of adults self-report inadequate sleep quality and duration, and millions of people are exposed to daily shift-work conditions in the United States alone [49–51]. Because the molecular clock sits at the absolute core of orchestrating an individual’s metabolism [34, 47, 52], disruption of normal circadian function has been reported to increase susceptibility to metabolic diseases, particularly T2DM [49, 53, 54, 55•, 56–60, 61•, 62, 63, 64•, 65, 66]. Next, we review current evidence associating circadian disruption, T2DM and beta-cell failure in humans.
Relationship Between Circadian Disruption, T2DM and Beta-Cell Failure
Insights From Human Studies
Numerous physiological functions are under direct control of the circadian system. In particular, regulation of glucose homeostasis in humans (glucose tolerance, insulin secretion, and insulin sensitivity) has been long known to exhibit robust circadian component [67–69]. The strongest evidence supporting the association between circadian disruption and T2DM comes from epidemiologic studies of individuals performing rotational shift-work across a variety of professional industries, who consistently show increased incidence of T2DM [53, 54, 55•, 56]. Epidemiologic studies also suggest that humans with disrupted/deficient sleep patterns exhibit an increased risk for T2DM, and sleep loss negatively impacts the treatment and management of diabetes in those already diagnosed [49, 57–60]. While these studies undoubtedly provide strong supporting evidence linking chronic exposure to circadian misalignment and T2DM in humans, a number of confounding environmental, socioeconomic, and health-related variables may have contributed to the observed positive associations. Furthermore, such studies are often unable to differentiate the mechanisms (ie, insulin secretion vs insulin sensitivity, etc) underlying increased susceptibility to T2DM under circadian disruption.
A number of studies examined the impact of sleep/circadian disruption on glucose homeostasis in humans under controlled laboratory settings [61•, 62, 63, 64•, 65, 66]. The classic study by Spiegel and colleagues [65] reported that 4 hours sleep deprivation per night for 5 days led to impaired glucose tolerance following the first breakfast meal, as well as in response to an intravenous glucose challenge (IVGTT). This data was the first clear demonstration that sleep loss may adversely affect beta-cell function and/or insulin sensitivity in humans [65, 70]. When beta-cell function was formally examined during the IVGTT, first phase insulin and c-peptide secretion were significantly diminished as reflected by ~40 % reduction in the disposition index (product of acute insulin repose and insulin sensitivity) [65]. A recent study also demonstrated glucose intolerance and decline in disposition index following 1 week of sleep restriction, although this effect was largely ascribed to a decrease in insulin sensitivity [62].
To gain insights into specific effects of circadian misalignment on glucose homeostasis, selected studies attempted to separate independent effects of behavioral and circadian cycles by exposing subjects to circadian misalignment, during which healthy adults ate, slept, and functioned 12 hours out of phase of their typical daily living schedules (shift-work paradigm) [61•, 64•]. It was reported that 1–3 weeks of circadian misalignment caused subjects to exhibit postprandial hyperglycemia and glucose intolerance, with a subset of individuals (~40 %) notably exhibiting glucose intolerance values classified as “prediabetic” according to current diagnostic criteria [64•]. More importantly, 3 weeks of circadian misalignment (with concurrent sleep restriction) resulted in deficient fasting and postprandial insulin secretion (despite significantly elevated prevailing glucose levels), which was normalized 9 days after circadian re-entrainment [61•]. Similar results were reported in another study where 3 weeks experimental exposure to nocturnal lifestyle resulted in loss of the positive association between postprandial glucose and insulin concentrations, characteristic of beta-cell failure [63].
Additional support for role of circadian system in glucose homeostasis and beta-cell health comes from GWAS studies. Recent GWAS studies have demonstrated an association between genetic variants in key circadian genes (eg, Cry2 and MTNR1B) and increased susceptibility to T2DM risk in humans [71, 72]. For example, a variant in a key component of the core circadian clock (Cry2) has been shown to be associated with increased fasting glucose levels and a decline in beta-cell function rendering increased risk of T2DM [71]. Perhaps the strongest genetic evidence supporting the role of circadian system in regulation of beta-cell health comes from GWAS studies that identified a link between a variance in the melatonin receptor 2 (encoded by MTNR1B gene) and increased risk for T2DM and impaired beta-cell function [72–74]. The gene linkage between MTNR1B and susceptibility to beta-cell failure has been extensively reproduced in many ethnically diverse cohorts worldwide and implies an important role for melatonin (key circadian hormone) in the regulation of beta cell function and/or mass in humans.
In summary, accumulating human studies suggest an association between prolonged occupational exposure to shift-work and T2DM. Additionally, individuals experiencing short or disrupted sleep patterns show increased susceptibility to T2DM. In controlled clinical studies, acute 1–3 weeks circadian misalignment (alone) or in combination with sleep restriction results in dysregulation of glucose homeostasis and consequent glucose intolerance attributed in part to loss of beta-cell function (as well as decline in insulin sensitivity). Moreover, presence of circadian gene variants increases T2DM risk through predisposition to beta-cell failure. Whether chronic exposure to circadian disruption (or presence of circadian gene variants) in humans can contribute to beta-cell failure through beta-cell loss is presently unknown, largely due to the inability to accurately assess beta-cell mass in vivo. Human studies are also unable to fully elucidate the potential molecular and cellular mechanisms that may be responsible for induction of beta-cell failure in response circadian misalignment. Such limitations can be partially addressed by utilizing animal research models [40•, 75–77, 78•, 79–81, 82•].
Insights From Animal Studies
Studies in rodent models support the premise that disruption of circadian rhythms leads to impaired glucose homeostasis and beta-cell failure, culminating in increased susceptibility to T2DM [40•, 75–77, 78•, 79–81, 82•] (Table 1)). The original observation that Clock mutant mice exhibit diurnal hyperglycemia provided the first direct evidence that disruption of the molecular circadian clock impairs glycemic control in vivo [80]. In addition to increased body weight and prevailing hyperlipidemia, these mice were hypoinsulinemic, which is indicative of beta-cell failure that was subsequently confirmed by examining glucose-stimulated insulin secretion (GSIS) in isolated islets [40•]. Subsequent studies confirmed that global (whole body) mutations or deletions of other key components of the molecular clock (ie, Bmal1, Per1, and Per2) disrupt normal glucose homeostasis through induction of hyperglycemia and glucose intolerance, the latter associated with diminished GSIS [76, 77].
Table 1.
Table summarizing current research in rodent models of genetic and environmental circadian disruption on glucose metabolism and β-cell phenotype
| Model | Circadian disruption | Glucose metabolism | Islet/β-cell phenotype | Reference |
|---|---|---|---|---|
| Mice | ClockΔ19/Δ19 | ↑ RPG | – | Turek FW, et al. [80] |
| Mice |
Per1−/− Per2−/− |
↓ GT | – | Lamia KA, et al. [76] |
| Mice | ClockΔ19/Δ19 | ↑ FPG ↑ RPG ↓ GT |
↓ GSIS (in vitro) ↓ islet size ↓ islet proliferation |
Marcheva B, et al. [40•] |
| Bmal1−/− | ↓ GSIS (in vitro) ↓ islet size |
|||
| Pdx<Bmal1−/−(Pancreas) | ↑ RPG ↓ GT |
↓ GSIS (in vitro) | ||
| Mice | Bmal1−/− | ↑ FPG ↓ GT |
↓ GSIS (in vivo) ↓ GSIS (in vitro) |
Lee J, et al. [77] |
| Mice | Pdx<Bmal1−/−(Pancreas) | ↓ GT | ↓ GSIS (in vitro) | Sadacca LA, et al. [79] |
| HIP Rats | Constant light (LL); | ↑ FPG | ↓ GSIS (in vivo) | Gale JE, et al. [75] |
| 6 h phase advance in LD,12:12 | ↓ β-cell area ↑ β-cell apoptosis |
|||
| Mice; MIN6 cells |
Rev-erbα siRNA | ↓ GSIS (in vitro) ↓ β-cell proliferation |
Vieira E, et al. [81] | |
| Mice | RIP<Bmal1−/− (β-cells) | ↓ GT | ↓ antioxidant genes | Lee J, et al. [82•] |
| 6 h phase advance in LD,12:12 | ↑ FPG | ↓ GSIS (in vivo) | ||
| Rats | Constant light (LL) | ↓ GSIS (in vitro) | Qian J, et al. [78•] |
FPG Fasting plasma glucose, RPG Random plasma glucose, GT Glucose tolerance, GSIS Glucose-stimulated insulin secretion
Since molecular clocks regulate a multitude of physiological functions, beta-cell and pancreas-specific mutants have been developed to investigate whether, and through which mechanisms, targeted disruption of beta-cell molecular clock results in beta-cell failure [40•, 82•]. Over the last few years, these studies have clearly demonstrated that, (1) pancreatic islets express self-sustained oscillations of clock genes, which are disrupted by clock gene mutations, (2) disruption of the beta-cell circadian clock leads to hyperglycemia and overt glucose-intolerance, and (3) genetic disruption of the beta-cell circadian clock leads to impaired GSIS both in vitro and in vivo (Table 1). The molecular mechanisms underlying beta-cell failure in clock mutants are under investigation and likely involve a combination of compromised beta-cell secretory function and mass [40•, 82•]. For instance, micro-array analysis revealed that islets isolated from Clock mutant mice exhibit altered expression of genes such as CyclinD1, Pdx1, and NeuroD1, known to regulate islet growth, survival, maturation, and proliferation, suggesting an involvement in the regulation of beta-cell mass and survival [40•]. Indeed, Clock mutant mice showed reduced islet size as well as a decline in beta-cell proliferation [40•]. Interestingly, primary beta-cells treated with siRNA for Rev-Erbα (another key component of the circadian clock) show diminished rate of beta-cell proliferation [81]. In addition to the potential abnormalities in beta-cell mass, Clock mutant mice exhibit distinct beta-cell secretory functional impairments in insulin vesicle trafficking, membrane fusion, and processing [40•].
Studies in beta-cell specific Bmal1 knockout mice indicate that beta-cell failure consequent to beta-cell clock disruption, is attributed to reduced antioxidant gene expression, mitochondrial dysfunction, and oxidative stress-induced mitochondrial uncoupling as evidenced by the upregulation of mitochondrial uncoupling protein 2 (UCP2) [77, 82•]. UCP2 is believed to function as a negative regulator of mitochondrial ROS production and its upregulation may be an adaptive response to increased ROS in the beta-cells [83]. Interestingly, Ucp2 mRNA shows daily oscillations in islets, indicating direct control by the circadian clock [82•].
Moreover, BMAL1 directly binds to E-box elements in the cis-promoter regions of nuclear factor erythroid 2-related factor 2 (Nrf2), a known master antioxidant regulatory factor [82•]. Therefore, BMAL1 and by extension the molecular clock in beta-cells, appears to orchestrate cellular antioxidant responses by regulating NRF2 levels, which subsequently activates a battery of downstream protective enzymes [84]. This would support the hypothesis that an intact circadian system upkeeps beta-cell homeostasis by providing resilience to oxidative stress, and scavenging excess ROS by the activation of several antioxidant genes [85]. It is, therefore, plausible to hypothesize that circadian disruption can increase the susceptibility to oxidative stress and consequent beta-cell failure, particularly in context of gluco-/lipotoxicity or increased expression of h-IAPP (the known inducers of beta-cell oxidative stress in T2DM). Whereas, an intact beta-cell circadian clock may play a protective role against beta-cell failure due to the robust antioxidant cellular defenses now known to be at least in part orchestrated by the cellular circadian clock [84].
It is currently unknown whether environment-induced circadian misalignment in humans (ie, shift-work, jetlag, and constant light) contributes to beta-cell failure through disruption of the beta-cell circadian clock. However, recent animal studies have provided evidence that simulation of light-induced circadian misalignment recapitulates metabolic and molecular defects observed in beta-cell specific clock gene mutants [75, 78•, 82•]. Specifically, circadian misalignment induced by 10 weeks exposure to constant light significantly alters the islet circadian clock function through impairment in the amplitude, phase, and inter-islet synchrony of clock gene oscillations [78•]. Consequently, exposure to constant light accelerated the development of hyperglycemia and beta-cell failure in diabetes-prone human islet amyloid transgenic (HIP) rats, observed independent of their weight gain [75]. Beta-cell failure in circadian-disrupted HIP rats was due to accelerated loss of both beta-cell secretory function and mass, as well as increased beta-cell apoptosis [75]. Since beta-cell circadian clock has been recently shown to orchestrate cellular response to oxidative stress [82•], loss of antioxidant cellular defenses due to circadian disruption would accelerate the cellular attrition associated with overexpression of h-IAPP. Similarly, the simulation of shift-work conditions for 8 weeks in mice also resulted in development of fasting hyperglycemia associated with the impaired GSIS [82•]. Importantly, as in beta-cell specific BMAL-1 knockout mice, simulation of shift-work conditions led to islet mitochondrial dysfunction and increased susceptibility to oxidative stress [82•]. Taken together, evidence from both genetic and environmental models of circadian disruption in rodents supports the premise that circadian misalignment leads to loss of glycemic control in vivo associated with diminished beta-cell secretory function and mass (mediated in part through increased susceptibility to oxidative stress).
Conclusions
The incidence of T2DM has reached an epidemic proportion worldwide. T2DM is a complex metabolic disease characterized by fasting and postprandial hyperglycemia partly due to induction of pancreatic beta-cell failure, which manifests as a deficit in beta-cell mass and function. Lifestyle factors such as lack of exercise and high-fat diet have long been known to increase the risk for T2DM. Emerging evidence suggests that disturbance of normal circadian rhythms is another lifestyle factor that may contribute to T2DM development. In recent years, environmental conditions associated with disruption of circadian rhythms (eg, shift-work, sleep loss, light at night, etc) have become increasingly prevalent and reported to significantly augment T2DM susceptibility partly through its effects on the beta-cell.
The circadian system is a fundamental property of nearly all living organisms including humans, and intracellular molecular clocks synchronize individuals’ biological processes to changes in LD cycles. Molecular clocks are present in most tissues (including beta-cells) and control a multitude of cellular functions, including the regulation of cellular metabolism and response to oxidative stress. A number of recent studies in rodent models with clock gene mutations as well as environment-induced circadian disruption were recently undertaken to delineate potential mechanisms of increased susceptibility to T2DM and beta-cell failure (Table 1). These studies report that mechanisms underlying circadian disruption-induced beta-cell failure likely include (1) defective insulin secretory function due to impairments in insulin vesicle trafficking, membrane fusion and processing, (2) altered rate of beta-cell growth, proliferation, and survival, and (3) increased beta-cell attrition due to augmented susceptibility to oxidative stress (Fig. 1). Understanding the molecular and physiological mechanisms responsible for circadian disruption-associated risk of T2DM warrants further research, and holds potential for contributing to the development of novel therapeutic and preventative strategies.
Fig. 1.
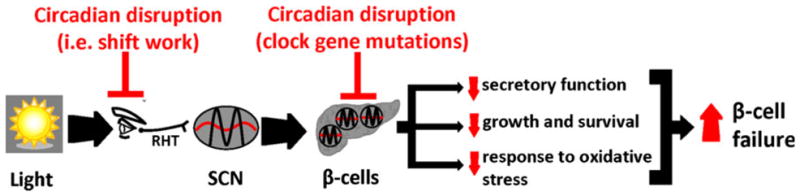
Schematic representation illustrating potential mechanisms by which circadian rhythm disruption increases susceptibility to beta-cell failure in T2DM. Changes in LD cycle are perceived by specialized ganglion cells in the retina, synchronizing the central pacemaker of the circadian system in the SCN to the solar day. The SCN subsequently functions to synchronize the transcription of peripheral circadian clocks (including beta-cell clocks) with changes in the LD cycle. Circadian rhythm disruption due to environment-induced circadian misalignment (eg, shift work) or mutations in genes comprising the core circadian oscillator network leads to beta-cell failure and T2DM onset. Mechanisms underlying circadian disruption-induced beta-cell failure likely include (1) defective insulin secretory function, (2) altered rate of beta-cell growth, proliferation, and plausibly survival, and (3) increased beta-cell attrition due to augmented susceptibility to oxidative stress
Footnotes
Conflict of Interest Kuntol Rakshit declares that he has no conflict of interest. Anthony P. Thomas declares that he has no conflict of interest. Aleksey V. Matveyenko declares that he has no conflict of interest.
Compliance with Ethics Guidelines
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.DeFronzo RA, Abdul-Ghani MA. Preservation of beta-cell function: the key to diabetes prevention. J Clin Endocrinol Metab. 2011;96:2354–66. doi: 10.1210/jc.2011-0246. [DOI] [PubMed] [Google Scholar]
- 2.Brunzell JD, Robertson RP, Lerner RL, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab. 1976;42:222–9. doi: 10.1210/jcem-42-2-222. [DOI] [PubMed] [Google Scholar]
- 3.Seltzer HS, Allen EW, Herron AL, Jr, et al. Insulin secretion in response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. J Clin Invest. 1967;46:323–35. doi: 10.1172/JCI105534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hojberg PV, Vilsboll T, Rabol R, et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia. 2009;52:199–207. doi: 10.1007/s00125-008-1195-5. [DOI] [PubMed] [Google Scholar]
- 5.Ward WK, Bolgiano DC, McKnight B, et al. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–28. doi: 10.1172/JCI111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porksen N, Hollingdal M, Juhl C, et al. Pulsatile insulin secretion: detection, regulation, and role in diabetes. Diabetes. 2002;51 (Suppl 1):S245–54. doi: 10.2337/diabetes.51.2007.s245. [DOI] [PubMed] [Google Scholar]
- 7.Pimenta W, Korytkowski M, Mitrakou A, et al. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA. 1995;273:1855–61. [PubMed] [Google Scholar]
- 8.Florez JC. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia. 2008;51:1100–10. doi: 10.1007/s00125-008-1025-9. [DOI] [PubMed] [Google Scholar]
- 9.Meier JJ, Bonadonna RC. Role of reduced beta-cell mass versus impaired beta-cell function in the pathogenesis of type 2 diabetes. Diabetes Care. 2013;36 (Suppl 2):S113–9. doi: 10.2337/dcS13-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler AE, Janson J, Bonner-Weir S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 11.Rahier J, Guiot Y, Goebbels RM, et al. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10 (Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 12.Sakuraba H, Mizukami H, Yagihashi N, et al. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 13.Yoon KH, Ko SH, Cho JH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–8. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 14.Yoneda S, Uno S, Iwahashi H, et al. Predominance of beta-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J Clin Endocrinol Metab. 2013;98:2053–61. doi: 10.1210/jc.2012-3832. [DOI] [PubMed] [Google Scholar]
- 15.Marchetti P, Del Guerra S, Marselli L, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535–41. doi: 10.1210/jc.2004-0150. [DOI] [PubMed] [Google Scholar]
- 16.Maedler K, Sergeev P, Ris F, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–60. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–4. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 18.Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–42. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 19.Haataja L, Gurlo T, Huang CJ, et al. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29:303–16. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu L, Sauter NS, Schulthess FT, et al. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57:645–53. doi: 10.2337/db07-0847. [DOI] [PubMed] [Google Scholar]
- 21.Newsholme P, Haber EP, Hirabara SM, et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and nonmitochondrial ROS production and activity. J Physiol. 2007;583:9–24. doi: 10.1113/jphysiol.2007.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuit F, De Vos A, Farfari S, et al. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem. 1997;272:18572–9. doi: 10.1074/jbc.272.30.18572. [DOI] [PubMed] [Google Scholar]
- 23.Matschinsky FM, Glaser B, Magnuson MA. Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes. 1998;47:307–15. doi: 10.2337/diabetes.47.3.307. [DOI] [PubMed] [Google Scholar]
- 24.Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39:647–52. doi: 10.2337/diab.39.6.647. [DOI] [PubMed] [Google Scholar]
- 25.Ishihara H, Wang H, Drewes LR, et al. Overexpression of mono-carboxylate transporter and lactate dehydrogenase alters insulin secretory responses to pyruvate and lactate in beta cells. J Clin Invest. 1999;104:1621–9. doi: 10.1172/JCI7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–42. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 27.Andersson SA, Olsson AH, Esguerra JL, et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol Cell Endocrinol. 2012;364:36–45. doi: 10.1016/j.mce.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Del Guerra S, Lupi R, Marselli L, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–35. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 29.MacDonald MJ, Longacre MJ, Langberg EC, et al. Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia. 2009;52:1087–91. doi: 10.1007/s00125-009-1319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414:807–12. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 31.Rosengren AH, Braun M, Mahdi T, et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61:1726–33. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo S, Dai C, Guo M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. 2013 doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva CM, Sato S, Margolis RN. No time to lose: workshop on circadian rhythms and metabolic disease. Genes Dev. 2010;24:1456–64. doi: 10.1101/gad.1948310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–54. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy AB, O’Neill JS. Healthy clocks, healthy body, healthy mind. Trends Cell Biol. 2010;20:36–44. doi: 10.1016/j.tcb.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–41. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 37.Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295:1070–3. doi: 10.1126/science.1067262. [DOI] [PubMed] [Google Scholar]
- 38.Hattar S, Liao HW, Takao M, et al. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–70. doi: 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buijs RM, Kalsbeek A. Hypothalamic integration of central and peripheral clocks. Nat Rev Neurosci. 2001;2:521–6. doi: 10.1038/35081582. [DOI] [PubMed] [Google Scholar]
- 40•.Marcheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–31. doi: 10.1038/nature09253. First demonstration that targeted disruption of beta-cell molecular clock results in beta-cell failure and T2DM. The study was also the first to show that (1) pancreatic islets express self-sustained oscillations of clock genes, (2) disruption of the beta-cell circadian clock leads to hyperglycemia and overt glucose-intolerance, and (3) genetic disruption of the beta-cell circadian clock leads to impaired GSIS both in vitro and in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saini C, Suter DM, Liani A, et al. The mammalian circadian timing system: synchronization of peripheral clocks. Cold Spring Harb Symp Quant Biol. 2011;76:39–47. doi: 10.1101/sqb.2011.76.010918. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi JS, Hong HK, Ko CH, et al. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gekakis N, Staknis D, Nguyen HB, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 44.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–90. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 45.Preitner N, Damiola F, Lopez-Molina L, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–60. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 46.Guillaumond F, Dardente H, Giguere V, et al. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J Biol Rhythm. 2005;20:391–403. doi: 10.1177/0748730405277232. [DOI] [PubMed] [Google Scholar]
- 47.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–42. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyse CA, Selman C, Page MM, et al. Circadian desynchrony and metabolic dysfunction: did light pollution make us fat? Med Hypotheses. 2011;77:1139–44. doi: 10.1016/j.mehy.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 49.Beihl DA, Liese AD, Haffner SM. Sleep duration as a risk factor for incident type 2 diabetes in a multiethnic cohort. Ann Epidemiol. 2009;19:351–7. doi: 10.1016/j.annepidem.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 50.US Congress OTA. Biological rythms: implications for the worker, OTA-BA-463. Washington DC: US Government Printing Office; 1991. [Google Scholar]
- 51.Basner M, Fomberstein KM, Razavi FM, et al. American time use survey: sleep time and its relationship to waking activities. Sleep. 2007;30:1085–95. doi: 10.1093/sleep/30.9.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rutter J, Reick M, McKnight SL. Metabolism and the control of circadian rhythms. Annu Rev Biochem. 2002;71:307–31. doi: 10.1146/annurev.biochem.71.090501.142857. [DOI] [PubMed] [Google Scholar]
- 53.Kroenke CH, Spiegelman D, Manson J, et al. Work characteristics and incidence of type 2 diabetes in women. Am J Epidemiol. 2007;165:175–83. doi: 10.1093/aje/kwj355. [DOI] [PubMed] [Google Scholar]
- 54.Mikuni E, Ohoshi T, Hayashi K, et al. Glucose intolerance in an employed population. Tohoku J Exp Med. 1983;141(Suppl):251–6. doi: 10.1620/tjem.141.suppl_251. [DOI] [PubMed] [Google Scholar]
- 55•.Pan A, Schernhammer ES, Sun Q, et al. Rotating night shift work and risk of type 2 diabetes: 2 prospective cohort studies in women. PLoS Med. 2011;8:e1001141. doi: 10.1371/journal.pmed.1001141. The largest and most extensive prospective cohort study to date with ~20-year follow-up, found an increased risk of T2DM following chronic exposure to rotating shift work in women. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suwazono Y, Dochi M, Oishi M, et al. Shiftwork and impaired glucose metabolism: a 14-year cohort study on 7104 male workers. Chronobiol Int. 2009;26:926–41. doi: 10.1080/07420520903044422. [DOI] [PubMed] [Google Scholar]
- 57.Mallon L, Broman JE, Hetta J. High incidence of diabetes in men with sleep complaints or short sleep duration: a 12-year follow-up study of a middle-aged population. Diabetes Care. 2005;28:2762–7. doi: 10.2337/diacare.28.11.2762. [DOI] [PubMed] [Google Scholar]
- 58.Meisinger C, Heier M, Loewel H. Sleep disturbance as a predictor of type 2 diabetes mellitus in men and women from the general population. Diabetologia. 2005;48:235–41. doi: 10.1007/s00125-004-1634-x. [DOI] [PubMed] [Google Scholar]
- 59.Nilsson PM, Roost M, Engstrom G, et al. Incidence of diabetes in middle-aged men is related to sleep disturbances. Diabetes Care. 2004;27:2464–9. doi: 10.2337/diacare.27.10.2464. [DOI] [PubMed] [Google Scholar]
- 60.Yaggi HK, Araujo AB, McKinlay JB. Sleep duration as a risk factor for the development of type 2 diabetes. Diabetes Care. 2006;29:657–61. doi: 10.2337/diacare.29.03.06.dc05-0879. [DOI] [PubMed] [Google Scholar]
- 61•.Buxton OM, Cain SW, O’Connor SP, et al. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med. 2012;4:129ra143. doi: 10.1126/scitranslmed.3003200. The first clear demonstration that exposure to circadian misalignment (with concurrent sleep restriction) for 3 weeks in otherwise healthy humans results in loss of appropriate beta-cell function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buxton OM, Pavlova M, Reid EW, et al. Sleep restriction for 1 week reduces insulin sensitivity in healthy men. Diabetes. 2010;59:2126–33. doi: 10.2337/db09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin LQ, Li J, Wang Y, et al. The effects of nocturnal life on endocrine circadian patterns in healthy adults. Life Sci. 2003;73:2467–75. doi: 10.1016/s0024-3205(03)00628-3. [DOI] [PubMed] [Google Scholar]
- 64•.Scheer FA, Hilton MF, Mantzoros CS, et al. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 2009;106:4453–8. doi: 10.1073/pnas.0808180106. Study reports that 10 days circadian misalignment caused the subjects to exhibit postprandial hyperglycemia and glucose intolerance, with a subset of individuals (~40 %) notably exhibiting glucose intolerance values classified as “prediabetic” according to the current diagnostic criteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–9. doi: 10.1016/S0140-6736(99)01376-8. [DOI] [PubMed] [Google Scholar]
- 66.Gonnissen HK, Rutters F, Mazuy C, et al. Effect of a phase advance and phase delay of the 24-hour cycle on energy metabolism, appetite, and related hormones. Am J Clin Nutr. 2012;96:689–97. doi: 10.3945/ajcn.112.037192. [DOI] [PubMed] [Google Scholar]
- 67.Boden G, Ruiz J, Urbain JL, et al. Evidence for a circadian rhythm of insulin secretion. Am J Physiol. 1996;271:E246–52. doi: 10.1152/ajpendo.1996.271.2.E246. [DOI] [PubMed] [Google Scholar]
- 68.Freinkel N, Mager M, Vinnick L. Cyclicity in the interrelationships between plasma insulin and glucose during starvation in normal young men. J Lab Clin Med. 1968;71:171–8. [PubMed] [Google Scholar]
- 69.Saad A, Dalla Man C, Nandy DK, et al. Diurnal pattern to insulin secretion and insulin action in healthy individuals. Diabetes. 2012;61:2691–700. doi: 10.2337/db11-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spiegel K, Knutson K, Leproult R, et al. Sleep loss: a novel risk factor for insulin resistance and type 2 diabetes. J Appl Physiol. 2005;99:2008–19. doi: 10.1152/japplphysiol.00660.2005. [DOI] [PubMed] [Google Scholar]
- 71.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–16. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lyssenko V, Nagorny CL, Erdos MR, et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet. 2009;41:82–8. doi: 10.1038/ng.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prokopenko I, Langenberg C, Florez JC, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41:77–81. doi: 10.1038/ng.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. 2009;41:89–94. doi: 10.1038/ng.277. [DOI] [PubMed] [Google Scholar]
- 75.Gale JE, Cox HI, Qian J, et al. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J Biol Rhythm. 2011;26:423–33. doi: 10.1177/0748730411416341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 2008;105:15172–7. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee J, Kim MS, Li R, et al. Loss of Bmal1 leads to uncoupling and impaired glucose-stimulated insulin secretion in beta-cells. Islets. 2011;3:381–8. doi: 10.4161/isl.3.6.18157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78•.Qian J, Block GD, Colwell CS, et al. Consequences of exposure to light at night on the pancreatic islet circadian clock and function in rats. Diabetes. 2013;62:3469–78. doi: 10.2337/db12-1543. This study first demonstrated that circadian misalignment induced by 10 weeks exposure to constant light significantly alters the islet circadian clock function through impairment in the amplitude, phase, and inter-islet synchrony of clock gene oscillations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sadacca LA, Lamia KA, de Lemos AS, et al. An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia. 2011;54:120–4. doi: 10.1007/s00125-010-1920-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science. 2005;308:1043–5. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vieira E, Marroqui L, Batista TM, et al. The clock gene Reverbalpha regulates pancreatic beta-cell function: modulation by leptin and high-fat diet. Endocrinology. 2012;153:592–601. doi: 10.1210/en.2011-1595. [DOI] [PubMed] [Google Scholar]
- 82•.Lee J, Moulik M, Fang Z, et al. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol Cell Biol. 2013;33:2327–38. doi: 10.1128/MCB.01421-12. This work was the first to report that beta-cell failure consequent to beta-cell clock disruption is attributed to reduced antioxidant gene expression, mitochondrial dysfunction, and oxidative stress-induced mitochondrial uncoupling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pi J, Collins S. Reactive oxygen species and uncoupling protein 2 in pancreatic beta-cell function. Diabetes Obes Metab. 2010;12 (Suppl 2):141–8. doi: 10.1111/j.1463-1326.2010.01269.x. [DOI] [PubMed] [Google Scholar]
- 84.Wilking M, Ndiaye M, Mukhtar H, et al. Circadian rhythm connections to oxidative stress: implications for human health. Antioxid Redox Signal. 2013;19:192–208. doi: 10.1089/ars.2012.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kondratov RV, Kondratova AA, Gorbacheva VY, et al. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006;20:1868–73. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]