

Abstract
Mammography is an important screening modality for the early detection of DCIS and breast cancer lesions. More specifically, high mammographic density is associated with an increased risk of breast cancer. However, the biological processes underlying this phenomenon remain largely unknown. Here, we re-interrogated genome-wide transcriptional profiling data obtained from low-density (LD) mammary fibroblasts (n = 6 patients) and high-density (HD) mammary fibroblasts (n = 7 patients) derived from a series of 13 female patients. We used these raw data to generate a “breast density” gene signature consisting of >1250 transcripts that were significantly increased in HD fibroblasts, relative to LD fibroblasts. We then focused on the genes that were increased by ≥ 1.5-fold (P < 0.05) and performed gene set enrichment analysis (GSEA), using the molecular signatures database (MSigDB). Our results indicate that HD fibroblasts show the upregulation and/or hyper-activation of several key cellular processes, including the stress response, inflammation, stemness, and signal transduction. The transcriptional profiles of HD fibroblasts also showed striking similarities to human tumors, including head and neck, liver, thyroid, lung, and breast cancers. This may reflect functional similarities between cancer-associated fibroblasts (CAFs) and HD fibroblasts. This is consistent with the idea that the presence of HD fibroblasts may be a hallmark of a pre-cancerous phenotype. In these biological processes, GSEA predicts that several key signaling pathways may be involved, including JNK1, iNOS, Rho GTPase(s), FGF-R, EGF-R, and PDGF-R-mediated signal transduction, thereby creating a pro-inflammatory, pro-proliferative, cytokine, and chemokine-rich microenvironment. HD fibroblasts also showed significant overlap with gene profiles derived from smooth muscle cells under stress (JNK1) and activated/infected macrophages (iNOS). Thus, HD fibroblasts may behave like activated myofibroblasts and macrophages, to create and maintain a fibrotic and inflammatory microenvironment. Finally, comparisons between the HD fibroblast gene signature and breast cancer tumor stroma revealed that JNK1 stress signaling is the single most significant biological process that is shared between these 2 data sets (with P values between 5.40E-09 and 1.02E-14), and is specifically associated with tumor recurrence. These results implicate “stromal JNK1 signaling” in the pathogenesis of human breast cancers and the transition to malignancy. Augmented TGF-β signaling also emerged as a common feature linking high breast density with tumor stroma and breast cancer recurrence (P = 5.23E-05). Similarities between the HD fibroblast gene signature, wound healing, and the cancer-associated fibroblast phenotype were also noted. Thus, this unbiased informatics analysis of high breast density provides a novel framework for additional experimental exploration and new hypothesis-driven breast cancer research, with a focus on cancer prevention and personalized medicine.
Keywords: EGF, FGF, JNK, PDGF, SAPK, TGF-beta, breast cancer, cancer-associated fibroblasts, fibrosis, gene signature, inflammation, mammographic density, mammography, microenvironment, stress signaling, tumor stroma, wound healing
Introduction
Apart from mutations in dominant breast cancer genes, high mammographic density is one of the strongest risk factors for breast cancer.1 Only advanced age and BRCA1/2 mutations are stronger risk factors. Women with >70% of the breast appearing dense have an increased relative risk of developing breast cancer of ≥ 6-fold, compared with women with low density. From twin studies, it is estimated that about two thirds of breast tissue is the result of inheritance: the other third is modifiable.2
Density is increased in women with known risk factors for breast cancer, such as a late first pregnancy and the use of hormone replacement therapy and is substantially decreased in some women who take the anti-estrogen, Tamoxifen. Risk of breast cancer is reduced in women who have a 10% or more reduction of density, over a period of a year or more.3
Tamoxifen's effect on breast stroma was demonstrated in mice, which showed a reduction of enzyme activity and of fragmentation of stromal components, such a fibronectin and collagen III. Whether this is a direct effect on the stroma or indirect by inhibition of epithelia cell activity is not clear, but this study indicates the marked activity of the stroma during mouse cycles and reduction by Tamoxifen.4
Dense tissue is mainly collagen associated, with increased proteoglycan deposition and immuno-staining for growth factors, such as insulin type growth factor 1, whereas non-dense tissue is largely fat.5 Some studies indicate that dense tissue contains a greater concentration of epithelial cells than non-dense tissue and this suggests one mechanism for increased risk.6 Another is that collagen increases the stiffness of the breast, inducing the proliferation and expansion of normal and abnormal epithelial cells and there is evidence that this effect is increased by the particular orientation and degree of cross-linking of collagen.7
There is not only a need to understand the mechanism(s) of the effect of mammographic density on breast cancer risk, but also to develop novel biomarkers which may indicate risk and the response to therapies, such as Tamoxifen. Not all women with high density develop breast cancer and not all respond to Tamoxifen. A potential marker of risk (CD36) was recently reported by the Tlsty laboratory.8
Yang et al., 2010 and colleagues9 reported a reduction in expression of the TGF-β gene and an increase in Cox-2 in high density tissue (with histologically normal epithelia) in dense tissue taken 5-cm or more away from an infiltrating carcinoma, compared with low density cancer associated stroma. However, it remains largely unknown what biological processes are involved in high mammographic density and its effects on breast cancer risk.
Here, we present a new bioinformatics analysis suggesting that high mammographic density fibroblasts promote breast cancer, by creating an inflammatory microenvironment, secondary to activated JNK1 stress kinase signaling.
These findings will now allow us to create: (1) new innovative cellular models; and (2) pre-clinical animal models of high mammographic density, to mimic the behavior of HD fibroblasts, and to explore their mechanistic role in breast cancer pathogenesis.
Consistent with our current findings in the breast, the stroma of human colonic adenomatous polyps shows the hyper-activation of both JNK and NFkB,10 while adjacent intestinal epithelial cells remain negative. These adenomatous polyps are thought to represent pre-cancerous lesions.
Results
High mammographic density is associated with an increased risk of breast cancer. Unfortunately, the biological processes underlying this phenomenon remain largely unknown. Understanding the biology underpinning high mammographic density has important implications for both breast cancer diagnosis and prevention.
Gene set enrichment analysis (GSEA) reveals the biological processes activated in HD fibroblasts: The stress response, inflammation, stemness, and growth factor signaling
To determine the biological processes that are activated in HD fibroblasts, we re-interrogated genome-wide transcriptional profiling data8 obtained from LD mammary fibroblasts (n = 6 patients) and HD mammary fibroblasts (n = 7 patients), from a series of 13 female patients (Table S1). These fibroblasts were derived from disease-free breast biopsies. In these gene sets, we focused on the gene transcripts that were increased by ≥ 1.5-fold (P < 0.05).
These up-genes (>1250 transcripts increased in HD fibroblasts, relative to LD fibroblasts) were then used to conduct gene set enrichment analysis (GSEA), to determine if there is significant overlap with existing gene sets deposited in the MSigDB (molecular signature database). HD fibroblast up-genes showed strong associations with gene sets related to cancer, the stress response, inflammation, stemness, and signal transduction (summarized in Table 1).
Table 1. Gene set enrichment analysis (GSEA) for HD vs. LD breast fibroblasts.
| Gene sets and description | P value |
|---|---|
| 1. Metastasis and cancer-related | |
| DODD_NASOPHARYNGEAL_CARCINOMA_UP Genes upregulated in nasopharyngeal carcinoma (NPC) compared with the normal tissue |
8.21E-16 |
| LIAO_METASTASIS Genes upregulated in the samples with intrahepatic metastatic hepatocellular carcinoma (HCC) vs. primary HCC |
6.69E-06 |
| DELYS_THYROID_CANCER_UP Genes upregulated in papillary thyroid carcinoma (PTC) compared with normal tissue |
1.67E-05 |
| CHARAFE_BREAST_CANCER_LUMINAL_VS_BASAL_UP Genes upregulated in luminal-like breast cancer cell lines compared with the basal-like ones |
1.74E-05 |
| SMID_BREAST_CANCER_NORMAL_LIKE_UP Genes upregulated in the normal-like subtype of breast cancer |
2.89E-05 |
| 2. Stress response and inflammation | |
|---|---|
| YOSHIMURA_MAPK8_TARGETS_UP Genes upregulated in vascular smooth muscle cells (VSMC) by MAPK8 (JNK1) |
5.20E-10 |
| MCLACHLAN_DENTAL_CARIES_UP Genes upregulated in pulpal tissue extracted from carious teeth |
5.16E-07 |
| ZHOU_INFLAMMATORY_RESPONSE_FIMA_UP Genes upregulated in macrophages by P.gingivalis FimA pathogen |
1.56E-06 |
| ZHOU_INFLAMMATORY_RESPONSE_LIVE_UP Genes upregulated in macrophage by live P.gingivalis |
1.41E-05 |
| MORF_NOS2A Neighborhood of NOS2A (iNOS) |
4.53E-05 |
| 3. Stemness | |
|---|---|
| MIKKELSEN_MEF_HCP_WITH_H3K27ME3 Genes with high-CpG-density promoters (HCP) bearing histone H3 trimethylation mark at K27 (H3K27me3) in MEF cells (embryonic fibroblast) |
1.76E-06 |
| CAGGTG_V$E12_Q6 Genes with promoter regions [-2kb,2kb] around transcription start site containing the motif CAGGTG which matches annotation for TCF3: transcription factor 3) |
3.18E-06 |
| HELLER_HDAC_TARGETS_SILENCED_BY_METHYLATION_UP Genes upregulated in multiple myeloma (MM) cell lines treated with both decitabine [PubChem = 451668] and TSA [PubChem = 5562] |
2.81E-05 |
| 4. Signaling | |
|---|---|
| SIGNAL_TRANSDUCTION Genes annotated by the GO term GO:0007165. The cascade of processes by which a signal interacts with a receptor, causing a change in the level or activity of a second messenger or other downstream target, and ultimately effecting a change in the functioning of the cell |
4.16E-07 |
| PLASMA_MEMBRANE Genes annotated by the GO term GO:0005886. The membrane surrounding a cell that separates the cell from its external environment. It consists of a phospholipid bilayer and associated proteins |
1.29E-06 |
| MORF_FLT1 Neighborhood of FLT1 (VGFR1) |
3.34E-06 |
| INTRINSIC_TO_MEMBRANE Genes annotated by the GO term GO:0031224. Located in a membrane such that some covalently attached portion of the gene product, for example part of a peptide sequence or some other covalently attached moiety such as a GPI anchor, spans or is embedded in one or both leaflets of the membrane |
8.90E-06 |
| REACTOME_PHOSPHOLIPASE_C_MEDIATED_CASCADE Genes involved in Phospholipase C-mediated cascade |
2.34E-05 |
| REACTOME_SIGNALING_BY_RHO_GTPASES Genes involved in Signaling by Rho GTPase |
5.22E-05 |
Cancer and metastasis
Interestingly, HD fibroblasts bear a striking resemblance to a number of human tumors, including head and neck (nasopharyngeal), liver, thyroid, and breast cancers (luminal and/or normal-like subtype). Similarities with metastatic liver cancer were also observed, relative to the primary tumor. Thus, the presence of HD fibroblasts may be a hallmark of a pre-cancerous phenotype, and this is consistent with the known predictive value of high density areas on mammography. HeatMaps for the HD fibroblast transcripts related to nasopharyngeal carcinoma, metastatic liver cancer and breast cancer sub-types are shown in Figures 1, 2, and 3. These results may reflect functional similarities between cancer-associated fibroblasts (CAFs) and HD fibroblasts; CAFs are a major part of the tumor stroma in most solid tumors, including human breast cancers, where the tumor stroma can represent 50% or more of the tumor mass.

Figure 1. HeatMaps for HD fibroblast transcripts related to nasopharyngeal carcinoma. For more details, see DODD_NASOPHARYNGEAL_CARCINOMA_UP listed in Table 1. This association has a P value of 8.21E-16. This association may be due to functional similarities between cancer-associated fibroblasts (CAFs) and HD fibroblasts; CAFs are relatively abundant in most solid tumors, where the tumor stroma can represent up to or greater than 50% of the tumor mass.
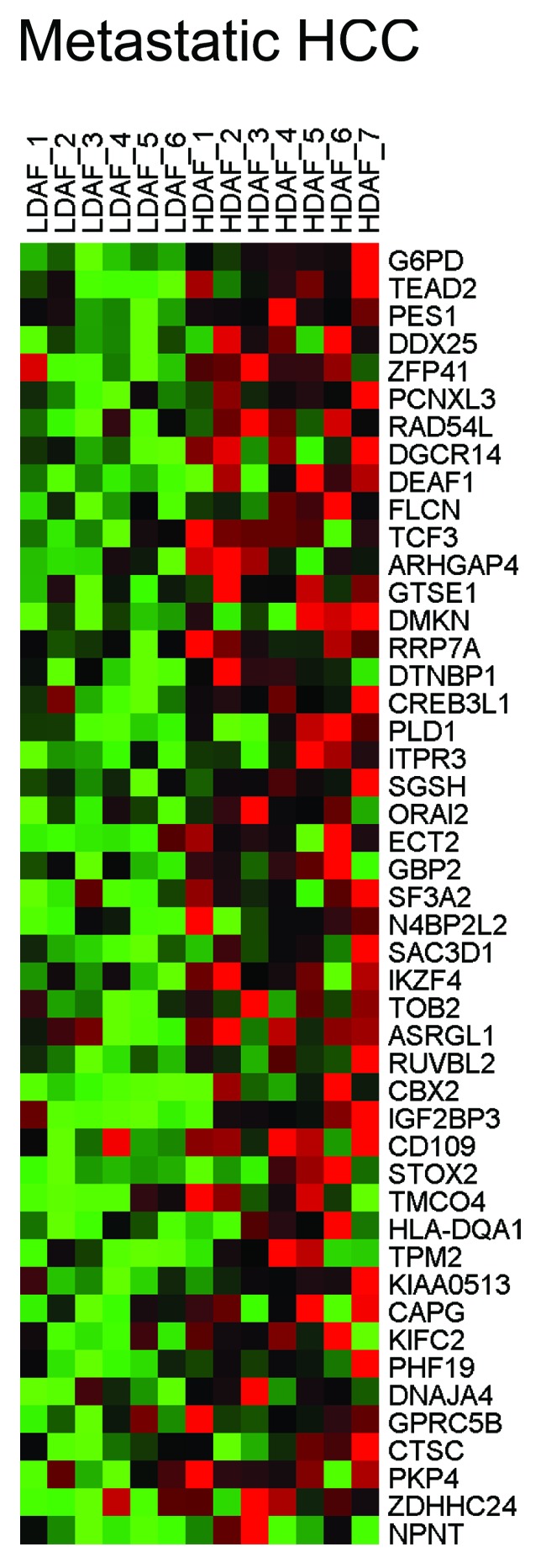
Figure 2. HeatMaps for HD fibroblast transcripts related to metastatic liver cancer. For more details, see LIAO_METASTASIS listed in Table 1. This association has a P value of 6.69E-06. HCC, hepatocellular carcinoma.
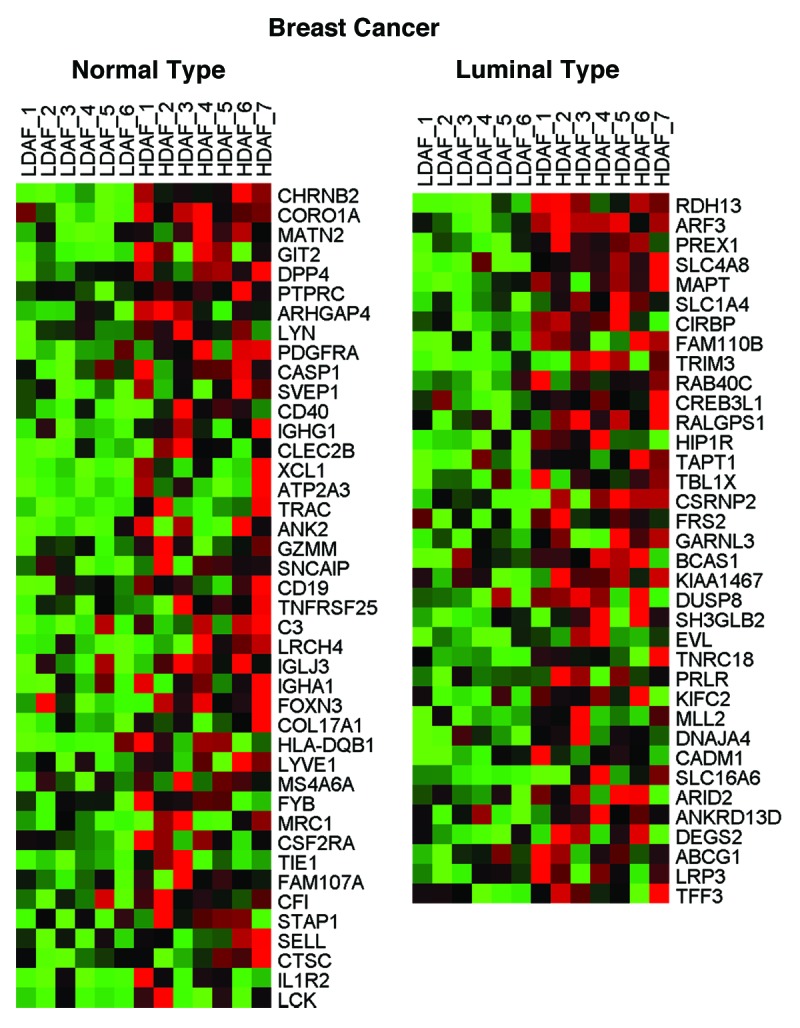
Figure 3. HeatMaps for HD fibroblast transcripts related to breast cancer sub-types. For more details, see CHARAFE_BREAST_CANCER_LUMINAL_VS_BASAL_UP and SMID_BREAST_CANCER_NORMAL_LIKE_UP listed in Table 1. These associations have P values of 1.74E-05 and 2.89E-05, respectively.
The stress response and inflammation
HD fibroblasts showed a strong association with the activation of the "stress response" mediated by JNK1, also known as MAPK8 (P = 5.20E-10). Consistent with the onset of a stress response, several gene sets associated with inflammation (dental caries) and activated macrophages, as well as iNOS (NOS2), showed striking similarities. Thus, external stimuli (from adjacent cells, such as abnormal breast epithelia or adipocytes) may induce a stress response, creating the HD fibroblast phenotype, leading to inflammation. Indeed, HD fibroblasts may behave like macrophages, driving and propagating inflammation, via the secretion of cytokines/chemokines and hydrogen peroxide, as well as via iNOS activation and NO production. This would be likely to generate an area or field of oxidative stress. HeatMaps for the HD fibroblast transcripts related to JNK1 signaling, the inflammatory response, and iNOS are shown in Figures 4, 5, and 6.
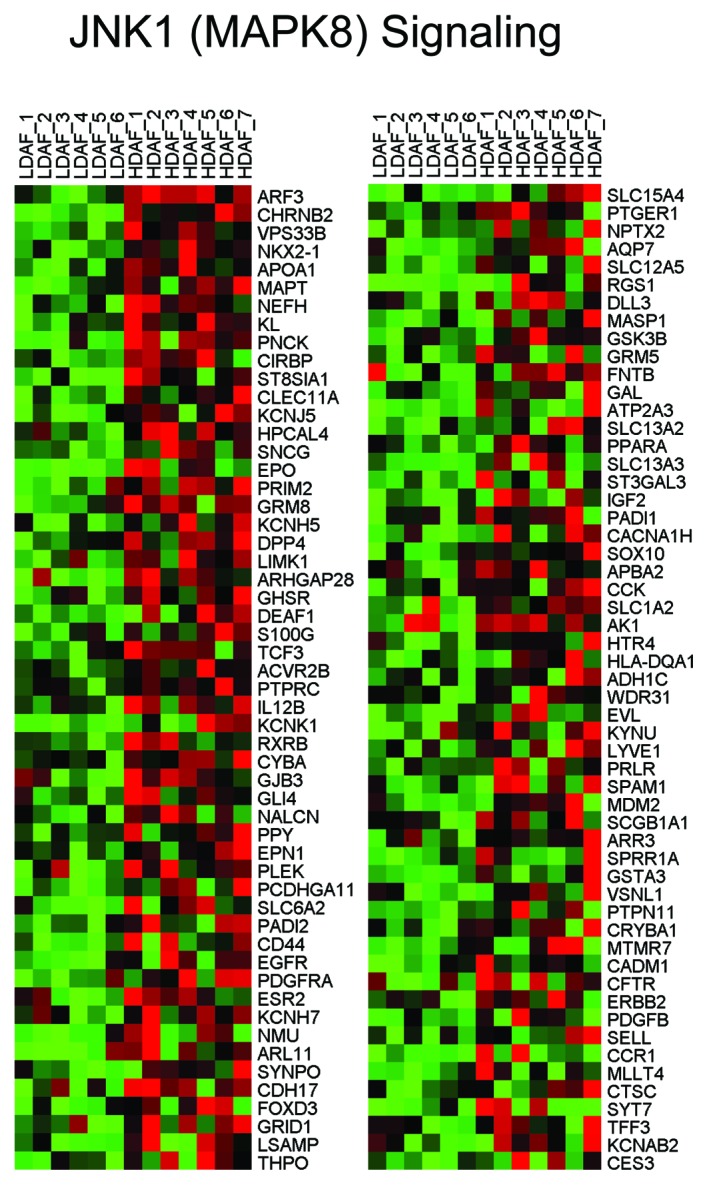
Figure 4. HeatMaps for HD fibroblast transcripts related to JNK1 (MAPK8) signaling. For more details, see YOSHIMURA_MAPK8_TARGETS_UP listed in Table 1. This association has a P value of 5.20E-10.

Figure 5. HeatMaps for HD fibroblast transcripts related to the macrophage inflammatory response and infection. For more details, see ZHOU_INFLAMMATORY_RESPONSE_FIMA_UP and MCLACHLAN_DENTAL_CARIES_UP listed in Table 1. These associations have P values of 1.56E-06 and 5.16E-07, respectively.

Figure 6. HeatMaps for HD fibroblast transcripts related to iNOS. For more details, see MORF_NOS2A listed in Table 1. This association has a P value of 4.53E-05. Other HeatMaps related to FLT1, phospholipase C, and Rho GTPases are also shown.
Increased stemness
Mesenchymal stem cells (MSCs) can express all three NOS isoforms, especially iNOS, under conditions of hypoxia. Consistent with an increase in stemness, we observed the genes upregulated in HD fibroblasts are closely related to TCF3, a known stem cell transcription factor, related to Wnt/β-catenin signaling. Also, 2 gene sets that are normally silenced by DNA methylation (which occurs during differentiation) are similar to the genes that are upregulated in HD fibroblasts, consistent with increased stemness. HeatMaps for the HD fibroblast transcripts related to TCF3 signaling are shown in Figure 7.

Figure 7. HeatMaps for HD fibroblast transcripts related to TCF3 signaling. For more details, see CAGGTG_V$E12_Q6 listed in Table 1. This association has a P value of 3.18E-06.
Signal transduction
Other signaling pathways also appeared to be activated, including those related to FLT1 (VGFR1; angiogenesis), PLC, and Rho GTPases. Similarities with GO TERM defined gene sets provided additional validation (Table S2). For example, HD fibroblast up-gene transcripts were associated with the MAPKKK cascade, Rho GTPase activation (including CDC42), cellular proliferation, extracellular matrix/secreted products, as well as FGF and EGF-receptor signaling. HeatMaps for the HD fibroblast transcripts related to various signaling pathways are shown in Figures 6 and 8.

Figure 8. HeatMaps for HD fibroblast transcripts related to FGF-R and EGF-R signaling. For more details, see the GO TERMS listed in Table S2. These associations have P values ranging from 2.22E-04 to 9.18E-06.
Cancer gene modules
The transcriptional profiles of HD fibroblasts also showed similarities to 14 distinct cancer gene modules (Table S3), including those related to signal transduction, the response to stress, the immune system, and inflammation, secreted proteins/extracellular space, liver cancer, and non-small cell lung cancer. Surprisingly, similarities were also noted with neurogenesis, which normally occurs during angiogenesis, as innervation normally follows bloods vessels and the vasculature. Thus, the similarities with various cancer gene modules provides further validation that certain key biological processes are involved or specifically activated in HD fibroblasts.
The HD fibroblast gene signature shows significant overlap with stromal signatures for breast cancer and tumor recurrence: Role of JNK1 signaling
To determine if the HD fibroblast gene signature is related to the pathogenesis of human breast cancers, we next intersected the HD fibroblast signature with 2 gene signatures derived from laser-captured tumor stroma, from a large series of human breast cancer patients, as described by Finak et al., 2008.11,12 More specifically, we re-interrogated the following two human breast cancer stromal gene lists:
1) Tumor stroma vs. normal stroma list. This signature compares the transcriptional profiles of tumor stroma (from n = 53 patients) to normal stroma (from n = 38 patients). Genes transcripts that were consistently upregulated in tumor stroma were selected and assigned a P value, with a cut-off of P < 0.05 (contains 6777 genes).
2) Recurrence stroma list. This signature compares the transcriptional profiles of tumor stroma obtained from patients with tumor recurrence (n = 11) to the tumor stroma of patients without tumor recurrence (n = 42). Genes transcripts that were consistently upregulated in the tumor stroma of patients with recurrence were selected and assigned a P value, with a cut-off of P < 0.05 (contains 3354 genes).
Remarkably, intersection of the HD fibroblast gene signature with these two breast cancer stromal signatures reveals significant similarities. The resulting Venn diagrams are shown in Figure 9. Note that the HD fibroblast gene signature is more closely related to breast cancer patients that undergo tumor recurrence. Importantly, these data indicate that certain key processes occurring in HD fibroblasts are also relevant for the pathogenesis of human breast cancers and for understanding the transition to malignancy.
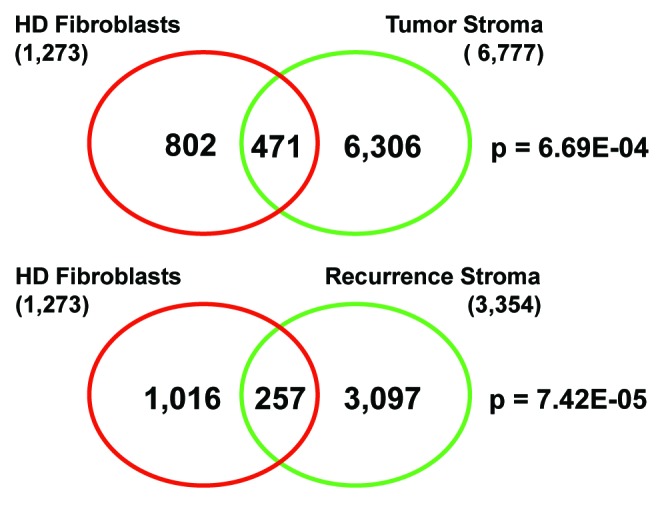
Figure 9. Venn diagrams for the intersection of HD fibroblast transcripts with tumor stroma from breast cancer patients. Intersection of the HD fibroblast gene signature with tumor stroma yields 471 common gene transcripts, with a P value of 6.69E-04. Intersection of the HD fibroblast gene signature with recurrence stroma yields 257 common gene transcripts, with a P value of 7.42E-05.
To better understand which biological processes are common between HD fibroblasts and human breast tumor stroma, we also performed gene-set enrichment analysis (GSEA) on the genes that are common between the various signatures. The results of this analysis are summarized in Tables 2 and 3.
Table 2. Biological processes that are common between HD fibroblasts and breast cancer tumor stroma.
| Gene sets and description | P value |
|---|---|
| 1. JNK1 and JUN signaling | |
| YOSHIMURA_MAPK8_TARGETS_UP Genes upregulated in vascular smooth muscle cells (VSMC) by MAPK8 (JNK1) |
1.02E-14 |
| JNK_DN.V1_DN Genes downregulated in JNK inhibitor-treated (SP600125 [PubChem = 8515]) keratinocytes |
8.42E-06 |
| TGANTCA_V$AP1_C Genes with promoter regions [-2kb,2kb] around transcription start site containing the motif TGANTCA which matches annotation for JUN: jun oncogene |
9.79E-06 |
| KEGG_MAPK_SIGNALING_PATHWAY MAPK signaling pathway |
1.28E-04 |
| 2. Stemness and DNA methylation | |
|---|---|
| CAGGTG_V$E12_Q6 Genes with promoter regions [-2kb,2kb] around transcription start site containing the motif CAGGTG which matches annotation for TCF3: transcription factor 3 |
1.04E-07 |
| BENPORATH_ES_WITH_H3K27ME3 Set 'H3K27 bound': genes posessing the trimethylated H3K27 (H3K27me3) mark in their promoters in human embryonic stem cells, as identified by ChIP on chip |
2.77E-07 |
| MIKKELSEN_MEF_HCP_WITH_H3K27ME3 Genes with high-CpG-density promoters (HCP) bearing histone H3 trimethylation mark at K27 (H3K27me3) in MEF cells (embryonic fibroblast) |
1.50E-06 |
| MEISSNER_NPC_HCP_WITH_H3K4ME2 Genes with high-CpG-density promoters (HCP) bearing histone H3 dimethylation mark at K4 (H3K4me2) in neural precursor cells (NPC). |
2.88E-05 |
| MIKKELSEN_MCV6_HCP_WITH_H3K27ME3 Genes with high-CpG-density promoters (HCP) bearing the tri-methylation mark at H3K27 (H3K27me3) in MCV6 cells (embryonic fibroblasts trapped in a differentiated state). |
3.19E-05 |
| MEISSNER_BRAIN_HCP_WITH_H3K4ME3_AND_H3K27ME3 Genes with high-CpG-density promoters (HCP) bearing histone H3 dimethylation at K4 (H3K4me2) and trimethylation at K27 (H3K27me3) in brain. |
7.32E-05 |
| ACEVEDO_LIVER_CANCER_WITH_H3K9ME3_UP Genes whose promoters display higher histone H3 trimethylation mark at K9 (H3K9me3) in hepatocellular carcinoma (HCC) compared with normal liver. |
8.41E-05 |
| HATADA_METHYLATED_IN_LUNG_CANCER_UP Genes with hypermethylated DNA in lung cancer samples |
1.53E-04 |
| CTTTGT_V$LEF1_Q2 Genes with promoter regions [-2kb,2kb] around transcription start site containing the motif CTTTGT which matches annotation for LEF1 |
2.62E-04 |
| 3. Cancer related | |
|---|---|
| DODD_NASOPHARYNGEAL_CARCINOMA_UP Genes upregulated in nasopharyngeal carcinoma (NPC) compared with the normal tissue |
2.17E-07 |
| LIAO_METASTASIS Genes upregulated in the samples with intrahepatic metastatic hepatocellular carcinoma (HCC) vs. primary HCC |
9.87E-06 |
| CHARAFE_BREAST_CANCER_LUMINAL_VS_MESENCHYMAL_UP Genes upregulated in luminal-like breast cancer cell lines compared with the mesenchymal-like ones |
1.51E-05 |
| CHARAFE_BREAST_CANCER_LUMINAL_VS_BASAL_UP Genes upregulated in luminal-like breast cancer cell lines compared with the basal-like ones |
2.40E-04 |
| 4. Angiogensis | |
|---|---|
| VART_KSHV_INFECTION_ANGIOGENIC_MARKERS_UP Angiogenic markers upregulated in lymph endothelial cells upon infection with KSHV (Kaposi sarcoma herpes virus). |
1.56E-06 |
| MORF_FLT1 Neighborhood of FLT1 (VGFR1) |
4.73E-05 |
| 5. NFkB-activation and cytokine/chemokine signaling | |
|---|---|
| MORF_MAP3K14 MAP3K14 |
6.32E-06 |
| PID_CXCR4_PATHWAY CXCR4-mediated signaling events |
4.75E-05 |
| MORF_TNFRSF25 Neighborhood of TNFRSF25 |
1.03E-04 |
| MORF_IL9 Neighborhood of IL9 |
1.25E-04 |
| MORF_NOS2A Neighborhood of NOS2A (iNOS) |
2.60E-04 |
| 6. FGFR signaling | |
|---|---|
| PID_FGF_PATHWAY FGF signaling pathway |
1.55E-05 |
| REACTOME_FGFR_LIGAND_BINDING_AND_ACTIVATION Genes involved in FGFR ligand binding and activation |
7.03E-05 |
| REACTOME_SIGNALING_BY_FGFR Genes involved in Signaling by FGFR |
7.75E-05 |
| REACTOME_FRS2_MEDIATED_CASCADE Genes involved in FRS2-mediated cascade |
8.36E-05 |
| REACTOME_DOWNSTREAM_SIGNALING_OF_ACTIVATED_FGFR Genes involved in downstream signaling of activated FGFR |
1.73E-04 |
| REACTOME_SIGNALING_BY_FGFR_IN_DISEASE Genes involved in Signaling by FGFR in disease |
2.14E-04 |
| 7. TGF-β signaling | |
|---|---|
| LABBE_TGFB1_TARGETS_UP Upregulated genes in NMuMG cells (mammary epithelium) after stimulation with TGFB1 |
4.75E-05 |
| 8. Other related signaling pathways | |
|---|---|
| REACTOME_PHOSPHOLIPASE_C_MEDIATED_CASCADE Genes involved in Phospholipase C-mediated cascade |
9.99E-05 |
| REACTOME_PI_3K_CASCADE Genes involved in PI-3K cascade |
1.13E-04 |
| REACTOME_SHC_MEDIATED_CASCADE Genes involved in SHC-mediated cascade |
2.37E-04 |
Table 3. Biological processes that are common between HD fibroblasts and breast cancer stroma, and are associated with tumor recurrence.
| Gene sets and description | P value |
|---|---|
| 1. JNK1 signaling | |
| YOSHIMURA_MAPK8_TARGETS_UP Genes upregulated in vascular smooth muscle cells (VSMC) by MAPK8 (JNK1) |
5.40E-09 |
| 2. Stemness and DNA methylation | |
|---|---|
| BENPORATH_ES_WITH_H3K27ME3 Set 'H3K27 bound': genes posessing the trimethylated H3K27 (H3K27me3) mark in their promoters in human embryonic stem cells, as identified by ChIP on chip |
1.65E-05 |
| MIKKELSEN_MEF_HCP_WITH_H3K27ME3 Genes with high-CpG-density promoters (HCP) bearing histone H3 trimethylation mark at K27 (H3K27me3) in MEF cells (embryonic fibroblast) |
1.24E-04 |
| 3. Cancer related | |
|---|---|
| LIAO_METASTASIS Genes upregulated in the samples with intrahepatic metastatic hepatocellular carcinoma (HCC) vs. primary HCC |
2.33E-05 |
| DODD_NASOPHARYNGEAL_CARCINOMA_UP Genes upregulated in nasopharyngeal carcinoma (NPC) compared with the normal tissue |
1.21E-04 |
| 4. TGF-β signaling | |
|---|---|
| TGFB_UP.V1_UP Genes upregulated in a panel of epithelial cell lines by TGFB1 |
5.23E-05 |
| LABBE_TGFB1_TARGETS_UP Upregulated genes in NMuMG cells (mammary epithelium) after stimulation with TGFB1 |
1.53E-04 |
| 5. Angiogenesis | |
|---|---|
| MORF_FLT1 Neighborhood of FLT1 (VGFR1) |
7.58E-05 |
| VART_KSHV_INFECTION_ANGIOGENIC_MARKERS_UP Angiogenic markers upregulated in lymph endothelial cells upon infection with KSHV (Kaposi's sarcoma herpes virus). |
1.02E-04 |
| 6. Aging | |
|---|---|
| LEE_AGING_MUSCLE_UP Upregulated in the gastrocnemius muscle of aged adult mice (30-mo) vs. young adult (5-mo) |
1.44E-04 |
| 7. Other signaling pathways | |
|---|---|
| PID_ERBB2ERBB3PATHWAY ErbB2/ErbB3 signaling events |
1.29E-04 |
| PID_CXCR4_PATHWAY CXCR4-mediated signaling events |
1.53E-04 |
Interestingly, biological processes that are common between HD fibroblasts and breast tumor stroma are: JNK1 stress signaling, stemness, angiogenesis, cytokine signaling, NFkB-activation, and iNOS, as well as FGF- and TGF-β signaling (Table 2).
Similarly, biological processes that are common between HD fibroblasts and the stroma of breast cancer patients with recurrence are: JNK1 stress signaling, stemness, angiogenesis, and aging, as well as TGF-β, ErbB2/3 and CXCR4 signaling (Table 3). It is of interest to note that aging is also one of the most significant risk factors for the development breast cancers, as well as many other types of human cancers.
Thus, activated JNK1 signaling appears to be the single most significant biological process that is common between HD fibroblasts and the tumor stroma (with P values between 5.40E-09 and 1.02E-14) (Tables 2 and 3). In further support of this notion, Table 4 lists the transcripts of JNK1 and other JNK1-interacting proteins that are upregulated in human tumor stroma, along with their P values, and associations with breast cancer tumor recurrence.
Table 4. JNK1 (MAPK8) in breast cancer derived tumor stroma.
| Gene symbol | Gene description | Tumor stroma | Recurrence stroma |
|---|---|---|---|
| Mapk8 | Mitogen-activated protein kinase 8 | 8.09E-15 | 3.08E-04 |
| Mapk8ip1 | Mitogen-activated protein kinase 8 interacting protein 1 | 1.22E-14 | 2.05E-02 |
| Mapk8ip2 | Mitogen-activated protein kinase 8 interacting protein 2 | 3.83E-14 | 2.20E-02 |
| Mapk8ip3 | Mitogen-activated protein kinase 8 interacting protein 3 | 7.00E-21 |
P values are as shown.
Augmented TGF-β signaling also emerged as a common feature linking high breast density, with tumor stroma and breast cancer recurrence (Tables 2 and 3). Importantly, TGF-β mediated signal transduction is known to converge on JNK1-signaling, via the activation of FAK and TAK1, driving the myofibroblast differentiation program and induction of α-smooth muscle actin (α-SMA) during fibrosis (see the "Discussion" section).
A diagram summarizing our most salient findings is presented in Figure 10. Interestingly, many of the key biological processes that are common between HD fibroblasts and breast cancer tumor stroma are also involved in wound healing, namely stress signaling, stemness, angiogesis, inflammation, and fibrosis. This is consistent with the concept that cancer behaves as a wound that does not heal.
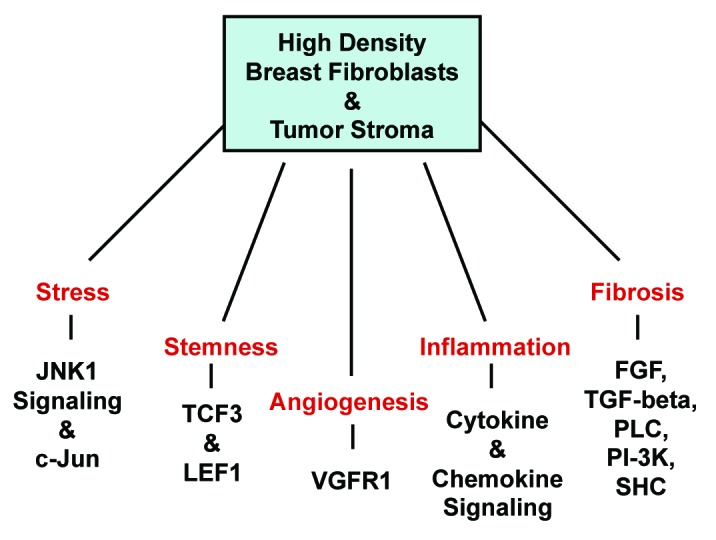
Figure 10. Biological processes that are shared between HD breast fibroblasts and the tumor stroma: Similarities to wound healing. These five shared biological processes include stress signaling, stemness, angiogenesis, inflammation and fibrosis. Many of these processes also commonly occur during wound healing. However, JNK1 stress kinase signaling is the single most significant signal transduction network that is activated (P = 1.02E-14; Table 2).
Interestingly, during wound healing, bFGF-signaling activates Rho GTPases (RhoA; Rac1), which in turn activate PI3-kinase and increase JNK activity. We see that all of the individual components of this signaling network are represented in the HD fibroblast gene signature13 (see Tables 1-3; Tables S1-3). In addition, genetic ablation of JNK (-/-) or pharmacological inhibition of JNK-activity (with SP-600125) are both sufficient to dramatically impair wound closure by dermal fibroblasts, by inhibiting cell motility/migration.14
The HD fibroblast gene signature shares many functional similarities with the cancer-associated fibroblast (CAF) phenotype
Consistent with the idea that HD fibroblasts may possess a CAF-like phenotype, HD fibroblasts overexpress the same classes of markers that are found in CAFs15-21 (Table 5), including PDPN and CDH11. More specifically, many of these CAF-markers are functionally related to matrix remodeling, autophagy, senescence, ketone production, hypoxia, oxidative stress, inflammation, DNA-damage, and stemness,22 as well as FGF/EGF/PDGF-signaling.23,24 These similarities also include molecular markers of the myofibroblast phenotype (such as, actin-binding proteins, the myosins, and other muscle proteins)22 (Table 5).
Table 5. Selected gene transcripts upregulated in HD fibroblasts: Functional similarities with cancer-associated fibroblasts.
| Symbol | P value | Fold-change | Gene description |
|---|---|---|---|
| 1. Cancer-associated fibroblast markers | |||
| PDPN | 4.54E-02 | 1.77 | podoplanin |
| CDH11 | 4.57E-02 | 1.54 | cadherin 11, type 2, OB-cadherin (osteoblast) |
| 2. Collagen(s) and matrix remodeling | |||
|---|---|---|---|
| COL6A5 | 3.23E-03 | 3.03 | collagen, type VI, α 5 |
| IMPG1 | 2.81E-02 | 2.76 | interphotoreceptor matrix proteoglycan 1 |
| P4HA3 | 1.07E-02 | 2.44 | prolyl 4-hydroxylase, α polypeptide III |
| ITGB1 | 3.14E-02 | 2.37 | integrin, β 1 (fibronectin receptor, β polypeptide) |
| ADAMTS17 | 2.77E-02 | 2.35 | ADAM metallopeptidase with thrombospondin type 1 motif, 17 |
| ELFN2 | 2.12E-02 | 1.79 | extracellular leucine-rich repeat and fibronectin type III domain containing 2 |
| COL17A1 | 3.28E-02 | 1.87 | collagen, type XVII, α 1 |
| MMP24 | 7.22E-03 | 1.63 | matrix metallopeptidase 24 (membrane-inserted) |
| THSD1 | 7.46E-03 | 1.61 | thrombospondin, type I, domain containing 1 |
| ECM1 | 5.09E-03 | 1.58 | extracellular matrix protein 1 |
| ADAMTSL4 | 4.45E-02 | 1.50 | ADAMTS-like 4 |
| 3. Autophagy and lysosome-related proteins | |||
|---|---|---|---|
| BECN1 | 2.67E-02 | 2.99 | beclin 1, autophagy related |
| ATP6V1E2 | 4.03E-04 | 2.01 | ATPase, H+ transporting, lysosomal 31kDa, V1 subunit E2 |
| ATP6V0A2 | 5.00E-02 | 1.74 | ATPase, H+ transporting, lysosomal V0 subunit a2 |
| CTSC | 4.73E-02 | 1.59 | cathepsin C |
| 4. Senescence-associated proteins | |||
|---|---|---|---|
| CDKN2D | 2.18E-02 | 2.27 | cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) |
| 5. Ketone production (indicative of mitochondrial dysfunction) | |||
|---|---|---|---|
| HMGCLL1 | 4.68E-03 | 2.63 | 3-hydroxymethyl-3-methylglutaryl-CoA lyase-like 1 |
| BDH1 | 4.07E-02 | 1.58 | 3-hydroxybutyrate dehydrogenase, type 1 |
| 6. Anti-oxidants and/or proteins that protect against mitochondrial oxidative stress | |||
|---|---|---|---|
| MTFR1 | 3.36E-02 | 2.82 | mitochondrial fission regulator 1 |
| GSTA3 | 3.96E-02 | 2.55 | glutathione S-transferase α 3 |
| GLDC | 1.45E-02 | 2.31 | glycine dehydrogenase (decarboxylating) |
| SOD2 | 2.02E-02 | 2.12 | superoxide dismutase 2, mitochondrial |
| G6PD | 1.24E-03 | 1.88 | glucose-6-phosphate dehydrogenase |
| HSPD1 | 2.17E-02 | 1.63 | heat shock 60kDa protein 1 (chaperonin) |
| 7. Hypoxia-related proteins and glucose-uptake | |||
|---|---|---|---|
| EPO | 5.41E-03 | 3.80 | erythropoietin |
| SLC5A9 | 2.16E-02 | 1.71 | solute carrier family 5 (sodium/glucose cotransporter), member 9 |
| HIGD1B | 1.59E-02 | 1.86 | HIG1 hypoxia inducible domain family, member 1B |
| 8. Inflammatory molecules, cytokines, and chemokines | |||
|---|---|---|---|
| IL4 | 2.23E-02 | 3.68 | interleukin 4 |
| CLNK | 6.98E-03 | 3.58 | cytokine-dependent hematopoietic cell linker |
| CCR1 | 4.68E-02 | 2.85 | chemokine (C-C motif) receptor 1 |
| S100A7 | 1.67E-02 | 3.07 | S100A7 (a.k.a., psoriasin; interacts with Jab1, c-jun activation domain binding protein 1) |
| IL1R2 | 4.77E-02 | 2.79 | interleukin 1 receptor, type II |
| CCL13 | 2.11E-02 | 2.68 | chemokine (C-C motif) ligand 13 |
| IL20RA | 4.38E-02 | 2.44 | interleukin 20 receptor, α |
| XCL1 | 2.30E-02 | 2.18 | chemokine (C motif) ligand 1 |
| CCR8 | 4.96E-02 | 1.96 | chemokine (C-C motif) receptor 8 |
| DMKN | 1.08E-02 | 1.92 | dermokine |
| CXCL6 | 2.82E-02 | 1.86 | chemokine (C-X-C motif) ligand 6 (granulocyte chemotactic protein 2) |
| IL4I1 | 4.46E-02 | 1.80 | interleukin 4 induced 1 |
| IL8 | 4.71E-02 | 1.76 | interleukin 8 |
| IL1RL1 | 3.62E-02 | 1.69 | interleukin 1 receptor-like 1 |
| CTF1 | 3.57E-02 | 1.57 | cardiotrophin 1 (IL6-like cytokine associated with cardiac hypertrophy) |
| CSF2RA | 3.92E-02 | 1.57 | colony stimulating factor 2 receptor, α, low-affinity (granulocyte-macrophage) |
| CCL17 | 3.38E-02 | 1.51 | chemokine (C-C motif) ligand 17 |
| GBP2 | 1.56E-02 | 1.50 | guanylate binding protein 2, interferon-inducible |
| 9. TNF-related genes | |||
|---|---|---|---|
| TNFAIP8L2 | 1.60E-02 | 2.30 | tumor necrosis factor, α-induced protein 8-like 2 |
| FBF1 | 2.34E-02 | 2.18 | Fas (TNFRSF6) binding factor 1 |
| CD40 | 2.03E-02 | 1.97 | CD40 molecule, TNF receptor superfamily member 5 |
| TNFRSF25 | 3.05E-02 | 1.88 | tumor necrosis factor receptor superfamily, member 25 |
| TRAF7 | 4.34E-02 | 1.87 | TNF receptor-associated factor 7, E3 ubiquitin protein ligase |
| TNFSF13 | 4.79E-02 | 1.76 | tumor necrosis factor (ligand) superfamily, member 13 |
| TRAF4 | 1.52E-02 | 1.52 | TNF receptor-associated factor 4 |
| 10. Components of the complement cascade | |||
|---|---|---|---|
| C4BPB | 2.55E-02 | 2.70 | complement component 4 binding protein, β |
| C9 | 4.11E-02 | 2.69 | complement component 9 |
| C3 | 3.06E-02 | 2.69 | complement component 3 |
| C1RL | 3.46E-02 | 1.79 | complement component 1, r subcomponent-like |
| CFI | 4.63E-02 | 1.71 | complement factor I |
| 11. Actin-binding proteins | |||
|---|---|---|---|
| CAPZA1 | 3.24E-02 | 3.64 | capping protein (actin filament) muscle Z-line, α 1 |
| TPM2 | 3.30E-02 | 2.43 | tropomyosin 2 (β) |
| CORO1A | 5.66E-04 | 2.74 | coronin, actin binding protein, 1A |
| 12. Myosin-related proteins | |||
|---|---|---|---|
| MYH7B | 3.63E-02 | 3.53 | heavy chain 7B, cardiac muscle, β |
| MYLK4 | 3.33E-02 | 3.28 | myosin light chain kinase family, member 4 |
| MYBPC2 | 1.09E-02 | 2.87 | myosin binding protein C, fast type |
| MYH14 | 4.81E-04 | 2.16 | myosin, heavy chain 14, non-muscle |
| MYO7A | 1.44E-02 | 2.06 | myosin VIIA |
| MYO7B | 2.65E-02 | 1.59 | myosin VIIB |
| 13. Other muscle proteins | |||
|---|---|---|---|
| SLMAP | 9.48E-03 | 2.19 | sarcolemma associated protein |
| SMPX | 1.76E-02 | 1.85 | small muscle protein, X-linked |
| MBNL3 | 1.21E-02 | 1.64 | muscleblind-like splicing regulator 3 |
| CTF1 | 3.57E-02 | 1.57 | cardiotrophin 1 (IL6-like cytokine associated with cardiac hypertrophy) |
| PHKA1 | 3.54E-02 | 1.53 | phosphorylase kinase, α 1 (muscle) |
| 14. Stem cell markers | |||
|---|---|---|---|
| SOX11 | 6.25E-03 | 4.31 | SRY (sex determining region Y)-box 11 |
| ALDH1A3 | 3.87E-02 | 3.21 | aldehyde dehydrogenase 1 family, member A3 |
| WNT8B | 3.49E-02 | 2.80 | wingless-type MMTV integration site family, member 8B |
| CD44 | 1.36E-02 | 2.66 | CD44 molecule (Indian blood group) |
| SOX21 | 2.18E-02 | 2.36 | SRY (sex determining region Y)-box 21 |
| WNT2 | 3.95E-02 | 2.05 | wingless-type MMTV integration site family member 2 |
| POU5F1 | 3.80E-02 | 2.05 | POU class 5 homeobox 1 (OCT4) |
| SPRR1A | 3.95E-02 | 1.97 | small proline-rich protein 1A |
| GSK3B | 2.03E-02 | 1.90 | glycogen synthase kinase 3 β |
| SOX10 | 2.88E-02 | 1.70 | SRY (sex determining region Y)-box 10 |
| SOX8 | 1.97E-02 | 1.70 | SRY (sex determining region Y)-box 8 |
| WNT2B | 3.91E-02 | 1.64 | wingless-type MMTV integration site family, member 2B |
| KLF8 | 3.24E-03 | 1.60 | Kruppel-like factor 8 |
| 15. FGF-R signaling | |||
| FGF6 | 1.60E-02 | 2.32 | fibroblast growth factor 6 |
| FGF9 | 4.10E-02 | 2.31 | fibroblast growth factor 9 (glia-activating factor) |
| FGFR2 | 1.45E-02 | 2.12 | fibroblast growth factor receptor 2 |
| FGFR1 | 4.29E-03 | 1.59 | fibroblast growth factor receptor 1 |
| FGFRL1 | 4.58E-03 | 1.56 | fibroblast growth factor receptor-like 1 |
| 16. EGF-R and PDGF-R signaling | |||
|---|---|---|---|
| EGFR | 1.38E-02 | 2.61 | epidermal growth factor receptor |
| PDGFB | 4.53E-02 | 2.22 | platelet-derived growth factor β polypeptide |
| PDGFRA | 1.41E-02 | 2.11 | platelet-derived growth factor receptor, α polypeptide |
| ERBB2 | 4.52E-02 | 1.68 | v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian) |
| 17. Kinases and kinase regulators | |||
|---|---|---|---|
| TTBK2 | 1.86E-02 | 4.16 | tau tubulin kinase 2 |
| TNK2 | 5.33E-03 | 4.00 | tyrosine kinase, non-receptor, 2 |
| JAK1 | 6.24E-03 | 3.60 | Janus kinase 1 |
| MYLK4 | 3.33E-02 | 3.28 | myosin light chain kinase family, member 4 |
| SRC | 3.33E-02 | 3.06 | v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (avian) |
| CDK6 | 8.11E-04 | 2.97 | cyclin-dependent kinase 6 |
| MAST4 | 7.84E-03 | 2.75 | microtubule associated serine/threonine kinase family member 4 |
| PRKAR1A | 2.11E-02 | 2.70 | protein kinase, cAMP-dependent, regulatory, type I, α (tissue specific extinguisher 1) |
| KSR1 | 2.10E-02 | 2.70 | kinase suppressor of ras 1 |
| BMX | 3.92E-02 | 2.62 | BMX non-receptor tyrosine kinase |
| MAP3K15 | 5.52E-03 | 2.55 | mitogen-activated protein kinase kinase kinase 15 |
| TEK | 1.03E-02 | 2.42 | TEK tyrosine kinase, endothelial |
| ADRBK1 | 2.50E-02 | 2.32 | adrenergic, β, receptor kinase 1 |
| AKAP14 | 4.25E-03 | 2.23 | A kinase (PRKA) anchor protein 14 |
| PNCK | 1.98E-03 | 2.13 | pregnancy upregulated non-ubiquitously expressed CaM kinase |
| TIE1 | 4.04E-02 | 2.06 | tyrosine kinase with immunoglobulin-like and EGF-like domains 1 |
| TOB2 | 1.86E-02 | 2.07 | transducer of ERBB2, 2 |
| LCK | 4.80E-02 | 2.01 | lymphocyte-specific protein tyrosine kinase |
| ETNK1 | 1.11E-02 | 1.98 | ethanolamine kinase 1 |
| MAPK8IP1 | 1.79E-02 | 1.98 | mitogen-activated protein kinase 8 interacting protein 1 |
| IPMK | 3.88E-02 | 1.92 | inositol polyphosphate multikinase |
| STK19 | 4.69E-02 | 1.91 | serine/threonine kinase 19 |
| DCLK3 | 9.34E-03 | 1.81 | doublecortin-like kinase 3 |
| ACVR2B | 9.09E-03 | 1.81 | activin A receptor, type IIB |
| DAPK2 | 1.37E-02 | 1.78 | death-associated protein kinase 2 |
| PLK1S1 | 1.36E-02 | 1.73 | polo-like kinase 1 substrate 1 |
| AKAP9 | 2.00E-02 | 1.73 | A kinase (PRKA) anchor protein (yotiao) 9 |
| MAP3K13 | 4.84E-02 | 1.66 | mitogen-activated protein kinase kinase kinase 13 |
| 18. DNA damage/repair and dna-binding proteins | |||
|---|---|---|---|
| CHD5 | 2.17E-03 | 3.08 | chromodomain helicase DNA binding protein 5 |
| PIF1 | 1.42E-02 | 2.74 | PIF1 5"-to-3" DNA helicase homolog (S. cerevisiae) |
| RAD52 | 4.15E-02 | 2.58 | RAD52 homolog (S. cerevisiae) |
| RAD54L | 7.84E-03 | 2.06 | RAD54-like (S. cerevisiae) |
| POLR2J | 3.20E-02 | 1.96 | polymerase (RNA) II (DNA directed) polypeptide J, 13.3kDa |
| WDHD1 | 1.67E-02 | 1.72 | WD repeat and HMG-box DNA binding protein 1 |
| TOP3B | 4.26E-02 | 1.66 | topoisomerase (DNA) III β |
| PRIM2 | 5.87E-03 | 1.60 | primase, DNA, polypeptide 2 (58kDa) |
| XRCC6 | 4.30E-02 | 1.60 | X-ray repair complementing defective repair in Chinese hamster cells 6 |
| 19. Breast cancer, tamoxifen-resistance or metastasis associated genes | |||
|---|---|---|---|
| SNCG | 5.10E-03 | 2.65 | synuclein, gamma (breast cancer-specific protein 1) |
| CYP19A1 | 4.87E-02 | 2.17 | cytochrome P450, family 19, subfamily A, polypeptide 1 (aromatase) |
| ESR2 | 1.46E-02 | 1.99 | estrogen receptor 2 (ER β) |
| PRLR | 3.61E-02 | 1.81 | prolactin receptor |
| BCAS1 | 2.46E-02 | 1.78 | breast carcinoma amplified sequence 1 |
| TFF3 | 4.88E-02 | 1.76 | trefoil factor 3 |
| MACC1 | 2.56E-02 | 1.55 | metastasis associated in colon cancer 1 |
| 20. Pregnancy associated genes | |||
|---|---|---|---|
| PSG1 | 1.13E-02 | 5.16 | pregnancy specific β-1-glycoprotein 1 |
| PSG4 | 1.35E-02 | 2.92 | pregnancy specific β-1-glycoprotein 4 |
| PNCK | 1.98E-03 | 2.13 | pregnancy upregulated non-ubiquitously expressed CaM kinase |
| PSG7 | 4.73E-02 | 2.05 | pregnancy specific β-1-glycoprotein 7 |
| 21. Glutamate receptors and transporters | |||
|---|---|---|---|
| GRIK2 | 1.80E-02 | 2.47 | glutamate receptor, ionotropic, kainate 2 |
| GRM8 | 6.07E-03 | 2.04 | glutamate receptor, metabotropic 8 |
| SLC1A7 | 8.13E-03 | 2.02 | solute carrier family 1 (glutamate transporter), member 7 |
| GRID1 | 1.58E-02 | 2.01 | glutamate receptor, ionotropic, delta 1 |
| GRIN2B | 2.93E-02 | 1.84 | glutamate receptor, ionotropic, N-methyl D-aspartate 2B |
| GRM5 | 2.10E-02 | 1.84 | glutamate receptor, metabotropic 5 |
| SLC1A4 | 1.92E-03 | 1.82 | solute carrier family 1 (glutamate/neutral amino acid transporter), member 4 |
| SLC1A2 | 3.16E-02 | 1.78 | solute carrier family 1 (glial high affinity glutamate transporter), member 2 |
| GRM7 | 1.46E-02 | 1.55 | glutamate receptor, metabotropic 7 |
Finally, HD fibroblast overexpression of glutamate receptors and transporters may suggest similarities with astrocytes, which are already known to share properties with CAFs.12,25
Discussion
Here, we re-interrogated previously published expression profiling data obtained from low-density (LD) and high-density (HD) mammary fibroblasts, derived from 13 female patients. Importantly, these fibroblasts were derived from disease-free breast biopsies. More specifically, we focused on the up-genes that were transcriptionally increased by ≥ 1.5-fold (P < 0.05) and performed gene-set enrichment analysis (GSEA), using the molecular signatures database (MSigDB). We now report that HD fibroblasts show the upregulation and/or hyper-activation of several key cellular processes, including the stress response, inflammation, stemness and various signal transduction pathways. GSEA predicts that certain stress and inflammation-related signaling networks may be involved, including those related to JNK1 and iNOS, as well as growth factor (FGF/EGF/PDGF) mediated signal transduction, driving an inflammatory, pro-proliferative, and cytokine-rich environment. Transcriptional profiles of HD fibroblasts showed significant overlap with the profiles derived from smooth muscle cells under oxidative stress (JNK1) and activated/infected macrophages (iNOS). As a consequence, HD fibroblasts may mimic the behavior of activated myofibroblasts and macrophages, to generate a pro-fibrotic and pro-inflammatory microenvironment, thereby increasing the risk of developing breast cancer. In addition, we compared the HD fibroblast gene signature with transcriptional profiling data derived from breast cancer tumor stroma. This analysis revealed that JNK1 signaling is the single most significant biological process that is common between these 2 data sets, and is associated with tumor recurrence. These results implicate "stromal JNK1-signaling" in disease pathogenesis and have important implications for breast cancer prevention.
JNK1 activation and inflammation in high breast density and the tumor stroma: Associations with myofibroblast differentiation, TGF-β signaling, and fibrosis
The stress-activated protein kinase/c-Jun N-terminal kinase (SAP/JNK) signaling network functions to regulate a diverse array of biological processes, including proliferation, programmed cell death, inflammation, and cellular metabolism, among others.26 Extracellular stressors (such as hydrogen peroxide and ROS), pro-inflammatory cytokines and/or even physical stress (including UV light), all activate JNK signaling.27-29 The JNK pathway mediates the immediate-early response to a wide-variety of cellular perturbations or stressors.30
Two dual-specificity kinases, namely MKK4 and MKK7, phosphorylate and activate JNK1 (also known as MAPK8).31,32 Then, activated JNK1 phosphorylates a number of key transcription factors, such as JUN, JDP2, and ATF2, thus regulating the activity of the AP-1 transcription complex. JNK1 also has a number of other targets, which it phosphorylates or activates that control survival and cell metabolism, including p53, YAP1, BCL2, and NFkB. JNK1-activation can induce autophagy, mitophagy, and aerobic glycolysis,33 which are all catabolic survival pathways.34,35 JNK1 also controls mitophagy by modulating FOXO3A and BNIP3 expression.36
JNK1 has been implicated in initiating the fibroblast-to-myofibrobast differentiation program during fibrosis,37 and controls the expression of α-smooth-muscle actin (α-SMA).38 In this context, TGF-β and CTGF signaling converge on JNK1-signaling via the activation of FAK and TAK1,39 which in turn activate JNK1.40 Thus, several JNK1 inhibitors (e.g., Leflunomide, CC-401, CC-930, XG-102, Bentamapimod)41 are being developed clinically for the treatment and prevention of various fibrotic and inflammatory disorders, such as idiopathic pulmonary fibrosis (IPF), and dermal scarring (keloids), as well as cardiac fibrosis/heart failure and inflammatory bowel disease (IBD).42 Other clinical uses for JNK1 inhibitors include the treatment of many other stress-related aging-associated and degenerative diseases,43 such as hearing loss, ischemic brain disease (stroke), retinal neo-vascularization, uveitis, and metabolic syndrome, as well as diabetes.44
Matrix stiffness has been implicated in the disease pathogenesis underlying high mammographic density.7,45-47 Interestingly, JNK1 signaling also senses and effectively responds to changes in matrix stiffness,48 as well as many other mechanical changes or stressors in the cellular microenvironment.49,50
Recently, we and others have demonstrated that cancer-associated fibroblasts (CAFs) have a myofibroblastic phenotype, and are characterized by the activation of a number of catabolic cellular processes, such as oxidative stress, autophagy, mitophagy, glycolysis, inflammation, and senescence.51 Cancer cells induce this CAF-phenotype in normal stromal cells by producing and secreting ROS/H2O2 and cytokines/chemokines.52-54 Thus, JNK1 activation in HD fibroblasts and the stromal microenvironment may confer a very similar "stress-response phenotype," with many of the same biological features that are characteristic of the CAF-phenotype (Table 5). This could mechanistically explain the predictive value of high mammographic density for detecting human breast cancers, and suggests that JNK1 inhibitors may be useful for cancer prevention as well, in patients with high mammographic density. New JNK1-based stromal models that mimic high mammographic density and the CAF-phenotype will be necessary to test this hypothesis experimentally.
Importantly, JNK inhibitors have been shown experimentally to ablate tumor growth in pre-clinical animal models,55 where they also effectively reduce tumor angiogenesis.56 Because JNK inhibitors have significant anti-angiogenic effects, this provides an indication they are also directly or indirectly targeting components of the tumor stroma, such as endothelial cells and/or cancer-associated fibroblasts. Thus, a JNK-based therapeutic strategy undoubtedly deserves further attention, for targeting the tumor stroma in breast cancer patients. Our findings would also suggest that a new JNK-based strategy could be used as an early intervention and/or for cancer prevention, especially in patients with high mammographic density. As JNK inhibitors behave both as anti-inflammatories and anti-fibrotics, this strategy would also prevent and/or ameliorate the pro-tumorigenic effects of cancer-associated inflammation in the tumor stroma.
High density regions of the breast functionally show increased PET avidity
18F-FDG-PET is another important imaging modality that is often used in breast cancer diagnosis and to follow the response to therapy in patients. FDG-PET avidity functionally measures glucose uptake and is thought to reflect a shift toward glycolysis. FDG-PET avidity also has been shown to be especially concentrated in areas of tissue fibrosis and inflammation, in a diverse array of human diseases. This is consistent with recent observations demonstrating that cancer-associated fibroblasts (CAFs) undergo myofibroblastic differentiation, are pro-inflammatory due to NFkB-activation, and show a metabolic shift toward aerobic glycolysis, with HIF1-stabilization.51,57 Thus, it has been proposed that cancer-associated fibroblasts58 are the preferred site of FDG-uptake in human tumors.59
Interestingly, areas of high mammographic density also show significantly increased FDG-PET avidity.60-62 These findings suggest that high mammographic density may also functionally represent areas of fibrosis, inflammation, and high glycolytic metabolism in stromal tissues. This interpretation would be consistent with our current bioinformatics analysis of HD breast fibroblasts, which provides transcriptional evidence of significant similarities with the CAF-phenotype (Table 5).
Dual role of JNK1/2 signaling in epithelial cancer cells and the tumor microenvironment
Recent studies from Roger Davis's laboratory directly support a dual role for JNK1/2 activation in tumor growth. For example, epithelial-specific deletion of JNK1/2 selectively in hepatocytes was sufficient to promote the development of hepatocellular carcinoma (HCC).63 Conversely, simultaneous deletion of JNK1/2 in both parenchymal and non-parenchymal liver cells dramatically inhibited tumor growth.63 Based on these results, Davis and colleagues concluded that JNK1/2 activation in the stromal microenvironment normally promotes carcinogenesis and tumor growth, by creating a pro-inflammatory niche.63 Interestingly, based on these genetic studies, inhibition of JNK1/2 in the tumor microenvironment appears to be dominant over loss of JNK1/2 in epithelial cancer cells.
Thus, systemic administration of JNK1/2 inhibitors would be predicted to prevent tumor growth, by inhibiting JNK1/2 in the tumor microenvironment. In support of this notion, systemic treatment with SP-600125 (a JNK1/2 inhibitor) experimentally suppresses tumor growth in several different pre-clinical xenograft models,55,56 likely by affecting the tumor microenvironment.
Similar conclusions were also reached using Drosophila as a genetic system, where the proliferation and metastasis of Ras-(G12V)-transformed cells was supported by JNK activation in adjacent neighboring cells, either via genetic manipulations or tissue injury.64
As such, we should consider using JNK1/2 inhibitors to clinically target myofibroblasts in: (1) high breast density; and (2) the tumor microenvironment, in a variety of different epithelial cancer types (Figure 11).
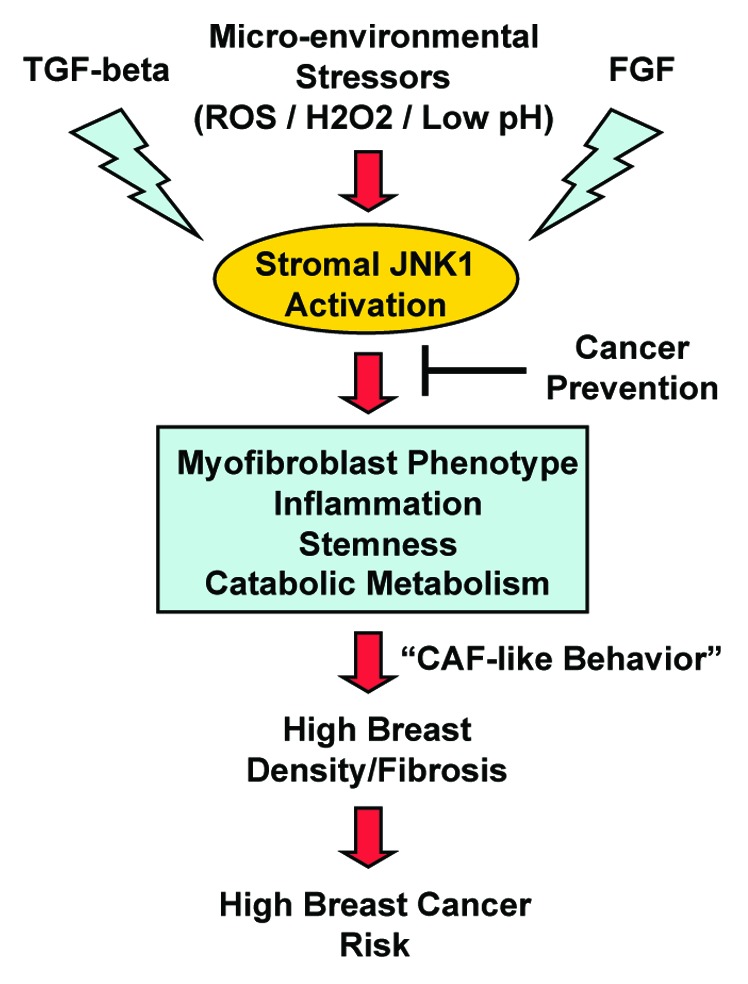
Figure 11. Understanding the role of JNK1 stress signaling in high mammographic density and breast cancer risk. Here, we postulate that microenviornmental stressors (ROS; hydrogen peroxide; acidic pH), TGF-β, and FGF signaling networks all converge on the JNK1 stress kinase, in mammary stromal fibroblasts. Then, hyper-activated JNK-signaling drives the onset of a myofibroblastic phenotype, characterized by chronic inflammation, stemness, and catabolic metabolism. This, in turn, leads to local fibrosis and high mammographic density. As such, HD fibroblasts with activated JNK1 signaling would generate a pro-tumorigenic environment, similar to what is currently observed in cancer-associated fibroblasts (CAFs) and the tumor stroma. HD fibroblasts would provide a "pre-fertilized" local microenvironment (the soil), for the successful engraftment and expansion of epithelial cancer cells (the seeds). Thus, "stromal" cancer prevention would involve the use of available JNK-inhibitors in new clinical trials, to reverse or prevent the HD fibroblast phenotype. The clinical response to JNK-therapy could be monitored by using FDG-PET-imaging, which allows the functional visualization of fibrotic and inflammatory areas of dense breast tissue.
Materials and Methods
Bioinformatics and data mining
Gene set enrichment analysis (GSEA)
For the gene set enrichment analysis we used MSigDB v3.1,65 after converting all the gene names in the database into RefSeq gene IDs. For each pair of gene sets X (e.g., set of upregulated genes) and Y (e.g., set of cell cycle genes according to MSigDB), we computed the probability (P value) that the observed overlap between sets X and Y is less than or equal to the overlap between set X and a randomly-chosen set of size equal to the size of set Y. This probability was calculated by applying the cumulative density function of the hypergeometric distribution on the size of set X, the size of set Y, the observed overlap between X and Y, and the total number of available genes. All computations, including correction for multiple hypothesis testing using the false discovery rate (FDR) were performed with the permutation test tool (permutation_test -kmin 5 -S n -p 1000 -f -a) included in the GenomicTools open-source package.66
Comparison of HD fibroblasts with gene signatures derived from the breast cancer tumor microenvironment
These comparisons were performed essentially as we have described previously, for other related data sets.12
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Dr Lisanti's and Dr Sotgia's current affiliation is the University of Manchester (United Kingdom), where they receive funding from Breakthrough Breast Cancer (BBC), The European Research Council (ERC), and the Manchester Cancer Research Centre (MCRC), as well as the University. Previously, Drs Lisanti and Federica Sotgia were supported by the resources of Thomas Jefferson University in Philadelphia, USA. Dr Ubaldo E Martinez-Outschoorn was supported by a Young Investigator Award from the Margaret Q Landenberger Research Foundation, and a K-08 Award from the NIH (K08-CA-175193).
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27379
References
- 1.McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1159–69. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- 2.Stone J, Dite GS, Gunasekara A, English DR, McCredie MR, Giles GG, Cawson JN, Hegele RA, Chiarelli AM, Yaffe MJ, et al. The heritability of mammographically dense and nondense breast tissue. Cancer Epidemiol Biomarkers Prev. 2006;15:612–7. doi: 10.1158/1055-9965.EPI-05-0127. [DOI] [PubMed] [Google Scholar]
- 3.Cuzick J, Warwick J, Pinney E, Duffy SW, Cawthorn S, Howell A, Forbes JF, Warren RM. Tamoxifen-induced reduction in mammographic density and breast cancer risk reduction: a nested case-control study. J Natl Cancer Inst. 2011;103:744–52. doi: 10.1093/jnci/djr079. [DOI] [PubMed] [Google Scholar]
- 4.Hattar R, Maller O, McDaniel S, Hansen KC, Hedman KJ, Lyons TR, Lucia S, Wilson RS, Jr., Schedin P. Tamoxifen induces pleiotrophic changes in mammary stroma resulting in extracellular matrix that suppresses transformed phenotypes. Breast Cancer Res. 2009;11:R5. doi: 10.1186/bcr2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, Khokha R, Boyd NF. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol Biomarkers Prev. 2001;10:243–8. [PubMed] [Google Scholar]
- 6.Hawes D, Downey S, Pearce CL, Bartow S, Wan P, Pike MC, Wu AH. Dense breast stromal tissue shows greatly increased concentration of breast epithelium but no increase in its proliferative activity. Breast Cancer Res. 2006;8:R24. doi: 10.1186/bcr1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–43. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, Berman HK, Gauthier ML, Zhao J, Hu D, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012;2:826–39. doi: 10.1158/2159-8290.CD-12-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang WT, Lewis MT, Hess K, Wong H, Tsimelzon A, Karadag N, Cairo M, Wei C, Meric-Bernstam F, Brown P, et al. Decreased TGFbeta signaling and increased COX2 expression in high risk women with increased mammographic breast density. Breast Cancer Res Treat. 2010;119:305–14. doi: 10.1007/s10549-009-0350-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardwick JC, van den Brink GR, Offerhaus GJ, van Deventer SJ, Peppelenbosch MP. NF-kappaB, p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene. 2001;20:819–27. doi: 10.1038/sj.onc.1204162. [DOI] [PubMed] [Google Scholar]
- 11.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 12.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, et al. Transcriptional evidence for the "Reverse Warburg Effect" in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer's disease, and "Neuron-Glia Metabolic Coupling". Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanazawa S, Fujiwara T, Matsuzaki S, Shingaki K, Taniguchi M, Miyata S, Tohyama M, Sakai Y, Yano K, Hosokawa K, et al. bFGF regulates PI3-kinase-Rac1-JNK pathway and promotes fibroblast migration in wound healing. PLoS One. 2010;5:e12228. doi: 10.1371/journal.pone.0012228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J Biol Chem. 2003;278:24624–8. doi: 10.1074/jbc.M301942200. [DOI] [PubMed] [Google Scholar]
- 15.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 16.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011;9:62. doi: 10.1186/1741-7015-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Genetic induction of the Warburg effect inhibits tumor growth. Oncotarget. 2012;3:1266–7. doi: 10.18632/oncotarget.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Cancer metabolism: new validated targets for drug discovery. Oncotarget. 2013;4:1309–16. doi: 10.18632/oncotarget.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Philp NJ, Pestell RG, Lisanti MP. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the "reverse Warburg effect" in positive lymph node tissue. Cell Cycle. 2012;11:1445–54. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, Sneddon S, Hulit J, Howell A, Lisanti MP. Mitochondria "fuel" breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. 2012;11:4390–401. doi: 10.4161/cc.22777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, Tanowitz HB, Sotgia F, Lisanti MP. Energy transfer in "parasitic" cancer metabolism: mitochondria are the powerhouse and Achilles' heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulsson J, Sjoblom T, Micke P, Ponten F, Landberg G, Heldin CH, Bergh J, Brennan DJ, Jirstrom K, Ostman A. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am J Pathol. 2009;175:334–41. doi: 10.2353/ajpath.2009.081030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saito RA, Micke P, Paulsson J, Augsten M, Peña C, Jonsson P, Botling J, Edlund K, Johansson L, Carlsson P, et al. Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer Res. 2010;70:2644–54. doi: 10.1158/0008-5472.CAN-09-3644. [DOI] [PubMed] [Google Scholar]
- 25.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, Birbe RC, Howell A, Pavlides S, Gandara R, et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabapathy K. Role of the JNK pathway in human diseases. Prog Mol Biol Transl Sci. 2012;106:145–69. doi: 10.1016/B978-0-12-396456-4.00013-4. [DOI] [PubMed] [Google Scholar]
- 27.Toone WM, Jones N. Stress-activated signalling pathways in yeast. Genes Cells. 1998;3:485–98. doi: 10.1046/j.1365-2443.1998.00211.x. [DOI] [PubMed] [Google Scholar]
- 28.Toone WM, Jones N. AP-1 transcription factors in yeast. Curr Opin Genet Dev. 1999;9:55–61. doi: 10.1016/S0959-437X(99)80008-2. [DOI] [PubMed] [Google Scholar]
- 29.Toone WM, Morgan BA, Jones N. Redox control of AP-1-like factors in yeast and beyond. Oncogene. 2001;20:2336–46. doi: 10.1038/sj.onc.1204384. [DOI] [PubMed] [Google Scholar]
- 30.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–95. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta. 2007;1773:1349–57. doi: 10.1016/j.bbamcr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Davies C, Tournier C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans. 2012;40:85–9. doi: 10.1042/BST20110641. [DOI] [PubMed] [Google Scholar]
- 33.Chambers JW, Pachori A, Howard S, Iqbal S, LoGrasso PV. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J Biol Chem. 2013;288:4000–11. doi: 10.1074/jbc.M112.406777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol. 2011;90:536–44. doi: 10.1016/j.ejcb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Guma M, Firestein GS. c-Jun N-Terminal Kinase in Inflammation and Rheumatic Diseases. Open Rheumatol J. 2012;6:220–31. doi: 10.2174/1874312901206010220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE, Hajjar RJ. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265. doi: 10.1038/cddis.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masamune A, Kikuta K, Suzuki N, Satoh M, Satoh K, Shimosegawa T. A c-Jun NH2-terminal kinase inhibitor SP600125 (anthra[1,9-cd]pyrazole-6 (2H)-one) blocks activation of pancreatic stellate cells. J Pharmacol Exp Ther. 2004;310:520–7. doi: 10.1124/jpet.104.067280. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Maruoka S, Horie T. Transforming growth Factor-beta1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med. 2001;163:152–7. doi: 10.1164/ajrccm.163.1.2005069. [DOI] [PubMed] [Google Scholar]
- 39.Greten FR. TAK1: Another mesh in the NF-κB - JNK controlled network causing hepatocellular carcinoma. J Hepatol. 2011;55:721–3. doi: 10.1016/j.jhep.2011.02.037. [DOI] [PubMed] [Google Scholar]
- 40.Liu S, Xu SW, Kennedy L, Pala D, Chen Y, Eastwood M, Carter DE, Black CM, Abraham DJ, Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18:2169–78. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plantevin Krenitsky V, Nadolny L, Delgado M, Ayala L, Clareen SS, Hilgraf R, Albers R, Hegde S, D'Sidocky N, Sapienza J, et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22:1433–8. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 42.Reinecke K, Eminel S, Dierck F, Roessner W, Kersting S, Chromik AM, Gavrilova O, Laukevicience A, Leuschner I, Waetzig V, et al. The JNK inhibitor XG-102 protects against TNBS-induced colitis. PLoS One. 2012;7:e30985. doi: 10.1371/journal.pone.0030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140–54. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–95. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barcus CE, Keely PJ, Eliceiri KW, Schuler LA. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. J Biol Chem. 2013;288:12722–32. doi: 10.1074/jbc.M112.447631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–32. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.You H, Padmashali RM, Ranganathan A, Lei P, Girnius N, Davis RJ, Andreadis ST. JNK regulates compliance-induced adherens junctions formation in epithelial cells and tissues. J Cell Sci. 2013;126:2718–29. doi: 10.1242/jcs.122903. [DOI] [PubMed] [Google Scholar]
- 49.Mengistu M, Brotzman H, Ghadiali S, Lowe-Krentz L. Fluid shear stress-induced JNK activity leads to actin remodeling for cell alignment. J Cell Physiol. 2011;226:110–21. doi: 10.1002/jcp.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daoud S, Schinzel R, Neumann A, Loske C, Fraccarollo D, Diez C, Simm A. Advanced glycation endproducts: activators of cardiac remodeling in primary fibroblasts from adult rat hearts. Mol Med. 2001;7:543–51. [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez-Outschoorn UE, Curry JM, Ko YH, Lin Z, Tuluc M, Cognetti D, Birbe RC, Pribitkin E, Bombonati A, Pestell RG, et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFκB target stromal MCT4. Cell Cycle. 2013;12:2580–97. doi: 10.4161/cc.25510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, Sotgia F. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs "fertilizer". Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, Sotgia F. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and "fibroblast addiction" are new therapeutic targets for drug discovery. Cell Cycle. 2013;12:2723–32. doi: 10.4161/cc.25695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gross ND, Boyle JO, Du B, Kekatpure VD, Lantowski A, Thaler HT, Weksler BB, Subbaramaiah K, Dannenberg AJ. Inhibition of Jun NH2-terminal kinases suppresses the growth of experimental head and neck squamous cell carcinoma. Clin Cancer Res. 2007;13:5910–7. doi: 10.1158/1078-0432.CCR-07-0352. [DOI] [PubMed] [Google Scholar]
- 56.Ennis BW, Fultz KE, Smith KA, Westwick JK, Zhu D, Boluro-Ajayi M, Bilter GK, Stein B. Inhibition of tumor growth, angiogenesis, and tumor cell proliferation by a small molecule inhibitor of c-Jun N-terminal kinase. J Pharmacol Exp Ther. 2005;313:325–32. doi: 10.1124/jpet.104.078873. [DOI] [PubMed] [Google Scholar]
- 57.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP. Cancer cells metabolically "fertilize" the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaudhri VK, Salzler GG, Dick SA, Buckman MS, Sordella R, Karoly ED, Mohney R, Stiles BM, Elemento O, Altorki NK, et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res. 2013;11:579–92. doi: 10.1158/1541-7786.MCR-12-0437-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lakhani P, Maidment AD, Weinstein SP, Kung JW, Alavi A. Correlation between quantified breast densities from digital mammography and 18F-FDG PET uptake. Breast J. 2009;15:339–47. doi: 10.1111/j.1524-4741.2009.00737.x. [DOI] [PubMed] [Google Scholar]
- 61.Mavi A, Cermik TF, Urhan M, Puskulcu H, Basu S, Cucchiara AJ, Yu JQ, Alavi A. The effect of age, menopausal state, and breast density on (18)F-FDG uptake in normal glandular breast tissue. J Nucl Med. 2010;51:347–52. doi: 10.2967/jnumed.109.068718. [DOI] [PubMed] [Google Scholar]
- 62.Vranjesevic D, Schiepers C, Silverman DH, Quon A, Villalpando J, Dahlbom M, Phelps ME, Czernin J. Relationship between 18F-FDG uptake and breast density in women with normal breast tissue. J Nucl Med. 2003;44:1238–42. [PubMed] [Google Scholar]
- 63.Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011;25:634–45. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463:545–8. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsirigos A, Haiminen N, Bilal E, Utro F. GenomicTools: a computational platform for developing high-throughput analytics in genomics. Bioinformatics. 2012;28:282–3. doi: 10.1093/bioinformatics/btr646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.