

Abstract
Chromosomal instability (CIN), as a common feature of tumors, represents a potential therapeutic target if ways can be found to specifically cause apoptosis in unstably dividing cells. We have previously shown that if signaling through the JNK pathway is reduced, apoptosis is triggered in models of chromosomal instability induced by loss of the spindle checkpoint. Here we identify components upstream and downstream of JNK that are able to mediate this effect, and test the involvement of p53 and DNA damage in causing apoptosis when JNK signaling is reduced in CIN cells. We show that cell cycle progression timing has a strong effect on the apoptosis seen when JNK signaling is reduced in genetically unstable cells: a shortened G2 phase enhances the apoptosis, while lengthening G2 rescues the JNK-deficient CIN cell death phenotype. Our findings suggest that chromosomal instability represents a significant stress to dividing cells, and that without JNK signaling, cells undergo apoptosis because they lack a timely and effective response to DNA damage.
Keywords: chromosomal instability, JNK, Drosophila, apoptosis, Mad2
Introduction
Chromosomal instability, the tendency to gain or lose significant amounts of DNA with each cell division, is now recognized to be a characteristic of many tumor types.1,2 These ongoing genetic changes, and the associated high mutation rate, result in a wide diversity of cells across the tumor, some of which may have a significant growth or survival advantage.3,4 This enhanced variation could, for example, explain the strong correlation between multi-drug resistance and chromosomal instability in late stage tumors.5 As there is no effective treatment for such tumors, there has been considerable interest in understanding the mechanisms by which chromosomal instability (CIN) is generated.
Gain or loss of whole chromosomes in CIN tumors has been attributed primarily to a defect in correcting improper attachments to the microtubule spindle during cell division.6 Merotelic attachments, in which one kinetochore is connected to both spindle poles, are normally removed before the sister chromatids are separated at anaphase. Changes to the rate at which merotelic attachments form, or are cleared, or the time available to remove them, can result in a lagging chromosome that fails to segregate at anaphase. Lagging chromosomes may not only lead to aneuploid progeny, but potentially also to DNA damage on the affected chromosome.7,8
The structural chromosomal instability that produces translocations has been associated with the formation of chromatin bridges during anaphase.9 Anaphase bridges can be caused by dicentric chromosomes, which are usually formed as a result of non-homologous end-joining DNA damage repair. Alternatively, they can result from chromosomes that have not entirely decatenated following replication or DNA damage repair, and thus are still linked when anaphase begins.10 In both cases, the anaphase bridge will be broken at some point before cytokinesis is completed, resulting in gain or loss of a chromosome section as well as a DNA double-stranded break that must be repaired.
With the incidence of CIN being extremely high in some tumor types,11 and the prognosis for CIN tumors being significantly worse than for non-CIN tumors,12 it is plausible that instability promotes cancer progression. On the other hand, chromosomal instability is not seen in normal dividing cells, so it is also a significant point of difference between normal and cancerous cells that can potentially be targeted for therapy. One approach to exploit this difference has been to increase the rate of mitotic errors, in an attempt to push already highly unstable cancer cells over their tolerance threshold.13 This may prove to be clinically useful, but has the drawback of increasing the mutation rate in normal dividing cells. Ideally we would like to identify interventions that have no significant effect on normal cells, but kill cells with chromosomal instability.
To identify genes that could be targeted to induce CIN-specific cell death, we used a Drosophila model to carry out a preliminary genetic screen for modifiers of the fate of CIN cells.14 We induced chromosomal instability by knocking down the spindle checkpoint protein Mad2, which reduces the time available to correctly orient the chromosomes at metaphase15 and leads to a significant rate of anaphase errors.14 We tested the set of kinases and phosphatases in Drosophila for those that caused apoptosis when knocked down in our induced CIN wing imaginal cells, but did not cause apoptosis when knocked down in control cells without CIN. A set of genes were identified that did not affect levels of chromosomal instability in normal cells, but were necessary for the survival of CIN cells and, as such, were of interest for anti-CIN therapy. Among these were Jun N-terminal kinase (JNK) and some of its potential regulators.
JNK, originally identified as a stress response kinase, has been implicated in many cellular responses to stress, including apoptosis, DNA damage repair, autophagy, and antioxidant production.16,17 Cell stresses can activate an upstream sensor such as p53, ATM, or one of the MAPKKKs, leading to signal transduction through kinases to produce activated JNK.18 JNK can be activated by a wide range of stimuli and regulates an equally wide range of targets directly by phosphorylation or indirectly through transcription (eg AP1 targets). In order to understand the mechanisms of CIN cell survival, we therefore wished to know which of these JNK signaling processes were required in CIN cells to avoid cell death.
Here we identify the JNK signaling pathway that is required for CIN cell survival, and show that the resulting cell death induced when JNK is reduced in CIN cells is caspase-mediated. We show that apoptosis in JNK-reduced CIN cells is partly p53-independent and identify a critical role for JNK signaling in G2 to avoid premature mitosis and consequent cell death.
Results
Our previous screen of kinases and phosphatases identified JNK as a target that could be knocked down to kill cells with induced chromosomal instability.14 In order to characterize the regulatory pathway involved, we tested a range of known mediators of JNK signaling for their effect on CIN tolerance (Table 1). We observed a significant reduction in CIN tolerance when we knocked down several JNK regulatory kinases, including JNKK (hemipterous), JNKKKs (slpr and tak1), and Ste-20-related regulators of JNKKKs (mbt, gckIII). To confirm that these effects on survival reflected CIN-specific cell death, we knocked down these JNK regulators in a proliferating epithelium (the developing wing disc) with or without inducing chromosomal instability (Fig. 1; Fig. S1A). We observed little cell death in wild-type third instar wing discs, or when half of the disc had reduced Mad2 to induce CIN (Fig. 1A and A’). Reduction of JNK or its regulators also induced little cell death in a wild-type background (Fig. 1B–D). In contrast, reduction of JNK signaling in cells with induced CIN gave significant levels of cell death (Fig. 1 B’–D’). Consistent with high levels of cell death and consequent loss of tissue, adult wings showed notching when JNK signaling was reduced in a CIN background (data not shown). Knockdown of the JNK targets FOXO and 14-3-3ζ also strongly induced cell death in CIN cells, but not normal dividing cells (Fig. 1E–F’), as did knockdown of Jun (Jra) and 14-3-3ε (data not shown).
Table 1. Survival of JNK pathway knockdowns in a CIN background.
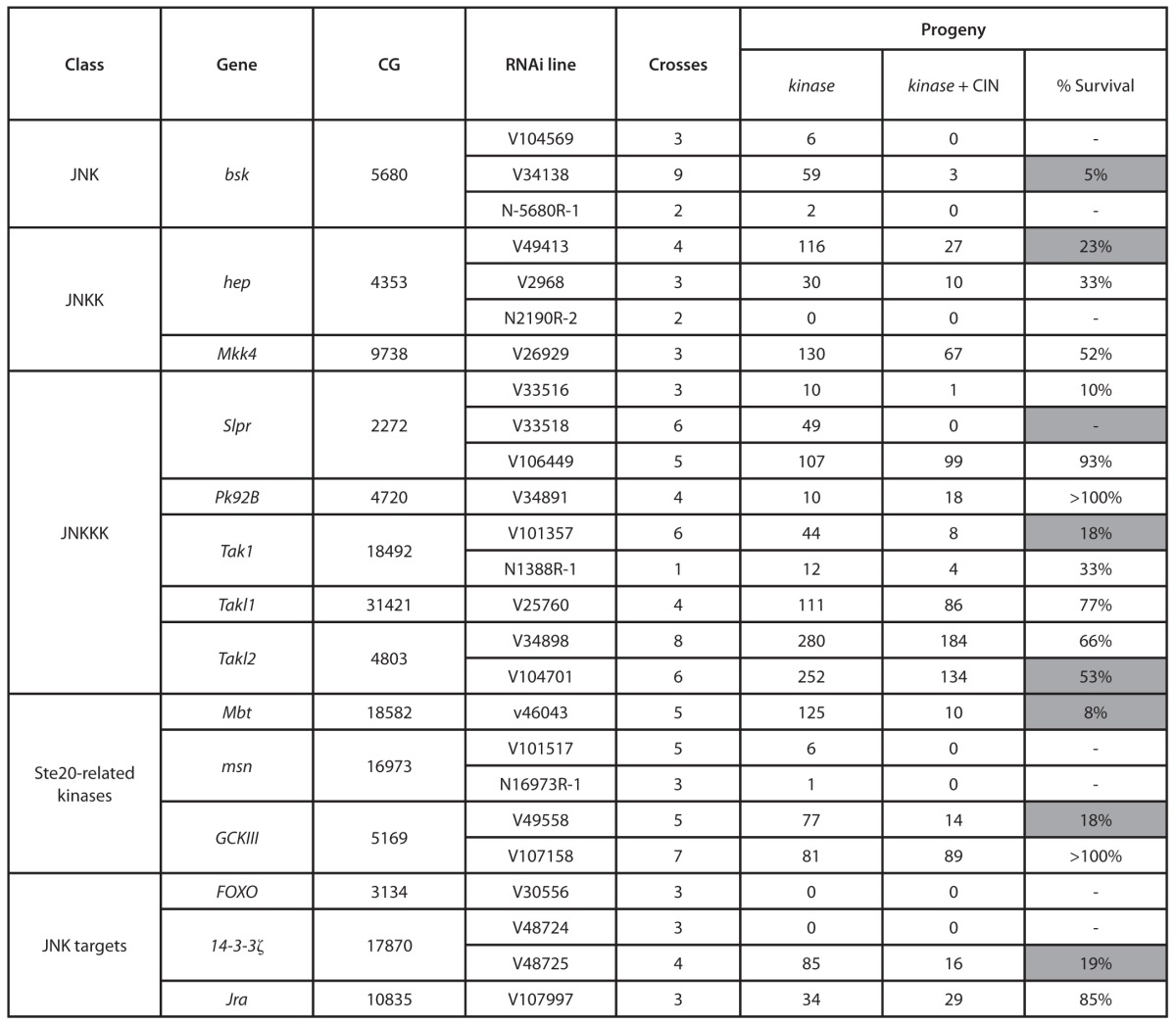
The indicated number of crosses of each RNAi line for JNK pathway members was set up with our test strain. This cross knocks down the JNK pathway member in all cells, with or without also knocking down Mad2 to generate CIN. The indicated number of sibling progeny were obtained. The percent survival shows the ratio of siblings surviving in the CIN background to those surviving in a wild-type background: 100% would indicate equal numbers with and without CIN. Shaded boxes show results that would not be expected to have occurred by chance if there were no CIN effect (P < 0.001) using the binomial distribution (see “Materials and Methods”).
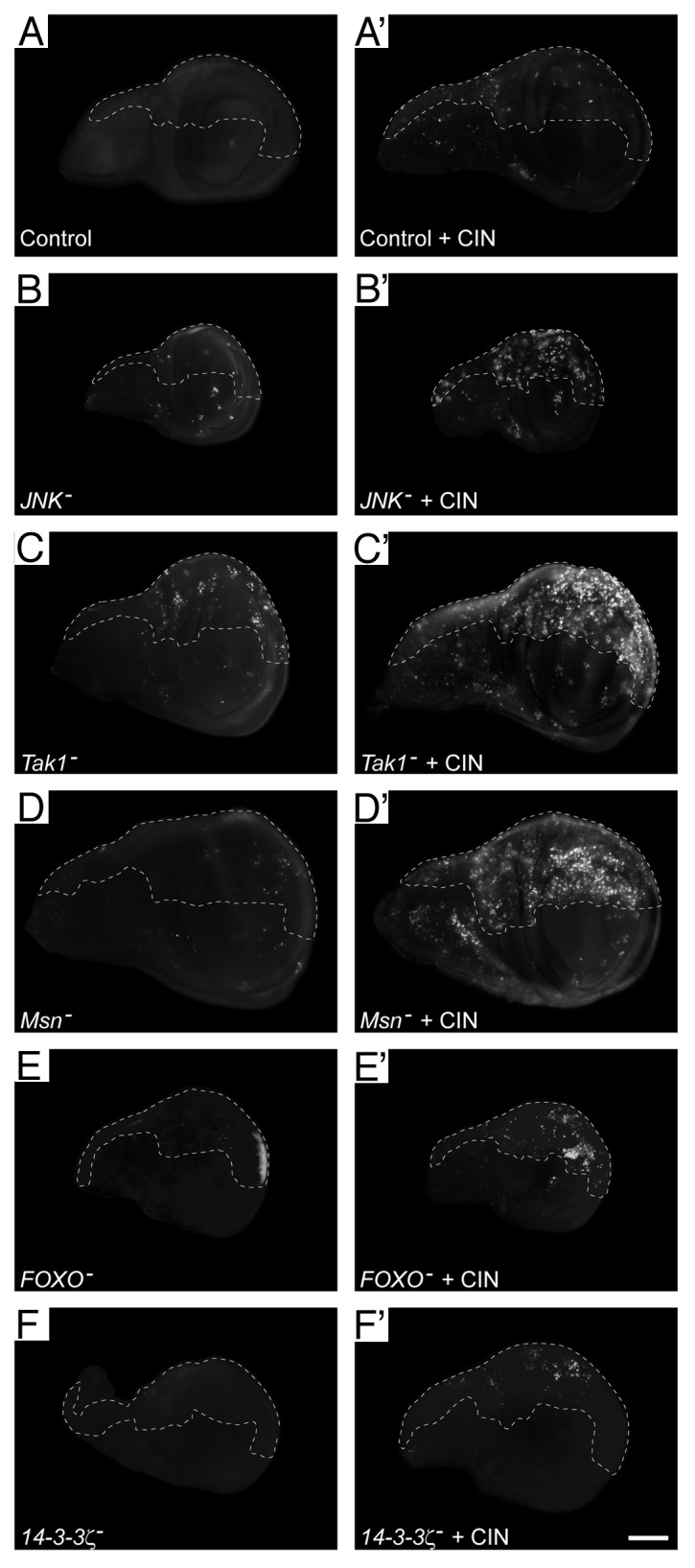
Figure 1. Knockdown of the JNK signaling pathway leads to cell death in CIN cells. Wing discs were stained with Acridine Orange to indicate cell death. In every disc, the unmarked region (anterior compartment) is the control tissue, in which no RNAi constructs are expressed, while the dashed line shows the posterior compartment in which JNK pathway members were knocked down by RNAi expression with or without CIN induced by mad2-RNAi expression. In control discs (A), induction of CIN caused little cell death (A’). For members of the JNK signaling pathway, the left column (B–F) shows discs in which JNK, Tak1, Msn, FOXO, or 14-3-3ζ have been reduced in the posterior compartment (dashed), with little cell death in each case. The right column (B’–F’) shows discs in which CIN has been induced (mad2-RNAi) in the posterior compartment along with knockdown of the indicated JNK pathway member. For each of these JNK pathway members, RNAi knockdown in CIN cells causes cell death that is not seen in the normally dividing control cells. The scale bar shows 100 μm
To confirm that the cell death observed was related to chromosomal instability rather than a specific interaction with Mad2, we tested knockdown of JNK signaling when BubR1 was knocked down. BubR1 is needed for the spindle assembly checkpoint, and its reduction leads to significant chromosomal instability.19 In this background, reduction of JNK also led to a significant increase in cell death (Fig. 4G and I; Fig. S1E). From these results, we conclude that signal transduction through the canonical JNK pathway is required for cells to be able to survive chromosomal instability induced by a weakened spindle checkpoint.
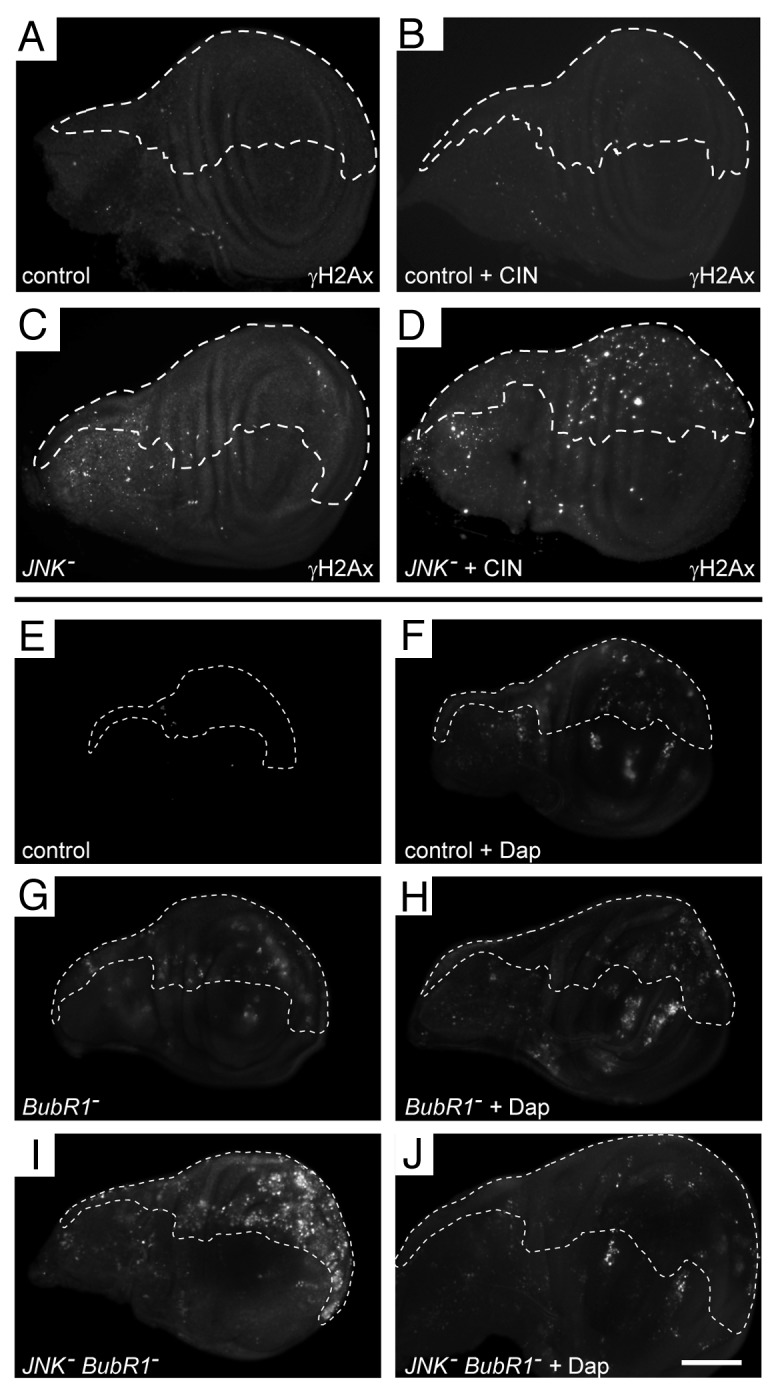
Figure 4. DNA damage is induced when JNK is reduced in CIN cells, and the cell death induced by knockdown of the JNK pathway in CIN cells is cell cycle-dependent. (A–D) Wing discs were stained for double-stranded DNA breaks (anti-P-H2AvD/γH2Ax). In every disc, the unmarked region does not express RNAi constructs, while the dashed line shows the posterior compartment cells that are affected by CIN (mad2-RNAi) and/or JNK knockdown. Control discs (A and B) show little DNA damage, even when CIN was induced (B). Knockdown of JNK also shows little DNA damage (C), but this is increased when JNK is reduced in CIN cells (D). (E–J) Wing discs were stained with Acridine Orange to show cell death. In each disc, the unmarked region does not express RNAi constructs, while the dashed line shows the area affected by overexpression of Dacapo and/or reduction of the indicated gene by RNAi. Control discs (E and F) show little cell death, although cell cycle arrest by overexpression of Dacapo (F) does cause some cell death, including death outside the overexpressing region (also seen in H and J). Induction of CIN by reducing BubR1 (G) causes some cell death, which is not altered by Dacapo overexpression (H). Knocking down JNK in CIN cells (I) gives considerable cell death, which is greatly reduced by overexpression of Dacapo (J). The scale bar shows 100 μm.
We next examined the mechanism by which death was induced in CIN cells with reduced JNK signaling. Inhibiting caspase signaling by expressing p3520 was able to prevent the cell death observed for knockdown of JNK, JNKKK (tak1) or PAK2 (mbt), suggesting that caspase-driven apoptosis was involved (Fig. 2; Fig. S1B). Consistent with this, knockdown of any of the canonical apoptotic mediators Hid, Bax (Debcl), and APAF (Ark) resulted in significantly reduced levels of cell death in JNK-reduced CIN cells (Fig. 2I). We conclude that the cell death observed corresponds to apoptosis mediated by the canonical caspase-mediated pathway. Elevated staining for activated Caspase-3 confirmed that JNK-reduced CIN cells die by caspase activation (data not shown). A typical activator of apoptosis through this pathway is p53, which is a problem clinically as so many cancers lack p53.21 We tested whether p53 was required for the CIN-specific apoptosis we had observed. Knockdown of either JNK or JNKKK (tak1) or PAK2 (mbt) in CIN cells gave apoptosis that was only modestly reduced by knockdown of p53 (Fig. 3; Fig. S1C). This mild effect was not simply due to ineffective depletion of p53, as this p53–RNAi construct caused complete loss of the apoptosis seen with other candidates from our screen, such as Pask.14 One alternative to p53 activation of apoptosis is E2F1, which can also activate the caspases through Hid.22 Knockdown of E2F1 was also able to reduce the level of apoptosis seen in JNK-reduced CIN cells (Fig. S2). We conclude from these results that reduced JNK signaling in CIN cells can lead to activation of the classical apoptotic response through p53, but can also use a p53-independent mechanism.
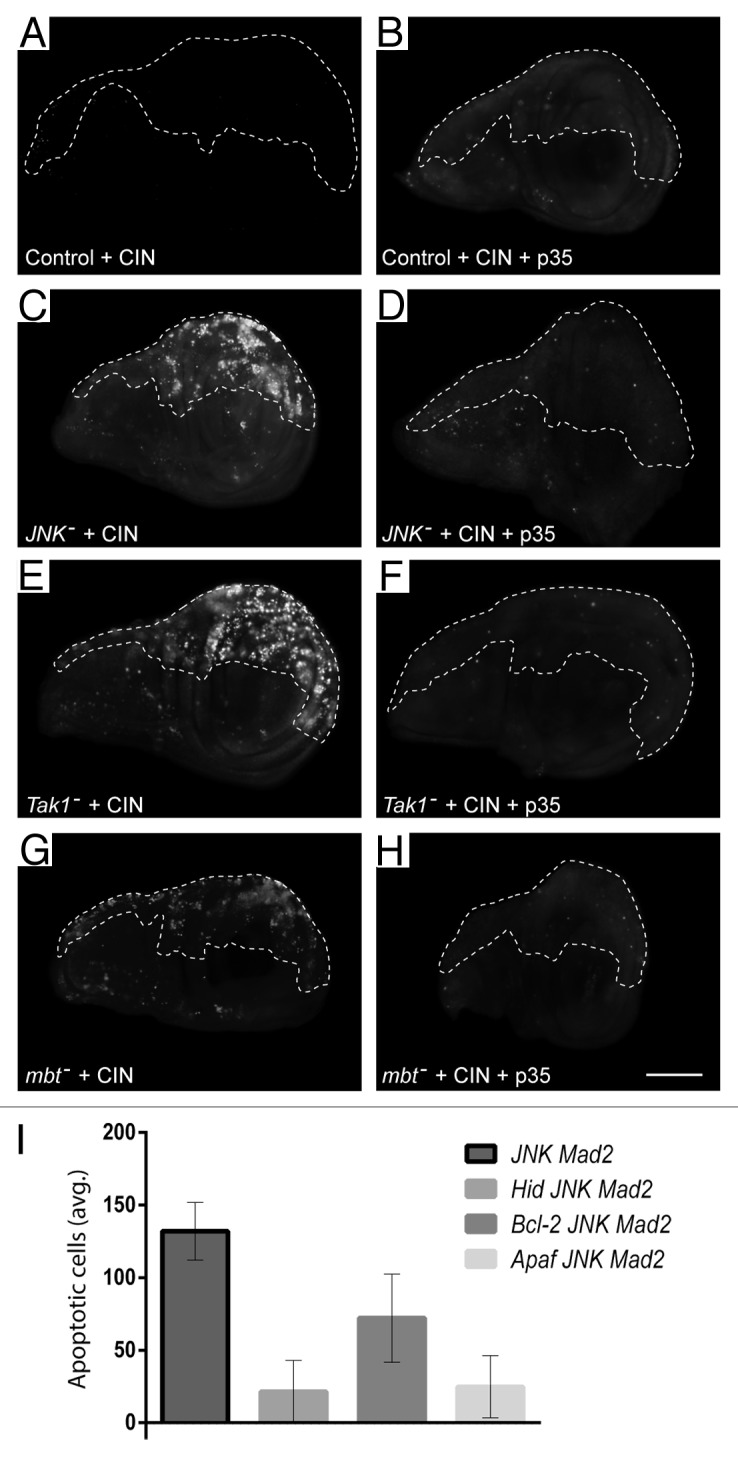
Figure 2. The cell death observed when the JNK pathway is knocked down in CIN cells can be greatly reduced by expressing the caspase inhibitor p35 or blocking the canonical apoptosis pathway. Wing discs were stained with Acridine Orange to show cell death. In every disc, the unmarked region does not express RNAi constructs, while the dashed line shows the posterior compartment affected by CIN (mad2-RNAi) and/or p35 overexpression. In control discs (A and B) CIN produced little cell death. When JNK was reduced in CIN cells (C), high levels of cell death were observed. This cell death was blocked by the caspase inhibitor p35 (D). The same inhibition of cell death by p35 was seen in CIN cells knocked down for the upstream JNK regulators Tak1 (E and F) and Mbt (G and H). The scale bar shows 100 μm. (I) The effect of knocking down the canonical apoptosis mediators Hid, Bcl-2, or Apaf on the amount of cell death seen in JNK-reduced CIN cells. The graph indicates the normalized average number of apoptotic cells per affected wing half for each genotype. Error bars indicate the 95% CI.
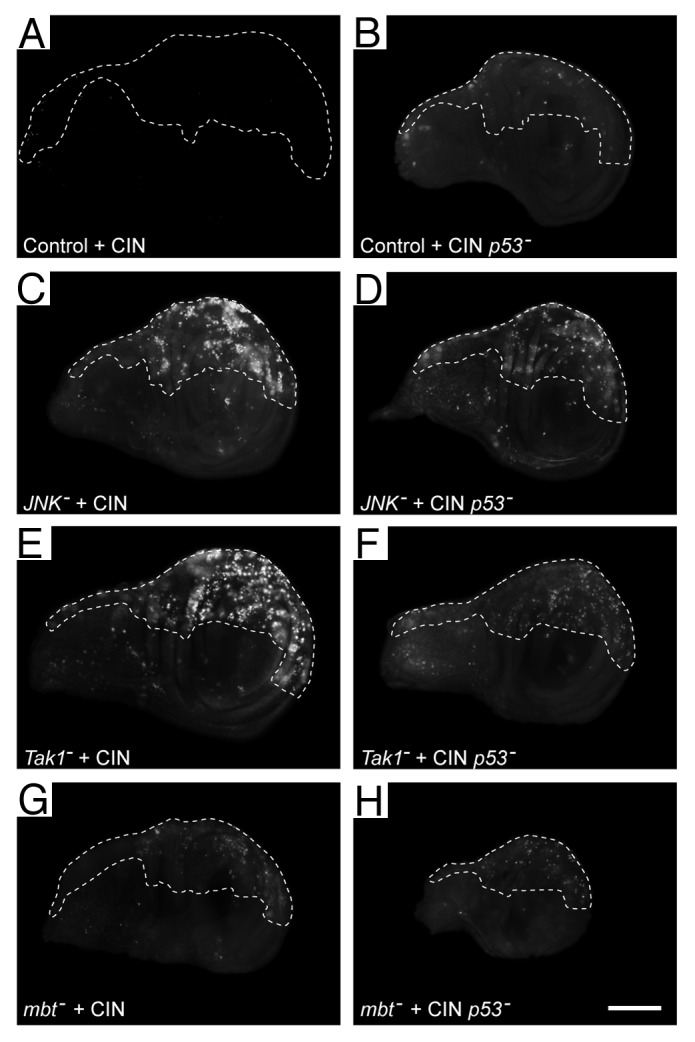
Figure 3. Some of the cell death observed when the JNK pathway is reduced in CIN cells is independent of p53. Wing discs were stained with Acridine Orange to show cell death. In every disc, the unmarked region does not express RNAi constructs, while the dashed line shows the area affected by CIN (induced by the expression of mad2-RNAi) and/or kncockdown of p53. Control wings (A and B) show little cell death when CIN is induced (A) or when p53 is knocked down in CIN cells (B). (C, E, and G) Imaginal discs in which members of the JNK signaling pathway, (JNK, Tak1, or Mbt) have been knocked down in CIN cells, giving rise to high levels of cell death. (D, F, and H) Imaginal discs showing that cell death is reduced but not eliminated by knockdown of p53 in CIN cells that are also knocked down for the indicated JNK pathway member. The scale bar shows 100 μm.
Both p53 and E2F1 promote apoptosis in response to DNA damage, so we tested for the presence of double-stranded DNA breaks in JNK-reduced CIN cells. We observed some DNA damage in cells with induced CIN, or reduced for JNK signaling (Fig. 4; Fig. S1D). Strikingly, however, cells with both CIN and reduced JNK signaling showed a significantly increased amount of DNA damage (Fig. 4D). It is known that passage through mitosis with a defective spindle checkpoint can be responsible for DNA damage, for example by failure to resolve late-replicating DNA before an early anaphase,23 or the late mitotic damage observed in Mad2-reduced vertebrate cells.7 Both of these sources of DNA damage require cells to pass through mitosis, so we tested whether blocking cell cycle progression would prevent the DNA damage and consequent apoptosis (Fig. 4; Fig. S1E). Overexpression of the cdk2 inhibitor Dacapo leads to a G1 arrest,24 and we found that inducing this cell cycle arrest was able to greatly reduce the amount of apoptosis seen in JNK-reduced CIN cells (Fig. 4J).
Having found that cell cycle progression was needed to observe apoptosis in JNK-reduced CIN cells, we considered how JNK might affect the process and interact with the spindle assembly checkpoint defect. One possibility was that loss of JNK signaling might cause defects in G2, as the processes of late replication and DNA repair must be completed in G2 if cells are to avoid a catastrophe in mitosis.25 To test whether CIN cells were particularly sensitive to G2 completion, we generated cells with a short G2 by overexpressing the M-phase entry factor Cdc25c (string).26 Shortening G2 in CIN cells led to an increase in the amount of DNA damage and cell death (Fig. 5; Figs. S1F and S3). Shortening G1 by overexpressing Cyclin E had less effect. We concluded from these experiments that CIN cells are particularly sensitive to an early onset of mitosis.

Figure 5. Cell death is increased in CIN cells when G2 is shortened and decreased when G2 is lengthened. Wing discs were stained with Acridine Orange to show cell death. In every disc, the unmarked region does not express RNAi constructs, while the dashed line shows the posterior compartment that is affected by CIN (mad2-RNAi) and/or gene overexpression. Control wings (A and B) show little cell death, even when CIN is induced (B). Cells with a shorter G1 phase from overexpression of Cyclin E (C and D) also show little cell death even when CIN is induced (D). Cells with a shorter G2 phase from overexpression of Cdc25 (stg-RNAi) (E and F) show some cell death, which is strongly enhanced by the induction of CIN (F). Cell death induced by JNK knockdown in CIN cells (G, H, and I) is reduced by lengthening G2 either by Myt1 overexpression (H) or loss of one copy of Cyclin B (I). The scale bar shows 100 μm.
Having observed that CIN cells were sensitive to early mitosis onset and that DNA damage was detected in JNK-reduced CIN cells before they went on to apoptose, we hypothesized that JNK was needed to delay mitotic onset following DNA damage, i.e., to trigger the DNA damage checkpoint. Consequently, we tested whether the DNA damage checkpoint remained intact in JNK-reduced cells. Ionizing radiation was used to generate a robust G2-phase arrest that could be removed by knockdown of the DNA damage checkpoint protein Chk1, but was unaffected by JNK knockdown (Fig. S4), with almost no cells escaping arrest. We conclude that JNK was not needed to arrest the cell cycle in G2 following massive externally induced DNA damage.
An alternative explanation for the effect of JNK knockdown on CIN cells is that JNK affects the efficiency with which DNA was repaired. JNK has been implicated in DNA damage repair27 and regulates FOXO and Gadd45, key mediators of DNA damage responses.28 If the G2 defect is in DNA repair, we would expect that CIN-specific apoptosis would also be seen when DNA damage-sensing and repair proteins are depleted. To test this, we examined CIN cells knocked down for either Grapes (Chk1) or Loki (Chk2), 2 key effectors of DNA repair.29 Induction of CIN when either of these proteins was knocked down led to a significant increase in apoptosis (Fig. S5), confirming that CIN cells are particularly sensitive to defects in DNA repair. If knockdown of JNK reduces the cells’ ability to repair DNA damage in the time available in G2, then delaying the onset of mitosis should be able to reduce the level of apoptosis seen in JNK-reduced CIN cells. To test this hypothesis we extended the length of G2 either by overexpressing the Cdk1 inhibitor Myt130 or by reducing the amount of cyclin B.31 In both cases we observed a reduction in the amount of apoptosis in JNK-reduced CIN cells (Fig. 5G–I; Fig. S1G), suggesting that JNK-reduced cells benefit from a longer G2 phase. We conclude from these results that although JNK does not affect the ability of the DNA damage checkpoint to arrest cells in G2 following massive damage, our data are consistent with a model in which reduced JNK signaling reduces the cell’s ability to effectively repair DNA damage before the onset of mitosis.
Discussion
Here we report studies exploring the surprising finding that suppression of JNK activity in wing imaginal disc cells exhibiting chromosomal instability (CIN) results in highly elevated levels of cell death not observed with either CIN or JNK knockdown alone.14 Our analysis suggests the involvement of a typical JNK signaling cascade from a MAPKKK through to the JNK targets FOXO and Jun. We show that the apoptosis observed in JNK-reduced CIN cells corresponds to canonical caspase-mediated apoptosis, and that JNK-reduced CIN cells exhibit high levels of DNA damage, as measured by γH2AX staining. The enhanced apoptosis and DNA damage phenotypes induced by JNK knockdown occur in cells exhibiting CIN but not in cells in which the cell cycle alone is deregulated. We also show that shortening the G2 phase (but not G1 phase) induces apoptosis in CIN cells and enhances the level of caspase in JNK-reduced CIN cells, while lengthening the G2 phase (but not G1 phase) suppresses the levels of apoptosis
In most cases, the analysis of the JNK pathway with respect to CIN has been limited to the use of JNK mutants or inhibitors (e.g., refs. 32 and 33), with little information available about what the upstream and downstream mediators might be. Our analysis suggests the involvement of a typical kinase signaling cascade from a MAPKKK through to the JNK targets FOXO and Jun. This kind of JNK response is induced by several stresses, for example in TNFα or heat shock responses.34,35
We have found that signaling through JNK is needed for cells to tolerate chromosomal instability induced by spindle checkpoint defects. This might appear surprising given the amount of data showing that signaling through JNK can lead to apoptosis.18 While there is no doubt that sustained activation of JNK leads to apoptosis, transient activation of JNK can instead promote cell survival following stress.36 Consistent with this, JNK is needed in Drosophila for effective stress tolerance.37 The stress involved in our case is imposed by the weakening of the spindle checkpoint, which leads to a high rate of anaphase errors and DNA damage. It has been shown in several organisms that even relatively minor aneuploidy can cause proteotoxic stress, JNK activation, and DNA damage sensitivity.38-40 In the context of cancer, loss of JNK has been reported to reduce the incidence of tumors in several mouse models,17 again consistent with a model in which signaling through JNK is necessary to tolerate the stresses of cellular transformation.
Nonetheless, given that activation of JNK is an essential feature of some apoptotic responses such as to irradiation,41,42 the question arises as to how cells that lack JNK are able to die in response to CIN, particularly as some CIN models generate JNK-dependent apoptosis.38 RNAi screening for knockdowns that cause apoptosis in normal wing discs has shown that although JNK activation was common when cells died, there were more than 200 knockdowns that could induce Caspase 3 activation with no JNK activation.43 Similarly, TNFα has been shown to induce JNK-independent cell death,44 so clearly JNK activation is only one of many methods for triggering apoptosis. In fact, reduced JNK signaling can even enhance cell death: knockdown of JNK sensitizes cells to CD95-mediated apoptosis,45 and phosphorylation of FOXO by JNK is needed for a wide range of stress survival responses.46
These results suggest a model in which cells require appropriate levels and timing of JNK activation in response to stress. Too much JNK signal may be interpreted as irreparable damage, triggering JNK-dependent apoptosis, which has been seen when hyperploidy is induced by centrosome defects.38 On the other hand, some JNK is needed to activate normal stress response genes, so without JNK the damage will accumulate until alternative mechanisms trigger apoptosis. We have shown that DNA damage is at least one of these alternative mechanisms: with reduced JNK signaling, there is a significant accumulation of unrepaired DNA damage in CIN cells. The observation that JNK knockdown in normal cells does not result in widespread DNA damage underlines that chromosomal instability is a significant stressor. As noted above, this may be partly through aneuploidy giving proteotoxic stress,39 while spindle checkpoint defects can also lead to DNA double-stranded breaks in telophase or S phase.7,8 The data reported here also support a model in which reduced mitotic timing caused by the checkpoint defect results in cells that are highly susceptible to unrepaired DNA damage. There is now strong evidence that sections of chromosomes are often still catenated even after anaphase onset, leading to ultrafine anaphase bridges that must be cleared by helicases during anaphase to avoid breaks.47 Treatments that increase the amount of still-catenated DNA in cells entering mitosis increase the amount of bridging and breakage, so we speculate that loss of Mad2 makes this worse, as it reduces the available time for repair.15 Consistent with this model, the majority of anaphase defects observed in our Mad2 knockdown CIN model are bridges (data not shown).
The functions of JNK in G2 are still unclear, particularly in Drosophila. A study in vertebrate cells has implicated JNK in the DNA damage checkpoint,48 though this study used an inhibitor of doubtful specificity.49 We also know that DNA damage checkpoint genes such as Chk2 are, like JNK, required for the survival of CIN cells,14 so it was plausible that loss of JNK might have been disrupting the DNA damage checkpoint. However, we saw no effect of JNK knockdown on the ability of cells to arrest in response to ionizing radiation. The DNA damage checkpoint thus appeared to be intact, at least in terms of responding to massive damage induced by γ-irradiation. JNK is also known to mediate the phosphorylation of histone 2A variants as part of DNA damage detection,50 so the primary problem in our JNK reduced cells could have been in detecting the DNA damage. We do not think this contributes significantly to the phenotype, however, as we detect elevated rather than reduced levels of phospho-histone2Av in JNK-reduced CIN cells.
In support of a third alternative explanation, there is clear evidence for a role for JNK signaling in promoting efficient DNA damage repair via AP1.51,52 Consistent with this model, we found that simply increasing the time available in G2 was able to significantly reduce the apoptosis in JNK-reduced CIN cells. This is a remarkable result, as it suggests that repair can be performed, given enough time, but that the DNA damage checkpoint does not give enough time in JNK-reduced CIN cells, which accumulate unrepaired DNA damage as the cells cycle. This is consistent with a model in which the role of JNK is primarily to ensure the timely production of repair enzymes in response to stress. Failure to repair damage before mitosis can lead to anaphase chromatin bridges and subsequent tetraploidy or bridge-breakage-fusion cycles that perpetuate the damage.53
Our observation that JNK signaling is needed to tolerate CIN is consistent with the elevated expression levels of JNK seen in tumors.17 This has been cited as surprising, as JNK can induce apoptosis. The explanation may be simply that although a tumor may be highly apoptosis resistant, it still has a need to manage high levels of DNA damage generated by CIN and ROS.54 The JNK levels must be high enough, then, to increase both the expression of antioxidants and to ensure effective DNA repair in G2. Whether this represents a therapeutic opportunity remains to be seen, but the development of specific and effective JNK inhibitors55 may permit testing in tumor cells of the JNK–CIN interaction described here.
Materials and Methods
Acridine Orange stains for cell death were performed as described.14 Briefly, third instar larval wing discs were dissected in PBS, then stained for 2 min in a 1 μM Acridine Orange solution (Invitrogen; http://products.invitrogen.com/ivgn/product/A1301), rinsed briefly, and imaged in PBS. Note that this is a live stain, so some variation in background levels is expected. Contrast, brightness and gamma levels were adjusted in Photoshop to most clearly reproduce the signal while retaining enough background to visualize the tissue. A representative disc was selected from at least 10 for each experiment shown. All original images are available on request. Quantitation of Acridine staining in Figure 2I was normalized by subtracting the number of Acridine Orange-positive cells in the wild-type anterior half of the wing from the number of Acridine Orange-positive cells in the posterior half (marked with mCD8-GFP) using ImageJ software (Cell Counter plugin). All other images were quantitated by measuring the average brightness in the anterior half and normalized by subtracting the average brightness of the wild-type posterior half. Genes were knocked down using either engrailed-Gal4 or hedgehog-Gal4 to drive RNAi expression in the posterior compartment of the larval wing imaginal disc. Note that it has been observed that disruption of the posterior compartment can potentially induce non-autonomous cell death in the anterior compartment,56 and this is clear in some of our genotypes (e.g., Fig. 4F, H, and J). Because we normalized using the anterior compartment, in these cases the quantification of apoptosis in the posterior compartment will be an underestimate.
Immunostainings were performed using standard protocols as described.14 Antibodies used were mouse anti-P-Histone3 (Cell Signaling 1:100; http://www.cellsignal.com/products/9701.html), Rabbit anti-P-H2AvD (Rockland 1:500; http://www.rockland-inc.com/Product.aspx?id=34929), goat anti-rabbit Alexa 568 (Invitrogen 1:100; http://products.invitrogen.com/ivgn/product/A11036), donkey anti-mouse Dylight 649 (Jackson 1:100). Ionizing radiation treatment was done by placing third instar larvae on sealed agar plates, then exposing them to Cobalt-60 for a dose of 40 Gy. They were allowed to rest for 1 h before dissecting and staining for mitotic cells with anti-P-Histone3. Quantification of DNA damage staining was normalized by counting the number of P-H2AvD-positive cells in the posterior wing half per 10 000 pixels and subtracting the number in the anterior half per 10 000 pixels. Imaging was performed on an Olympus BX60 microscope with DP71 camera or a Zeiss Axioplan2 with AxioCam MRm camera.
Drosophila stocks
All stocks were raised at 25 °C, and most were sourced from the Bloomington stock center or the Vienna Drosophila Resource Center. Additional stocks were UAS-dacapo,24 UAS-CyclinE,57 and UAS-Myt1.30 The cyclin B allele used was CycB.2 Table 1 shows the results of crossing our test strain (UAS-Mad2RNAi/CyO; da-Gal4/TM6b Gal80ts) to each candidate RNAi line. Note that half of the progeny will carry TM6b instead of Gal4 and hence be wild-type (not shown in Table 1). All crosses listed in Table 1 produced more than 20 TM6b progeny, ensuring that when low numbers of their mutant siblings were seen, it was not due to problems with parental fertility. Statistical analysis of knockdown progeny numbers was assessed by assuming a frequency of 0.5 for CIN (Mad2-RNAi, candidate-RNAi) vs. non-CIN (candidate-RNAi alone) progeny and testing the likelihood of obtaining at least one such result by chance from the approximately 500 lines tested in this paper and Shaukat et al.,14 using the binomial distribution (1-BINOMDIST(0,500,BINOMDIST(CIN#, CIN+NONCIN#, 0.5, FALSE),FALSE)). The level of knockdown is expected to vary between RNAi lines, with a significant false-negative rate but low false-positive rate.58 Insertion site effects cannot be ruled out and may explain some of the variation between RNAi constructs targeting the same gene. KK lines with the same insertion point on the second chromosome that should not be affected by insertion site effects were V104569, V106449, V101357, V104701, V101517, V107158, and V107997.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Bloomington and VDRC stock centers, H Richardson and L Quinn for providing fly stocks, and the University of Adelaide and the University of Melbourne for student scholarships. Helpful comments and advice were given by M Bogoyevitch and M Murray. This work was funded in part by National Health and Medical Research Council grants 525477 and 1027878.
Glossary
Abbreviations:
- CIN
chromosomal instability
- JNK
Jun N-terminal kinase
- RNAi
RNA mediated gene interference
- ROS
reactive oxygen species
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27484
References
- 1.Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122:1138–43. doi: 10.1172/JCI59954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–8. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 3.Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010;103:1139–43. doi: 10.1038/sj.bjc.6605912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolodner RD, Cleveland DW, Putnam CD. Cancer. Aneuploidy drives a mutator phenotype in cancer. Science. 2011;333:942–3. doi: 10.1126/science.1211154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71:1858–70. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 2012;13:501–14. doi: 10.1038/embor.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–8. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 8.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–8. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–95. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strunnikov AV. One-hit wonders of genomic instability. Cell Div. 2010;5:15. doi: 10.1186/1747-1028-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 12.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–8. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 13.Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A. 2009;106:19108–13. doi: 10.1073/pnas.0904343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaukat Z, Wong HW, Nicolson S, Saint RB, Gregory SL. A screen for selective killing of cells with chromosomal instability induced by a spindle checkpoint defect. PLoS One. 2012;7:e47447. doi: 10.1371/journal.pone.0047447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buffin E, Emre D, Karess RE. Flies without a spindle checkpoint. Nat Cell Biol. 2007;9:565–72. doi: 10.1038/ncb1570. [DOI] [PubMed] [Google Scholar]
- 16.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–95. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 17.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 18.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–9. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Basu J, Bousbaa H, Logarinho E, Li Z, Williams BC, Lopes C, Sunkel CE, Goldberg ML. Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila. J Cell Biol. 1999;146:13–28. doi: 10.1083/jcb.146.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lannan E, Vandergaast R, Friesen PD. Baculovirus caspase inhibitors P49 and P35 block virus-induced apoptosis downstream of effector caspase DrICE activation in Drosophila melanogaster cells. J Virol. 2007;81:9319–30. doi: 10.1128/JVI.00247-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 22.Wichmann A, Uyetake L, Su TT. E2F1 and E2F2 have opposite effects on radiation-induced p53-independent apoptosis in Drosophila. Dev Biol. 2010;346:80–9. doi: 10.1016/j.ydbio.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–60. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 24.Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87:1225–35. doi: 10.1016/S0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- 25.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–6. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehman DA, Patterson B, Johnston LA, Balzer T, Britton JS, Saint R, Edgar BA. Cis-regulatory elements of the mitotic regulator, string/Cdc25. Development. 1999;126:1793–803. doi: 10.1242/dev.126.9.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes KJ, Meares GP, Chambers KT, Corbett JA. Repair of nitric oxide-damaged DNA in beta-cells requires JNK-dependent GADD45alpha expression. J Biol Chem. 2009;284:27402–8. doi: 10.1074/jbc.M109.046912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr., DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 29.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price DM, Jin Z, Rabinovitch S, Campbell SD. Ectopic expression of the Drosophila Cdk1 inhibitory kinases, Wee1 and Myt1, interferes with the second mitotic wave and disrupts pattern formation during eye development. Genetics. 2002;161:721–31. doi: 10.1093/genetics/161.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu W, Jiang F, Bi X, Zhang YQ. Drosophila FMRP participates in the DNA damage response by regulating G2/M cell cycle checkpoint and apoptosis. Hum Mol Genet. 2012;21:4655–68. doi: 10.1093/hmg/dds307. [DOI] [PubMed] [Google Scholar]
- 32.Chen P, O’Neal JF, Ebelt ND, Cantrell MA, Mitra S, Nasrazadani A, Vandenbroek TL, Heasley LE, Van Den Berg CL. Jnk2 effects on tumor development, genetic instability and replicative stress in an oncogene-driven mouse mammary tumor model. PLoS One. 2010;5:e10443. doi: 10.1371/journal.pone.0010443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Atayar C, Rosati S, Bosga-Bouwer A, Kluin P, Visser L. JNK is constitutively active in mantle cell lymphoma: cell cycle deregulation and polyploidy by JNK inhibitor SP600125. J Pathol. 2009;218:95–103. doi: 10.1002/path.2521. [DOI] [PubMed] [Google Scholar]
- 34.Geuking P, Narasimamurthy R, Lemaitre B, Basler K, Leulier F. A non-redundant role for Drosophila Mkk4 and hemipterous/Mkk7 in TAK1-mediated activation of JNK. PLoS One. 2009;4:e7709. doi: 10.1371/journal.pone.0007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonda RL, Garlena RA, Stronach B. Drosophila heat shock response requires the JNK pathway and phosphorylation of mixed lineage kinase at a conserved serine-proline motif. PLoS One. 2012;7:e42369. doi: 10.1371/journal.pone.0042369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ventura JJ, Hübner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–10. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Biteau B, Hochmuth CE, Jasper H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell. 2008;3:442–55. doi: 10.1016/j.stem.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dekanty A, Barrio L, Muzzopappa M, Auer H, Milán M. Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc Natl Acad Sci U S A. 2012;109:20549–54. doi: 10.1073/pnas.1206675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–5. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–30. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132:3935–46. doi: 10.1242/dev.01949. [DOI] [PubMed] [Google Scholar]
- 42.Luo X, Puig O, Hyun J, Bohmann D, Jasper H. Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J. 2007;26:380–90. doi: 10.1038/sj.emboj.7601484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Umemori M, Habara O, Iwata T, Maeda K, Nishinoue K, Okabe A, Takemura M, Takahashi K, Saigo K, Ueda R, et al. RNAi-mediated knockdown showing impaired cell survival in Drosophila wing imaginal disc. Gene Regul Syst Bio. 2009;3:11–20. doi: 10.4137/grsb.s2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma X, Huang J, Yang L, Yang Y, Li W, Xue L. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res. 2012;22:425–31. doi: 10.1038/cr.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuntzen C, Sonuc N, De Toni EN, Opelz C, Mucha SR, Gerbes AL, Eichhorst ST. Inhibition of c-Jun-N-terminal-kinase sensitizes tumor cells to CD95-induced apoptosis and induces G2/M cell cycle arrest. Cancer Res. 2005;65:6780–8. doi: 10.1158/0008-5472.CAN-04-2618. [DOI] [PubMed] [Google Scholar]
- 46.Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal. 2011;14:593–605. doi: 10.1089/ars.2010.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan KL, Hickson ID. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin Cell Dev Biol. 2011;22:906–12. doi: 10.1016/j.semcdb.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 48.Gutierrez GJ, Tsuji T, Cross JV, Davis RJ, Templeton DJ, Jiang W, Ronai ZA. JNK-mediated phosphorylation of Cdc25C regulates cell cycle entry and G(2)/M DNA damage checkpoint. J Biol Chem. 2010;285:14217–28. doi: 10.1074/jbc.M110.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melino M, Hii CS, McColl SR, Ferrante A. The effect of the JNK inhibitor, JIP peptide, on human T lymphocyte proliferation and cytokine production. J Immunol. 2008;181:7300–6. doi: 10.4049/jimmunol.181.10.7300. [DOI] [PubMed] [Google Scholar]
- 50.Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, Bode AM, Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christmann M, Tomicic MT, Aasland D, Kaina B. A role for UV-light-induced c-Fos: Stimulation of nucleotide excision repair and protection against sustained JNK activation and apoptosis. Carcinogenesis. 2007;28:183–90. doi: 10.1093/carcin/bgl119. [DOI] [PubMed] [Google Scholar]
- 52.MacLaren A, Black EJ, Clark W, Gillespie DA. c-Jun-deficient cells undergo premature senescence as a result of spontaneous DNA damage accumulation. Mol Cell Biol. 2004;24:9006–18. doi: 10.1128/MCB.24.20.9006-9018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol. 2012;199:871–81. doi: 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glorieux C, Dejeans N, Sid B, Beck R, Calderon PB, Verrax J. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem Pharmacol. 2011;82:1384–90. doi: 10.1016/j.bcp.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 55.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;1804:463–75. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 56.Pérez-Garijo A, Fuchs Y, Steller H. Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. Elife. 2013;2:e01004. doi: 10.7554/eLife.01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richardson H, O’Keefe LV, Marty T, Saint R. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development. 1995;121:3371–9. doi: 10.1242/dev.121.10.3371. [DOI] [PubMed] [Google Scholar]
- 58.Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–6. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.