

Abstract
We examined the relationship of clinical differences among sickle cell disease (SCD) patients in order to understand the major contributors to early mortality in a contemporary cohort.
Survival data were obtained for 542 adult subjects who were enrolled since 2002 at three university hospitals in the southeast United States. Subjects were followed for a median of 9.3 years. At enrollment, clinical parameters were collected, including hemoglobin (Hb) genotype, baseline laboratory values, comorbidities, and medication usage. Levels of soluble adhesion molecules were measured for a subset of 87 subjects. The relationship of clinical characteristics to survival was determined using regression analysis.
Median age at enrollment was 32 years. Median survival was 61 years for all subjects. Median survival for Hb SS and Sβ0 was 58 years and for Hb SC and Sβ+ was 66 years. Elevated white blood count, lower estimated glomerular filtration rate, proteinuria, frequency of pain crises, pulmonary hypertension, cerebrovascular events, seizures, stroke, sVCAM-1 and short-acting narcotics use were significantly associated with decreased survival. 42% of subjects were on hydroxyurea therapy, which was not associated with survival.
SCD continues to reduce life expectancy for affected individuals, particularly those with Hb Sβ0 and SS. Not only were comorbidities individually associated with decreased survival, but an additive effect was observed, so that those with a greater number of negative endpoints had worse survival (p<0.0001). The association of higher sVCAM-1 levels with decreased survival suggests that targeted therapies to reduce endothelial damage and inflammation may also be beneficial.
Keywords: sickle, anemia, survival, mortality, adhesion, phenotypes
BACKGROUND
While it is clear that survival of sickle cell disease (SCD) patients has improved over the last 40 years, the factors that portend positive and negative prognoses need be readdressed given the development of new treatment modalities, such as stem cell transplantation, that hold promise for cure but also carry considerable risk. SCD patients are now living long enough that many patients, families, and physicians might not wish to incur further risk in order to obtain a chance at cure. Thus, in order to give patients and physicians the opportunity to make informed decisions, more detailed data are needed to identify truly favorable and unfavorable phenotypic traits and thus help better identify individualized treatment options for patients with this disease.
Sickle cell disease was long a disease of children and young adults due to its devastating natural progression. In the 1970s, studies estimated the median survival of homozygotes in the United States to be only 14.3 years.[1]Since that time, a number of interventions have been implemented to improve quality and duration of life.[2–5] With modern advances, the survival of patients with SCD was estimated in the 1990’s to be 42 years for males and 48 years for females.[6]
Clearly, SCD is manifested by diverse presentations, and its prognosis varies across the patient population. Most notably, hemoglobin (Hb)genotype influences severity of disease. Hemoglobin SS has a significantly lower survival than Hb SC disease and Sβ+ thalassemia.[2, 6] Within these groups, therefore, research has focused on identifying phenotypes that predict an unfavorable prognosis. Prior studies have shown renal failure, seizures, acute chest syndrome (ACS), low fetal Hb level, and baseline white blood cell (WBC) count greater than 15,000 cells per cubic millimeter to be associated with decreased survival.[6]Among SCD patients on hydroxyurea therapy, it has been shown that higher total Hb, fetal Hb, reticulocyte counts, and fewer instances of acute chest syndrome were all associated with improved survival.[7]
Molecular phenotypes and biomarkers have become a new topic of investigation.[8] In particular, endothelial proteins that contribute to adhesion—such as VCAM-1, ICAM-1, alphaVbeta3 integrin, P-selectin, and E-selectin—have been described as contributors to the pathophysiology of vaso-occlusive crises.[9–18]Some of these proteins have been shown to circulate in free, soluble form at elevated levels in SCD patients. In addition, in SCD patients with crises, increased expression of these molecules on endothelial cells has been demonstrated.[19, 20]Some studies have even suggested that the ability of hydroxyurea to decrease vaso-occlusive crises, long thought to be mediated by upregulation of fetal Hb, may actually also be related to downregulation of adhesion proteins and their ligands.[21–23]These correlations suggest that adhesion proteins are potentially useful targets for prognostication as well as novel treatments for SCD.
The goal of this study was to identify associations between the presence of certain clinical phenotypes at enrollment, such as comorbid conditions and laboratory data, and decreased survival among SCD patients in a contemporary cohort.
METHODS
Subjects
Institutional Review Board approval was obtained and subjects were enrolled at their respective institutions. The present analysis included 542 adult subjects (≥ 18 years at the time of enrollment) diagnosed with SCD(Hb SS, SC, Sβ0, or Sβ+) by hemoglobin electrophoresis, globin synthesis gene studies, or genetic analysis who were regularly followed in clinic at Duke University Medical Center, the University of North Carolina Health Care System, and Grady Health System. Subjects who had received blood transfusion within 90 days prior to enrollment were excluded.
Study Design
At the time of enrollment, we evaluated subjects for characteristics suspected to influence mortality of patients with SCD using a standardized validated patient history questionnaire,[24] laboratory data, and medical chart review. Included subjects were followed for a median of 9.3 years(minimum 2.7, maximum 10.5 years). The primary endpoint was death during the follow-up period. Using a cross-sectional design, associations between baseline characteristics present at enrollment and decreased survival were determined. Baseline characteristics included comorbid conditions, baseline laboratory values, pain, and medication use, as shown in Tables I, II, and III.
TABLE I.
Prevalence of baseline clinical characteristics at the time of enrollment.
| CHARACTERISTIC | N WITH CHARACTERISTIC | N TOTAL | % WITH CHARACTERISTIC |
|---|---|---|---|
| COMORBIDITIES | |||
|
| |||
| Pulmonary Hypertension* | 73 | 222 | 32.8 |
| ACS | 354 | 475 | 74.5 |
| CHF | 28 | 470 | 6.0 |
| AVN | 150 | 461 | 32.5 |
| Leg Ulcer | 112 | 464 | 24.1 |
| Stroke | 73 | 465 | 15.7 |
| Seizure | 57 | 474 | 12.0 |
| TIA | 18 | 455 | 4.0 |
| Retinopathy | 103 | 443 | 23.3 |
| Proteinuria | 124 | 460 | 27.0 |
| Gallstones | 293 | 471 | 62.2 |
| Chronic Transfusion | 30 | 469 | 6.4 |
| Priapism | 80 | 215 | 37.2 |
|
| |||
| SURGICAL HISTORY | |||
|
| |||
| Splenectomy | 48 | 378 | 12.7 |
| Cholecystectomy | 257 | 383 | 67.1 |
|
| |||
| MEDICATIONS | |||
|
| |||
| Iron Chelator | 24 | 463 | 5.2 |
| Hydroxyurea | 184 | 441 | 41.7 |
| Daily Long Acting Narcotics | 120 | 374 | 32.1 |
| Antihypertensives | 73 | 447 | 16.3 |
| Antidepressants | 43 | 447 | 9.6 |
| Bronchodilators | 27 | 448 | 6.0 |
Defined as tricuspid regurgitant velocity ≥2.5 m/s by echocardiography
TABLE II.
Hazard ratios for clinical characteristics reported as dichotomous variables. Hazard ratios > 1 reflect increased risk of death.
| CHARACTERISTIC | N WITH CHARACTERISTIC | N TOTAL | HAZARD RATIO | 95% CONFIDENCE INTERVAL | p-VALUE |
|---|---|---|---|---|---|
| MEDICAL HISTORY | |||||
|
| |||||
| Pulmonary Hypertension* | 73 | 222 | 2.34 | 1.32, 4.14 | 0.0036 |
| Diastolic Hypertension** | 96 | 374 | 0.86 | 0.53, 1.39 | 0.5308 |
| Systolic Hypertension** | 97 | 374 | 1.11 | 0.66, 1.85 | 0.6964 |
| ACS | 354 | 475 | 1.27 | 0.81, 1.99 | 0.2945 |
| CHF | 28 | 470 | 1.51 | 0.81, 2.84 | 0.1949 |
| O2 saturation < 92% | 21 | 333 | 0.88 | 0.38, 2.06 | 0.7716 |
| Leg Ulcer | 112 | 464 | 1.66 | 1.12, 2.47 | 0.0118 |
| AVN | 150 | 461 | 0.84 | 0.57, 1.25 | 0.3939 |
| Proteinuria | 124 | 460 | 1.86 | 1.27, 2.73 | 0.0014 |
| CVE | 122 | 458 | 1.95 | 1.34, 2.83 | 0.0005 |
| Stroke | 73 | 465 | 1.64 | 1.04, 2.58 | 0.0335 |
| Seizure | 57 | 474 | 2.54 | 1.62, 3.98 | <0.0001 |
| TIA | 18 | 455 | 1.85 | 0.93, 3.70 | 0.0803 |
| Retinopathy | 103 | 443 | 0.78 | 0.50, 1.22 | 0.2815 |
| Pain Crisis in Past Year*** | 256 | 459 | 1.74 | 1.18, 2.58 | 0.0054 |
| Priapism | 80 | 215 | 1.15 | 0.64, 2.07 | 0.6348 |
|
| |||||
| SURGICAL HISTORY | |||||
|
| |||||
| Splenectomy | 48 | 378 | 1.51 | 0.87, 2.64 | 0.1453 |
| Cholecystectomy | 257 | 383 | 1.03 | 0.67, 1.59 | 0.8862 |
|
| |||||
| MEDICATIONS | |||||
|
| |||||
| Hydroxyurea**** | 172 | 386 | 0.82 | 0.55, 1.22 | 0.3228 |
| Bronchodilators | 27 | 448 | 1.08 | 0.53, 2.23 | 0.8309 |
| Desferol | 21 | 447 | 1.27 | 0.61, 2.67 | 0.5271 |
| Antihypertensives | 73 | 447 | 1.74 | 1.10, 2.73 | 0.0168 |
| Antidepressants | 43 | 447 | 1.55 | 0.81, 2.97 | 0.1827 |
| Narcotics (< daily) | 379 | 477 | 1.30 | 0.81, 2.09 | 0.2807 |
| Long Acting Narcotics (daily) | 120 | 374 | 1.20 | 0.80, 1.82 | 0.3811 |
| Short Acting Narcotics (daily) | 144 | 345 | 1.55 | 1.02, 2.36 | 0.0395 |
| Long Acting Narcotics Only (daily) | 42 | 342 | 1.07 | 0.58, 1.98 | 0.8268 |
| Short Acting Narcotics Only (daily) | 66 | 342 | 1.60 | 1.01, 2.56 | 0.0475 |
| Long AND Short Acting Narcotics(daily) | 75 | 342 | 1.15 | 0.72, 1.86 | 0.5571 |
| Long OR Short Acting Narcotics (daily) | 189 | 348 | 1.68 | 1.08, 2.61 | 0.0204 |
| Acetaminophen | 23 | 372 | 1.69 | 0.87, 3.31 | 0.1244 |
| Migraine Therapy | 4 | 446 | 1.17 | 0.16, 8.52 | 0.8783 |
Defined as tricuspid regurgitant velocity ≥2.5 m/s by echocardiography
Trait is defined as the top quartile of patients in our sample.
Defined as pain crisis requiring inpatient admission within 1 year.
Calculated in Hb SS and Hb Sβ0 patients only
TABLE III.
Hazard ratios for characteristics measured as continuous variables including all genotypes.
| TRAIT | N | HAZARD RATIO | p-VALUE |
|---|---|---|---|
| Hemoglobin (off HU*) | 362 | 1.21 | 0.0043 |
| Hemoglobin (off HU) adjusted for GFR | 319 | 0.93 | 0.3277 |
| Hemoglobin (on HU) | 168 | 1.36 | 0.0107 |
| Hemoglobin (on HU) adjusted for GFR | 157 | 0.83 | 0.1785 |
| WBC (off HU) | 351 | 1.06 | 0.0419 |
| WBC (on HU)** | 168 | 1.63 | 0.3181 |
| Platelets (off HU) | 350 | 1.00 | 0.9402 |
| Platelets (on HU)** | 167 | 0.65 | 0.3066 |
| Fetal Hemoglobin (off HU)** | 180 | 1.00 | 0.9954 |
| Fetal Hemoglobin (on HU)** | 124 | 0.79 | 0.3199 |
| Ferritin** | 153 | 1.27 | 0.0628 |
| Mean Corpuscular Volume (off HU) | 361 | 1.01 | 0.5220 |
| Mean Corpuscular Volume (on HU) | 168 | 1.02 | 0.2582 |
| Lactate Dehydrogenase** | 382 | 0.82 | 0.1621 |
| Reticulocytes (off HU)** | 313 | 1.01 | 0.9663 |
| Hemolytic Index*** | 313 | 1.10 | 0.3206 |
| Total Bilirubin | 410 | 1.01 | 0.8887 |
| GFR | 414 | 1.07 | <0.0001 |
| Body Mass Index | 343 | 0.98 | 0.3138 |
| Body Mass Index adjusted for HU | 317 | 0.98 | 0.2711 |
| NT-proBNP** | 87 | 1.62 | 0.0004 |
| sICAM-1 | 87 | 0.1376 | |
| sVCAM-1 | 87 | 2.03 | 0.0003 |
| E-Selectin | 87 | 0.1513 | |
| P-Selectin | 87 | 0.8673 | |
| sCD40L | 87 | 0.8235 | |
| IL-6 | 87 | 0.6459 | |
| IL-8 | 87 | 0.9505 | |
| IL-10 | 87 | 0.5447 | |
| TNF-a | 87 | 0.5876 |
HU is hydroxyurea.
denotes variables converted to logarithmic scale.
calculated in Hb SS and Hb Sβ0 subjects only
For comorbidities at enrollment, any history of the event, whether ongoing or resolved, was considered as positive. Pulmonary hypertension (pHTN) was defined as a tricuspid regurgitant jet (TR jet) ≥2.5 m/s. When available, laboratory values included the average of three recent steady state values to account for outliers. Laboratory data obtained during acute illness were excluded. Using these data, we calculated and included the hemolytic index (defined as the first principal component of LDH, AST, bilirubin and reticulocyte count).[25]In a subset of 87 subjects from Duke University and the University of North Carolina, we also obtained levels of plasma markers, including soluble adhesion molecules. Using the patient history questionnaire, pain was evaluated based on the number of hospitalizations in the past 12 months and use of narcotic analgesics at home. Medication use was also evaluated with the questionnaire, and our final analysis examined use of hydroxyurea, migraine medications, bronchodilators, iron chelators, antihypertensives, and antidepressants.
Using a previously described model,[26] an end organ severity score was generated using the following parameters: pulmonary (pHTN or O2 saturation < 92%), renal (proteinuria or creatinine > 1), cerebrovascular (stroke, TIA or seizures), AVN of hip or shoulder, and leg ulcers. One point was recorded for organ damage in each category. Points were then summed to create the severity score ranging from 0–5.
Statistical Methods
Contingency tables for categorical variables and means for continuous variables were generated using PROC FREQ and PROC MEANS in SAS v9.2 (SAS Systems, Cary, NC), respectively. Kaplan-Meier survival curves were created and hazard ratios for mortality during the study period were calculated to determine baseline characteristics associated with decreased survival. Kaplan-Meier survival analyses were performed using PROC LIFETEST; survival curves were created using Prism (GraphPad, Inc.). Regression analysis based on the Cox proportional hazards model was employed to determine the effect of clinical phenotypes on survival time using PROC PHREG. All models were adjusted for gender and age at enrollment. Several diagnostic metrics were employed to confirm that the assumption of proportional hazards for each model was met, including examination of log-negative-log survival curves and plots of Schoenfeld residuals (http://support.sas.com/resources/papers/proceedings13/428-2013.pdf) as well as testing the interaction between each clinical predictor and survival time. Only LDH was found to violate the proportional hazards assumption.
RESULTS
Baseline Characteristics
Our study included 542 subjects. The median age at enrollment was 32 years (range 18 to 84 years). Of these, 427 (78.8%) had Hb SS, 70 (12.9%) had Hb SC, 23 (4.2%) had Sβ+, and 22 (4.1%) had Sβ0. Median follow-up time was 9.3 years (range 2.7–10.5 years). At the conclusion of the study, 128 participants (23.6%) had died. The median age at death was 47 years for females, 44 years for males, and 45 years overall (range 20 to 86 years), as shown in supplemental Table I. Median survival was 61 years of age for all subjects, with no difference between males and females.
Table I and supplemental Table II show baseline clinical characteristics of our cohort. Among our population, 79.4% had graduated high school. Tobacco use was reported by 34.2%. Approximately 42% of all subjects and 45% of HbSS and Sβ0 subjects were on hydroxyurea at the onset of the study. Eighteen percent of HbSC and 31% of Hb Sβ+ thalassemia subjects were also taking hydroxyurea.
Survival Data
Hemoglobin Genotype
Among sickle cell genotypes, survival curves demonstrated that HbSS and Sβ0 portended the worst prognoses, followed by Hb SC and Sβ+ (Figure 1A). Overall median survival for HbSS and Sβ0 was 58 years and for Hb SC and Sβ+ was 66 years, a difference that was statistically significant (p=0.0031). In subjects with Hb SS, the median age at death was 44.5 years and 25.3% of the subjects had died by the conclusion of the study, while Hb Sβ0 subjects had a median age at death of 43 years and 40.9% mortality. However, these differences were not statistically significant (p=0.2188).
FIGURE 1.
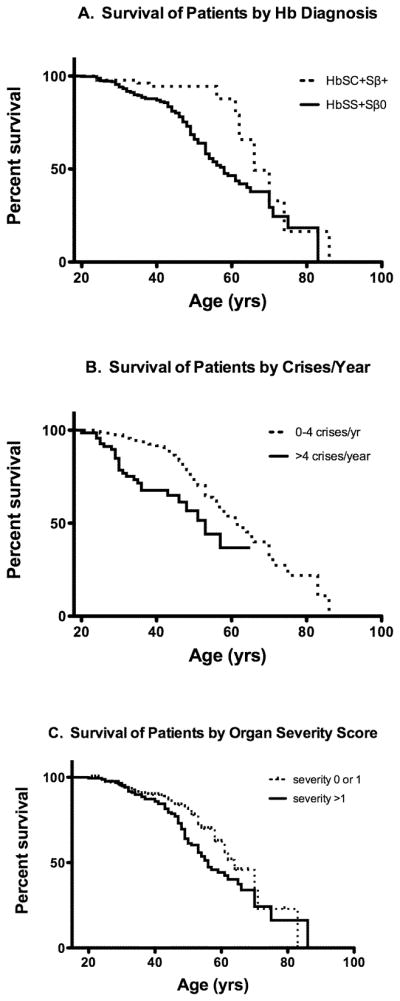
FIGURE 1A: Survival curves by genotype.
Hb SS and Sβ0 thalassemia subjects (solid line) had a median survival of 58 years. Hb SC and Sβ+ thalassemia subjects (dotted line) had a median survival 66 years. The difference between curves is significant (p=0.0031).
FIGURE 1B: Survival curves by frequency of vaso-occlusive pain crises defined as a crisis requiring inpatient admission. For subjects with 0–4 pain crises/year (dotted line), median survival was 61 years. For subjects with >4 crises/year, median survival was 53 years. Hazard ratio was 3.6982 (p<0.0001).
FIGURE 1C: Survival curves by organ severity score.
Degree to which subjects had organ-specific sequelae of SCD was rated on a scale of 0–5, as described; no subject had a score of >4. Median survival for subjects with a score of 0–1 (dotted line) was 64 years and for subjects with a score >1 was 56 years. Hazard ratio between the two curves was 1.502 (p=0.0263).
Table II shows hazard ratios for dichotomous variables(reported as “present” or “not present”) and Table III shows hazard ratios for continuous variables.
Hematologic
As expected, low hemoglobin was associated with poorer survival: for every g/dL decrease in Hb, the HR increased by 1.2077 (p=0.0043). However, this association disappeared when correcting for GFR. In addition, with every 103/μ Lincrease in WBC, the calculated hazard ratio increased by 1.059 (p=0.0419). Hemolytic index was not a statistically significant factor in survival among HbSS and HbSβ0 subjects (Table III).
Cardiopulmonary
Cardiopulmonary disease is known to be the primary etiology of death in adult SCD subjects.[27] In our sample, pHTN was present in 32% of SCD subjects. In our survival analysis, this proved to be a poor prognostic factor (HR=2.3386, p=0.0036). Similarly, elevated NT-proBNP was a marker of early death (HR=1.617, p=0.0004). While a diagnosis of hypertension did not correlate with early mortality, SCD subjects who were on chronic antihypertensive therapy did suffer earlier death than their counterparts.
Neurologic
A history of neurologic disease was present in 32% of subjects and correlated with early mortality. A history of any cerebrovascular event (CVE), defined as at least one stroke, seizure, or transient ischemic attack (TIA), was associated with decreased survival (HR=1.9464, p=0.0005). When analyzed individually, stroke (HR=1.6373, p=0.0335) and seizure (HR=2.5369, p<0.0001) were still associated with poor prognosis.
Renal
Presence of ≥1+ proteinuria by dipstick, observed in 26% of our sample, was an indicator of decreased longevity (HR=1.8618, p=0.0014). An association between renal function and decreased survival was corroborated with estimated GFR,[28] which demonstrated that the risk of death increased with worsening GFR (HR=1.068 per ml/min decrease in GFR, p<0.0001).
Hepatobiliary
As shown in Table I, gall bladder disease and cholecystectomy were among the most frequent comorbid conditions found in our SCD subjects. However, none of the hepatobiliary disease measures evaluated in our study were associated with survival.
Musculoskeletal/Cutaneous
Leg ulcers were a predictor of early mortality (HR=1.6631, p=0.0118). A vascular necrosis, while common with a prevalence of 32%, had no impact on prognosis.
Pain
As shown in Table II, having at least one crisis in the last year was associated with decreased survival (HR=1.744, p=0.0054, CI=1.18 to 2.58). In our cohort, 56% had suffered at least one pain crisis in the prior year, and 15% of those were hospitalized greater than four times. Survival was significantly lower in those who had experienced greater than four crises compared to those with less than four (Figure 1B, HR=2.1849, p=0.0006). These results were consistent when restricting the analysis to only subjects with Hb SS or Sβ0. Similarly, some patterns of narcotic use were related to decreased survival as shown in Table II.
Biomarkers
Of the evaluated adhesion molecules and inflammatory markers shown in Table III, only sVCAM-1 levels were associated with decreased survival (HR=2.032, p=0.0003). This association was also observed when including only subjects with SS or Sβ0 (HR=1.949, p=0.0009).
Hydroxyurea
Hydroxyurea has previously been shown to decrease mortality in SCD subjects.[29]Survival of Hb SS or Sβ0 subjects using hydroxyurea therapy was compared to those subjects not using hydroxyurea, and no difference in survival was observed (p=0.3228). This analysis was extended to compare hydroxyurea among subjects with the previously mentioned comorbidities, and no significant improvement in survival was observed among subjects with specific comorbidities.
Organ System Severity Score
For 493 subjects with SCD, a severity score of 0–5 was calculated to assess damage to multiple end organs, as defined in the methods section. One hundred forty-four subjects had a severity score of “0,” 175 scored “1,” 104 scored “2,” 56 scored “3” and 14 subjects scored “4.” No subjects had a severity score of “5.” Subjects with a score of 0–1 had a favorable prognosis compared to those with scores of >1 (Figure 1C, HR=1.502, p=0.0263).
DISCUSSION
Previous studies have reported an expected survival of 42 to 48 years in Hb SS subjects and 60 to 68 years in Hb SC subjects.[6] Based on our hazard estimates, median survival in our population is now 58 years for Hb SS and Sβ0 and 66 years for Hb SC and Sβ+. These data suggest that medical progress over the last 10–20 years has led to improvement in the overall survival for patients with Hb SS and Sβ0.
However, these data must be considered with caution. Our study only enrolled adult sickle cell patients. This exclusion of pediatric sickle cell patients, a small number of whom die in childhood, may have caused some inflation in our survival estimates. However, recent data have shown that SCD-related survival to adulthood is nearly 94%[5], suggesting that exclusion of the pediatric population should not have drastically improved our survival estimates. Second, 6.18% of our original study population was lost to follow up when we performed our “look back” for survival. However, it is the impression of the centers involved that most subjects lost to follow up had simply moved away, although this cannot be rigorously confirmed. Additionally, the patient population studied was limited to a cohort of patients from the southeastern United States. Thus, regional variations in comorbid conditions, such as rates of obesity or tobacco use, might limit the generalizability of our findings to the entire country. Finally, our subject population, consisting of patients from three referral centers, may be skewed toward a higher severity of disease than exists in the community.
In our evaluation of hematologic parameters, Hb level predicted survival, with the risk of early death inversely correlated to the baseline Hb level. The degree of WBC elevation was similarly related to survival, with a higher level predicting a shorter survival. However, when hazard ratios for Hb were corrected for GFR, the detected relationship was no longer statistically significant. This may suggest that hemoglobin is a biologic marker for the effect of kidney disease on survival and not independently associated with survival. In contrast to prior studies,[2, 6] Hb F level did not correlate with mortality in our study, even after controlling for hydroxyurea use.
With improvements in antibiosis and vaccination, the primary cause of death among SCD patients has shifted from infection to cardiopulmonary disease.[27] Our study was not intended to identify cause of death, but cardiopulmonary disease was clearly a risk factor for early death. Specifically, elevated tricuspid regurgitant jet velocity (≥2.5 m/s)and pro-BNP levels were strongly associated with poor survival. Although our study used echocardiography rather than right heart catheterization to identify subjects with possible or presumed pulmonary hypertension, thus likely identifying some patients without true pulmonary arterial hypertension,[30, 31]the use of tricuspid regurgitant jet velocity is a less invasive surrogate for evaluation with approximately a 25% positive predictive value in SCD patients.[31]Moreover, our results are consistent with prior studies showing an association between presumptive pulmonary hypertension, defined similarly by tricuspid regurgitant jet velocity ≥2.5 m/sec, and early mortality, and are furthermore corroborated by the significance of an elevated pro-BNP in our analysis.[32]
Treatment with antihypertensive medications was a poor prognostic factor. This is not surprising, because patients who require antihypertensive medications likely had more severe hypertension and potentially other secondary comorbidities. However, to further evaluate this finding, we compared subjects in the highest quartile for both systolic and diastolic blood pressure to the rest of our sample and found no difference in survival. Finally, the relationship of antihypertensive medication use to survival was no longer significant when analyzed with GFR as a covariate. Hence, as some antihypertensives(specifically ACE inhibitors) are used for renal protection in the setting of proteinuria, the prognostic significance of these medications may reflect underlying kidney disease. Nevertheless, our findings do support the need for future studies addressing therapeutic goals for blood pressure levels and preferred approaches to antihypertensive therapy and proteinuria in SCD patients.
Pulmonary comorbidities were not related to survival in our sample. The most notable of these was history of acute chest syndrome, which has been a well-documented prognostic factor in prior studies.[6, 32]In our methodology, history of ACS was determined by patient report of ACS or pneumonia, thus leading to some inherent inaccuracy in the reported prevalence of ACS. Patients who died from ACS prior to enrollment could not be included in the analysis. Hence, our results cannot be used to show that ACS does not influence survival. Instead, our findings suggest that there is no change in the long-term survival of SCD patients who survive an episode of ACS. In addition, asthma, as identified by use of bronchodilators, also did not affect survival.
During episodes of inflammation and endothelial injury, such as vaso-occlusive crises, endothelial adhesion molecules are released into the plasma.[20] Hence, we anticipated that elevations in soluble adhesion molecules would also be related to severity of SCD and would prove useful in predicting early mortality. Indeed, in this respect, we have confirmed the findings of Kato, et al.[33] that elevated sVCAM-1 is clearly related to poor prognosis. This raises the possibility that underlying vascular damage and inflammation is a primary facilitator of early death in SCD patients, and that future treatments targeting these inflammatory processes may prove to be life prolonging.
Our results also underscore the significance of pain in the natural history of SCD. We have shown associations between survival and pain frequency, hospitalizations due to pain, and narcotic use, consistent with prior studies.[6]However, the effects of pain on survival may not be reversible by improvements in pain management alone. Greater frequency and severity of pain may reflect more severe underlying disease. Further, the relationship between narcotic use and decreased survival may reflect direct adverse effects of the medications, such that more aggressive analgesia could worsen survival. Still, adequate management of SCD pain could yield improvements to patient quality of life,[34]and prevention of acute pain episodes might also be an effective strategy for decreasing healthcare costs.[35]
Assessment using our previously developed organ severity score,[26] based on number of affected organ systems, also showed that, with each additional organ system affected, survival decreased. The largest incremental risk increase occurred as patients moved from two involved organ systems to three. Escalation of the severity score from three organ systems to four did not affect prognosis. However, due to the small number of patients with a severity score of four, we likely lacked adequate statistical power to detect significant differences in that analysis.
As we used a cross-sectional design, there are many limitations to our study. Our analysis could only assess associations between survival and characteristics present at baseline without proving causality. Additionally, we cannot determine whether subjects developed new comorbid conditions during the follow-up period nor whether these influenced prognosis. Our design also cannot be used to determine risk of death from acute complications of SCD such as ACS, strokes, or seizures. Instead, we hope our results will help predict the expected survival of SCD patients who have survived such complications previously. Finally, use of hydroxyurea was neither randomized nor monitored, so findings regarding the effects of this medication may not reflect ideal use nor comparable populations.
In conclusion, our survival data highlight improvements in overall survival in SCD patients over the last decade. Despite this progress, SCD patients continue to experience decreased quality of life and, ultimately, a shortened life span compared to the general population. Thus, there is a dire need for new therapeutic options. While we have identified several predictors of decreased survival, the question remains whether treatments targeted at these factors would improve mortality. As emerging but riskier therapies such as bone marrow transplant and gene therapy begin to offer hope for a potential cure,[36] our results may be useful to help identify high risk patients most in need of these therapies. Furthermore, while we have identified phenotypes associated with prognosis, it nevertheless remains unclear why the overall course of SCD is so varied across the patient population. Future studies identifying genetic variations associated with more severe phenotypes and overall survival could provide a dramatic improvement in our understanding of SCD, as well as unveil novel therapeutic targets.
Supplementary Material
Acknowledgments
We would like to thank our subjects for participating in this study. This work was supported by grants HL068959 and HL079915 from the National Heart, Lung, and Blood Institute (NHLBI) of the National Institute of Health (NIH) and a Duke University Faculty Resident Research Grant. HE is a recipient of an American Society of Hematology HONORS Award.
References
- 1.Diggs L. Sickle Cell Disease: Diagnosis, Management, Education and Research. Mosby; 1973. pp. 189–229. [Google Scholar]
- 2.Leikin SL, Gallagher D, Kinney TR, et al. Mortality in children and adolescents with sickle cell disease. Cooperative study of sickle cell disease. Pediatrics. 1989;84:500–508. [PubMed] [Google Scholar]
- 3.Davis H, Schoendorf K, Gergen P, et al. National trends in the mortality of children with sickle cell disease, 1968 through 1992. Am J Public Health. 1997;87:1317–1322. doi: 10.2105/ajph.87.8.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.From the Centers for Disease Control and Prevention. Mortality among children with sickle cell disease identified by newborn screening during 1990–1994: California, Illinois, and New York. JAMA. 1998;279:1059–1060. [PubMed] [Google Scholar]
- 5.Quinn C, Rogers Z, Buchanan G. Survival of children with sickle cell disease. Blood. 2004;103:4023–4027. doi: 10.1182/blood-2003-11-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platt O, Brambilla D, Rosse W, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg M, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 8.Johnson C, Telen MJ. Adhesion molecules and hydroxyurea in the pathophysiology of sickle cell disease. Haematoligica. 2008;93:481–486. doi: 10.3324/haematol.12734. [DOI] [PubMed] [Google Scholar]
- 9.Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gee B. Sickle reticulocytes adhere to VCAM-1. Blood. 1995;85:268–274. [PubMed] [Google Scholar]
- 11.Telen MJ. Role of adhesion receptors molecules and vascular endothelium in the pathogenesis of sickle cell disease. Hematology. 2007:84–90. doi: 10.1182/asheducation-2007.1.84. [DOI] [PubMed] [Google Scholar]
- 12.Kaule D, Liu X-d, Zhang X, et al. Peptides based on alphaV-binding domains of erythrocyte ICAM-4 inhibit sickle red cell -endothelial interactions and vaso-occlusion in the microcirculation. Am J Physiol Cell Physiol. 2006:291. doi: 10.1152/ajpcell.00639.2005. [DOI] [PubMed] [Google Scholar]
- 13.Zennadi R, Moeller B, Whalen E, et al. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood. 2004;10:2708–2717. doi: 10.1182/blood-2006-11-056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zennadi R, Hines P, DeCastro L, et al. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-aVB3 interactions. Blood. 2004;104:3774–3781. doi: 10.1182/blood-2004-01-0042. [DOI] [PubMed] [Google Scholar]
- 15.Okpali I, Daniel Y, Haynes R, et al. Relationship between the clinical manifestations of sickle cell disease and the expression of adhesion molecules on white blood cells. Eur J Haematol. 2002;69:135–144. doi: 10.1034/j.1600-0609.2002.02775.x. [DOI] [PubMed] [Google Scholar]
- 16.Fadlon E, Vordermeier S, Pearson T, et al. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91:266–274. [PubMed] [Google Scholar]
- 17.Finnegan E, Turhan A, Golan D, et al. Adherent leukocytes capture sickle erythrocytes in an in vitro flow model of vaso-occlusion. American Journal of Hematology. 2007;82:266–275. doi: 10.1002/ajh.20819. [DOI] [PubMed] [Google Scholar]
- 18.Turhan A, Weiss L, Mohandas N, et al. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci USA. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown M, Wick T, Eckman J. Activation of vascular endothelial cell adhesion molecule expression by sickle blood cells. Pediatr Pathol Mol Med. 2001;20:47–72. [PubMed] [Google Scholar]
- 20.Solovey A, Lin Y, Browne P, et al. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997:337. doi: 10.1056/NEJM199711273372203. [DOI] [PubMed] [Google Scholar]
- 21.Styles L, Lubin B, Vichinsky E, et al. Decrease of very late activation antigen-4 and CD36 on reticulocytes in sickle cell patients treated with hydroxyurea. Blood. 1997;89:2554–2559. [PubMed] [Google Scholar]
- 22.Covas D, de Lucena A, Vianna Bonini Palma P, et al. Effects of hydroxyurea on the membrane of erythrocytes and platelets in sickle cell anemia. Haematoligica. 2004;89:273–280. [PubMed] [Google Scholar]
- 23.Brun M, Bourdoulous S, Couraud P, et al. Hydroxyurea downregulates endothelin-1 gene expression and upregulates ICAM-1 gene expression in cultured human endothelial cells. Pharmacogenomics J. 2003;3:215–226. doi: 10.1038/sj.tpj.6500176. [DOI] [PubMed] [Google Scholar]
- 24.Adam S, Jonassaint J, Kruger H, et al. Surgical and obstetric outcomes in adults with sickle cell disease. The American Journal of Medicine. 2008;121:916–921. doi: 10.1016/j.amjmed.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minniti CP, Sable C, Campbell A, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. haematologica. 2009;94:340–347. doi: 10.3324/haematol.13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afenyi-Annan A, Kail M, Combs M, et al. Lack of Duffy antigen expression is associated with organ damage in patients with sickle cell disease. Transfusion. 2008;48:917–924. doi: 10.1111/j.1537-2995.2007.01622.x. [DOI] [PubMed] [Google Scholar]
- 27.Fitzhugh CD, Lauder N, Jonassaint JC, et al. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. American Journal of Hematology. 2009;85:36–40. doi: 10.1002/ajh.21569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levey A, Berg R, Gassman J, et al. Creatinine filtration, secretion and excretion during progressive renal disease. Modification of Diet in Renal Disease (MDRD) Study Group. Kidney Int Suppl. 1989;27:S273–280. [PubMed] [Google Scholar]
- 29.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17. 5 year follow-up. Am J Hematol. 2010;85:403–408. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gladwin M, Sachdev V, Jison M, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 31.Florence Parent MD, Dora Bachir MD, Jocelyn Inamo MD, et al. A Hemodynamic Study of Pulmonary Hypertension in Sickle Cell Disease. N Engl J Med. 2011;365:44–53. doi: 10.1056/NEJMoa1005565. [DOI] [PubMed] [Google Scholar]
- 32.Powars DR, Chan LS, Hiti A, et al. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 33.Kato G, Martyr S, Blackwelder W, et al. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. British journal of haematology. 2005;130:943–953. doi: 10.1111/j.1365-2141.2005.05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClish D, Penberthy L, Bovbjerg V, et al. Health related quality of life in sickle cell patients: The PiSCES project. Health and Quality of Life Outcomes. 2005:3. doi: 10.1186/1477-7525-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kauf T, Coates T, Huazhi L, et al. The cost of health care for children and adults with sickle cell disease. American Journal of Hematology. 2009;84:323–327. doi: 10.1002/ajh.21408. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh M, Kang E, Fitzhugh C, et al. Allogeneic Hematopoietic Stem-Cell Transplantation for Sickle Cell Disease. N Engl J Med. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.