

Abstract
Macrophages play diverse roles in tissue homeostasis and immunity, and canonically activated macrophages are critically associated with acute inflammatory responses. It is known that activated macrophages undergo cell death after transient activation in some settings, and the viability of macrophages impacts on inflammatory status. Here we report that TGFβ- activated kinase (TAK1) activators, TAK1-binding protein 1 (TAB1) and TAK1-binding protein 2 (TAB2), are critical molecules in the regulation of activated macrophage survival. While deletion of Tak1 induced cell death in bone marrow derived macrophages even without activation, Tab1 or Tab2 deletion alone did not profoundly affect survival of naïve macrophages. However, in lipopolysaccharide (LPS)-activated macrophages, even single deletion of Tab1 or Tab2 resulted in macrophage death with both necrotic and apoptotic features. We show that TAB1 and TAB2 were redundantly involved in LPS-induced TAK1 activation in macrophages. These results demonstrate that TAK1 activity is the key to activated macrophage survival. Finally, in an in vivo setting, Tab1 deficiency impaired increase of peritoneal macrophages upon LPS challenge, suggesting that TAK1 complex regulation of macrophages may participate in in vivo macrophage homeostasis. Our results demonstrate that TAB1 and TAB2 are required for activated macrophages, making TAB1 and TAB2 effective targets to control inflammation by modulating macrophage survival.
Introduction
Macrophages are characterized by phagocytic activity, and play diverse roles in different tissue types. While resident macrophages participate in morphogenesis and tissue homeostasis, resident and recruited macrophages also play a major role in acute inflammatory responses [1]. Upon tissue injury or invasion by microorganisms, circulating inflammatory monocytes are recruited and differentiated toward mature macrophages. These macrophages are canonically activated by necrotic debris and bacterial moieties through Toll-like receptor signaling pathway, developing into so-called M1 polarized macrophages [2]. Activated macrophages clean dead cells and microorganisms by phagocytosis and produce inflammatory cytokines resulting in amplification of inflammation. Subsequently, these activated macrophages are deactivated or killed to terminate inflammatory conditions. In some experimental settings, it is known that lipopolysaccharide (LPS)-induced activation of macrophages reduces macrophage viability [3]–[5]. However, the mechanism by which activated macrophages undergo cell death is still largely elusive.
TGFβ- activated kinase (TAK1) is a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, and is an indispensable intermediate of cytokine and Toll-like receptor pathways [6]–[8]. TAK1 is recruited to and activated by the receptor proximal complex of TNF, IL-1, and Toll-like receptors through a poly-ubiquitin chain-mediated mechanism [9]. TAK1-binding protein 2 (TAB2) and its closely related protein, TAK1-binding protein 3 (TAB3), have ubiquitin binding domains and tether between TAK1 and the poly-ubiquitin chain resulting in activation of TAK1 [10]–[14]. TAB2 and TAB3 may redundantly function in innate immune pathways, but TAB2 plays an indispensable role at least during development [15]. Additionally, it has recently been shown that Tab3 deletion does not impair innate or adaptive immunity [16]. Thus, TAB2 is the major adaptor between TAK1 and activating poly-ubiquitin chains in immune cells. TAK1 is also activated through another binding partner, TAK1-binding protein 1 (TAB1), which is structurally unrelated to TAB2/3 and binds to TAK1 at a site different from the TAB2/3-binding site [17], [18]. TAB1 is found to be constantly associated with TAK1, and we recently demonstrated that TAB1 is involved in stress-dependent TAK1 activation [19] and activity of TAK1 in epithelial tissues [20]. Major known downstream molecules of TAK1 are IκB-kinases (IKKs) and mitogen-activated protein kinases (MAPKs) including p38 and JNK, which in turn activate transcription factors NF-κB and AP-1, respectively.
In vivo, TAK1 signaling is found to be important for immune responses in T and B cells through regulation of NF-κB and MAPK pathways in a mouse model [21]–[24], which is anticipated from the results in the tissue culture system described above. However, unexpectedly, the most overt phenotype caused by Tak1 deficiency in vivo is tissue damage associated with cell death in the epidermis, intestinal epithelium and liver [25]–[31]. Since Tak1 deficiency does not cause cell death in primary culture fibroblasts or keratinocytes, the cell death must be induced depending on the in vivo environment. TAK1 has been found to be integral to prevent tissue-derived TNF-induced cell death in vivo, which is evidenced by the fact that Tnfr1 deletion can rescue cell death and tissue damage in these tissues [26], [27], [29]. Single deletion of Tab1 or Tab2 does not cause any abnormalities in the epidermis and intestinal epithelium but double deletion of Tab1 and Tab2 phenocopies Tak1 deficiency [20], suggesting that TAB1 and TAB2 redundantly function in TAK1 regulation in these tissues. However, the specific roles of TAB1 and TAB2 in adult tissues are still largely elusive.
Recent studies have demonstrated that Tak1 deficiency in myeloid cells results in hyper-proliferation of neutrophils and increased inflammatory conditions [32], [33]. Bone marrow derived macrophages (BMDMs) generated from myeloid-specific Tak1-deficient mice have been reported to undergo spontaneous cell death under normal culture conditions [32], [34]. These studies have determined that TAK1 is required for proper differentiation of myeloid lineage and survival of macrophage precursors and/or mature macrophages. However, it is not clear whether TAK1 is important for maintenance of progenitors or mature macrophages and what the role of TAK1 is in activation of macrophages. Here we investigate the role of TAK1 and its binding partners, TAB1 and TAB2, in both mature naïve and activated macrophages, and have determined that, TAB1 and TAB2 are essential modulators of TAK1 activity and cell survival in LPS-activated macrophages.
Materials and Methods
Bone marrow cell isolation and macrophage differentiation
Tak1flox/flox, Tab1flox/flox, and Tab2flox/flox C57BL/6 mice were described previously [15], [22], [35]. Rosa26.CreERT and Tnfr-/- mice were purchased from Jackson Laboratories, and bred in our lab to produce the indicated genotypes [36]–[38]. Experiments performed in vitro required isolating bone marrow cells from Tak1, Tab1 and Tab2 mutant mice with flox/flox (WT), flox/+ Rosa26.CreERT (F+Cre) or flox/flox Rosa26.CreERT (iKO). Bone marrow derived macrophages (BMDMs) were generated by the standard procedure culturing bone marrow cells in 30% L929 cell-conditioned medium. To achieve gene deletion, cells were treated with 0.3 µM 4-hydroxytamoxifen (4-OHT) for 4 days. All animal experiments including in vivo LPS treatment described later were conducted with the approval of the North Carolina State University Institutional Animal Care and Use Committee (IACUC protocol # 11-138B). All efforts were made to minimize animal suffering.
Crystal violet assay
BMDMs were plated onto 12-well plates at a concentration of 2×105 cells per well and treated with 0.3 µM 4-OHT. In some experiments, BMDMs were subsequently treated with LPS (1 µg/ml) for 1 and 3 days. Cells were fixed using 10% formalin, and stained with 0.1% crystal violet. The dye was eluted and analyzed at 595 nm.
Reagents and antibodies
Specific monoclonal and polyclonal antibodies against the following antigens were used: CD3ε (145-2C11), CD11b (M1/70), and B220 (RA3-6B2) (eBioscience); F4/80 (BM8)(BioLegend), phospho-TAK1, TAK1, TAB1, and TAB2 described previously; phospho-IκB, phospho-p38, IκB, and p38 (Cell Signaling). Necrostatin-1 (Nec-1) was purchased from Santa Cruz and applied to the culture at the final concentration of 50 µM. LPS is derived from source strain Salmonella minnesota ATCC 9700 (Sigma-Aldrich, catalog number L6261).
Flow cytometry
BMDMs were detached from culture dishes and incubated with annexin V-Pacific Blue (BioLegend) and Fixable Viability Dye eFluor 780 (eBiosicence) for cell death analysis. Stained cells were analyzed on flow cytometer (BD Biosciences LSR II), and data were analyzed using FlowJo software (Tree Star). Events were gated to exclude debris based on forward scatter (FSC) and side scatter (SSC) profile, then gated on Pacific Blue (annexin V) or APC-Cy7 (fixable viability dye) when compared to unstained control.
Western blotting
BMDMs were lysed in extraction buffer (20 mM HEPES [pH 7.4], 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 2 mM DTT, 1 mM Na3VO4, 1 mM PMSF, 20 µM aprotinin, 0.5% Triton X-100) in ice for 15 minutes. Cells and debris were then pelleted by centrifugation at 20,000 G for 10 at 4°C. Cell extracts were resolved on SDS-PAGE and transferred to Hybond-P membranes (GE Healthcare). The membranes were immunoblotted with various antibodies, and the bound antibodies were visualized with horseradish peroxidase-conjugated antibodies against rabbit or mouse IgG using the ECL Western blotting system (GE Healthcare).
Mouse model (in vivo)
For in vivo experiments, deletion of Tab1 was achieved in Tab1iKO mice by intraperitoneal injection of 50 mg per kg tamoxifen on 3 consecutive days. After a period of 3-5 weeks, to reduce effects of Cre toxicity, whole blood was isolated and gene deletion was verified by Western Blot. To exclude the effect of Cre toxicity, we included F+Cre (Rosa26.CreERT2 Tab1flox/+) mice as controls. Mice were intraperitoneally injected with 8 mg/kg LPS, and were euthanized at 72 hours and dissected. Peritoneal leukocytes were collected by peritoneal lavage and collected in phosphate buffered saline (PBS), and splenocytes were harvested and prepared in a single cell suspension. Red blood cells were lysed by suspending cells in 0.83% NH4Cl lysis buffer and washed once with PBS. Cells were incubated with anti-CD16/32 in ice for 20 minutes to block FcγRII/III, followed by incubation with fluorophore-conjugated monoclonal antibodies (CD11b, B220 CD3ε, and F4/80) to evaluate cell type. Cells were washed in PBS and characterized on a BD LSRII flow cytometer (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star).
Statistical analysis
Cell counts were normalized to control and compared using a two-tailed Student's t-test. Values shown are means ± standard deviation with results considered significant if a probability of Type I error was <.05.
Results
Deletion of Tak1 or double deletion of Tab1 or Tab2 spontaneously kills bone marrow derived macrophages
We characterized bone marrow derived macrophages (BMDMs) in adult mice having deletions of Tak1, Tab1 or Tab2 single or Tab1 and Tab2 double gene using the ubiquitously-expressed inducible Cre recombinase system, Rosa26.CreERT deleter mice. Rosa26.CreERT Tak1flox/flox (referred to as Tak1iKO), Rosa26.CreERT Tab1flox/flox (referred to as Tab1iKO), Rosa26.CreERT Tab2flox/flox (referred to as Tab2iKO), and Rosa26.CreERT Tab1flox/flox Tab2flox/flox (referred to as diKO) were compared with littermate or age matched controls, no-Cre flox/flox (referred to as WT or control). In some experiments, mice having heterozygous gene deletion, Rosa26.CreERT flox/+ were also used as a control, which did not show any abnormality nor did WT. Bone marrow cells were differentiated to macrophages and gene deletion was subsequently induced by treatment of 4-hydroxytamoxifen (4-OHT) in macrophage culture medium, and vehicle (ethanol) was treated as control. The amounts of TAK1, TAB1 and TAB2 proteins were determined by immunoblotting at 4 days with 4-OHT or vehicle treatment. For Tak1iKO and diKO macrophages, we additionally treated with necrostatin-1 (Nec-1), an inhibitor of receptor interacting protein 1 (RIP1), which is known to block Tak1-deficient macrophage death [34]. Nec-1 blocked cell death and allowed us to recover sufficient protein for Western blotting. We observed fluctuations in the amount of proteins among different litters and with Nec-1 treatment. Nonetheless, TAK1, TAB1, and TAB2 were greatly reduced within 4 days with 4-OHT treatment in Tak1iKO macrophages (Figure 1A). Reduction of TAB1 and TAB2 in Tak1iKO macrophages is presumably due to destabilization of unbound TAB1 and TAB2. In contrast, TAB1 or TAB2 but not TAK1 was reduced in Tab1iKO or Tab2iKO macrophages, respectively. Both TAB1 and TAB2 but not TAK1 were reduced in diKO macrophages. These indicate that TAK1 is intact in Tab1iKO, Tab2iKO, and diKO macrophages, which allow us to investigate the specific roles of TAB1 and TAB2 without altering the protein level of TAK1 in these macrophages. The number of macrophages declined at 8 days with 4-OHT treatment in Tak1iKO macrophages, whereas the number of control macrophages, including WT with 4-OHT and Tak1iKO with vehicle treatment, were not altered during the period of experiment (Figure 1B). Thus, TAK1 is essential for cultured macrophage integrity. These results are consistent with the previously reported results in myeloid-specific deletion of Tak1 [34]. Thus, TAK1 seems to be important for survival of not only precursors but also mature naïve macrophages. In contrast to Tak1 deletion, either Tab1 or Tab2 single deletion only moderately or slightly decreased the number of macrophages (Figure 1B). This indicates that TAB1 or TAB2 may partly contribute to but is not required for naïve macrophage survival. Importantly, we found that double deletion of Tab1 and Tab2 reduced the number of macrophages to a level similar to Tak1 deletion (Figure 1B). Collectively, these results suggest that TAB1 and TAB2 may redundantly function to maintain TAK1 activity in naïve macrophages and TAK1 basal activity is required for survival of naïve macrophages.
Figure 1. TAK1 is required for macrophage survival.

(A) Western blotting analysis of TAK1, TAB1 and TAB2 in control, Tak1iKO, Tab1iKO, Tab2iKO and diKO BMDMs. Bone marrow cells were cultured in macrophage medium and treated with 0.3 µM 4-OHT or vehicle (ethanol) for 4 days. Tak1iKO and diKO BMDMs were additionally treated with 50 µM Necrostatin-1 (Nec-1). Anti-β-actin Western blotting was used as a loading control. The numbers beside each panel denote the size and the position of molecular weight markers. (B) Viability of WT, Tak1iKO, Tab1iKO, Tab2iKO, and inducible double-deficient (diKO) BMDMs. Cells were cultured for 8 days with 0.3 µM 4-OHT and stained with 0.1% Crystal Violet. Data are mean percentages of attached macrophages compared to ethanol-treated +/− SD for 3 independent experiments. Asterisks indicate p-values: ** = P<0.005; *** = p<0.0005.
TAB1 and TAB2 are required for survival of LPS-stimulated macrophages
To determine the role of TAK1 complex in activated macrophages, we treated Tab1iKO or Tab2iKO macrophages with bacterial moiety, lipopolysaccharide (LPS). Wild type macrophages treated with LPS did not exhibit reduced cell viability in our experimental setting (Figure S1). Even in this condition, we found that activation of macrophages by LPS treatment noticeably reduced the numbers of Tab1iKO macrophages within 3 days (Figure 2A). Tab1iKO macrophages exhibited both increased annexin V binding and loss of plasma membrane integrity (Figures 2B). The percentage of cells showing a necrotic feature, cell permeability dye-positive, was greatly increased by LPS treatment (Figure 2C). Apoptotic cells characterized by annexin V binding-positive but permeability dye-negative were also increased by LPS treatments (Figure 2D). Thus, LPS-activated macrophages require TAB1 for their survival, and Tab1 deficiency causes cell death having both necrotic and apoptotic features. Similarly, we found that Tab2iKO macrophages were significantly declined upon LPS treatment (Figure 3A). In contrast to Tab1 deletion, Tab2iKO macrophage underwent cell death only one day after LPS treatment. LPS-treatment of Tab2iKO macrophages increased the frequency of annexin V-positive and cell permeability dye-positive population (Figure 3B). Similar to Tab1iKO, both necrotic and apoptotic cells were increased by LPS treatment in Tab2iKO macrophages (Figure 3C and 3D). These results demonstrate that, in contrast to naïve macrophages, both TAB1 and TAB2 are important for activated macrophage survival.
Figure 2. TAB1 is required for LPS-activated macrophage survival.

(A) Viability of LPS-treated Tab1iKO macrophages. Tab1iKO and control BMDMs were cultured for 8 days with 0.3 µM 4-OHT followed by 3 days 1 µg/ml LPS. Viability was measured by Crystal Violet Assay, and data shown are mean percentages of attached macrophages compared to 8 days treated with vehicle +/− SD of 3 independent experiments. (B) Flow cytometry analysis of Tab1iKO BMDMs. Tab1iKO or Tab1F+ Cre BMDMs were cultured in macrophage medium with 0.3 µM 4-OHT or vehicle (ethanol) for 4 days. All cells including attached and floating cells were collected and stained with annexin V-Pacific Blue and Fixable viability dye eFlour 780, then analyzed on flow cytometer. Events were gated to exclude dead cells and debris, then gated on events positive for annexin V and fixable viability dye compared with unstained controls. Shown is representative figure of 3 independent experiments. (C and D) Tab1iKO and controls including WT and F+Cre BMDMs were cultured 3 days in 0.3 µM 4-OHT-containing macrophage medium, then 4 days with the addition of 1 µg/ml LPS. (C) Viability Dye-positive cells as a percentage of total cells is shown. (D) Graph shows Annexin V-positive but viability dye-negative cells. Graph represents results of four independent experiments +/− SD.
Figure 3. TAB2 is required for LPS-activated macrophage survival.

(A) Viability of LPS-activated Tab2-deficient macrophages. Tab2iKO BMDMs were cultured for 8 days with 0.3 µM 4-OHT, then 1 µg/ml LPS was added to culture medium for 1 day. Data are mean percentages of attached macrophages compared to 8 days treated with 4-OHT alone +/− SD between 3 independent experiments as measured by Crystal Violet Assay. (B) Percentages of annexin V and viability dye positive cells. Tab2iKO and control (WT) BMDMs were cultured with 0.3 µM 4-OHT for 3 days then with 1 µg/ml LPS for 1 additional day in 3 independent experiments. Cells were stained with viability dye and annexin V and analyzed by flow cytometry. Percentages of single positive and double positive cells based on unstained controls in a representative experiment are shown. (C and D) Necrotic and apoptotic LPS-activated macrophages with Tab2 deficiency. Tab2iKO, and controls including WT and F+Cre BMDMs were cultured 3 days in 0.3 µM 4-OHT-containing macrophage medium, then 1 day in medium containing 0.3 µM 4-OHT and 1 µg/ml LPS. (C) Necrotic cells are shown as percentage positive for viability dye. (D) Annexin V positive but viability dye-negative cells as a percentage of total cells is shown. Graphs indicate results of four independent experiments +/− SD.
LPS activates TAK1 through TAB1 and TAB2 in macrophages
We hypothesize that TAB1 and TAB2 mediate LPS-induced TAK1 activation, which may be required for LPS-activated macrophage survival. To test this, we examined the levels of TAK1 activation and subsequent downstream events, activation of NF-κB and p38, following LPS stimulation. We note that, since Tak1 or Tab1 and Tab2 double deletion spontaneously kills macrophages, we treated macrophages with Nec-1 to obtain live macrophages with Tak1 or Tab1 and Tab2 double deletion. It is known that while RIP1 participates in NF-κB and p38 pathways, RIP1 catalytic activity is dispensable [39]. Consistent with this notion, NF-κB and p38 were activated in wild type macrophages even with Nec-1 treatment. We found that LPS activated TAK1 and its downstream IKK and p38 in macrophages (Figure 4A, middle 4 lanes), and Tak1 deficiency reduced activation of both IKK and p38 (Figure 4A, left 4 lanes). TAK1 activity was monitored by phosphorylation of Thr 187 (Figure 4A, top panel), which is known to be associated with activation of TAK1 [40] However, non-specific bands were detected around the phosphorylated TAK1 in macrophages protein extracts, which were seen even in unstimulated macrophagse (Figure 4A, asterisks). Thus, we also utilized retardation of TAK1 band on SDS-PAGE to monitor TAK1 activation, which is caused by phosphorylation of several sites associated with TAK1 activation [41], [42]. While TAK1 exhibited migration shift upon LPS stimulation in wild type macrophages, Tab1 and Tab2 double deficiency abolished migration shift (Figure 4A, second panel), and reduced activation of IKK and p38 (Figure 4A, right 3 lanes) suggesting that LPS-induced activation of IKK and p38 is largely mediated by TAK1 and that TAB1 and TAB2 are essential for TAK1 activation in response to LPS. Deletion of either Tab1 or Tab2 impaired LPS-induced migration shift of TAK1, suggesting some impairment of TAK1 activation in response to LPS (Figures 4B and C, top panels). However, a single deletion of either Tab1 or Tab2 had a marginal effect on the LPS-induced degradation of IκB and phosphorylation and p38, suggesting that one of these proteins can activate TAK1 sufficiently at least in the pathways leading to activation of IKK and p38. These results collectively suggest that LPS-induced activation of TAK1 is mediated by TAB1 and TAB2, and that either deletion of Tab1 or Tab2 reduces TAK1 activation. However, LPS-induced activation of NF-κB and p38 seems to require at a minimum TAB1 or TAB2.
Figure 4. LPS activates TAK1 through TAB1 and TAB2.

(A) Western blotting analysis of Tak1iKO and Tab1Tab2diKO and control BMDMs. Cells were cultured for 4 days with 0.3 µM 4-OHT in the presence of 50 µM Nec-1, followed by 1 µg/ml LPS treatment for the indicated period of time. Anti-βactin was used as a loading control. Asterisks indicate non-specific bands. (B) Tab1iKO or control BMDMs were cultured with 0.3 µM 4-OHT for 3 days then treated with 1 µg/ml LPS for the indicated period of time. Whole cell extracts were analyzed by Western blotting using the indicated antibodies. Asterisks indicate non-specific bands. (C) Tab2iKO or control BMDMs were cultured with 0.3 µM 4-OHT for 3 days then treated with 1 µg/ml LPS for the indicated period of time. Whole cell extracts were analyzed by Western blotting using the indicated antibodies. Asterisks indicate non-specific bands.
LPS-induced cell death in Tab1- or Tab2-deficient macrophages is partially rescued by inhibition of RIP1
Tak1 deletion is reported to cause RIP1-dependent cell death upon TNF treatment in several cell types [26], [27], [29]. Earlier study demonstrates that Tak1-deficient naïve macrophages die in a RIP1-dependent manner [34]. We hypothesize that activated macrophages die with the mechanism similar to that in naïve macrophages due to insufficient activity of TAK1. To test this, we examined the involvement of RIP1 kinase activity in Tab1iKO and Tab2iKO macrophage death by using Nec-1. In Tab1iKO macrophages, Nec-1 significantly and marginally reduced LPS-induced necrotic and apoptotic cells, respectively (Figure 5A and 5B). Nec-1 treatment exhibited some trends of reduction of cell death in Tab2iKO macrophages, although these did not reach to a statistic significance (Figure 5C and 5D). Both necrotic and apoptotic cells were decreased. These results suggest that RIP1 kinase activity may be at least partially involved in LPS-induced cell death in Tab1iKO and Tab2iKO macrophages.
Figure 5. TAB1 or TAB2 deletion causes RIP1-dependent cell death.

(A) Tab1iKO or control BMDMs were cultured with 0.3 µM 4-OHT with or without 50 µM Nec-1 for 8 days then treated with 1 µM LPS for 3 days, and viability was measured by Crystal Violet Assay. * = p<.05. (B) Viability of Tab2iKO BMDMs treated with RIP1 inhibitor. Tab2iKO BMDMs were treated with 0.3 µM 4-OHT for 8 days with our without 50 µM Necrostatin-1 (Nec-1), then treated with 1 µM LPS for one day. Viability was measured by Crystal Violet Assay. * = p<.05.
Tab1-deficient macrophages were reduced upon LPS injection in vivo
We next examined the role of TAK1 complex in macrophages in vivo. We anticipate LPS injection to recruit and activate monocytes from the bone marrow and circulation to the peritoneal cavity. Germline deletion of Tak1 gene causes multiple defects in embryogenesis [43], [44], and deficiency of Tak1 in adult mice also causes acute severe liver injury and mortality [25], [26]. We were not able to analyze Tak1 deficient macrophages in adult mice due to these multiple acute effects. Tab2iKO mice did not exhibit overt abnormality upon gene deletion by tamoxifen injection; however, LPS stimulation caused acute liver dysfunction within 6 h (unpublished observations). Tab2-deficient macrophages were difficult to analyze in LPS-challenged mice for this reason. We then focused on Tab1iKO mice. Tab1 gene was deleted in Tab1iKO mice by tamoxifen injection for 3 consecutive days. Although germline deletion of Tab1 causes embryonic lethal phenotype [35], [45], we found that deletion of Tab1 gene in adult mice did not cause overt abnormalities even though TAB1 proteins were greatly diminished (Figure 6A). To rule out the effect of Cre toxicity described previously [46], [47], the experiments were performed after more than 3 weeks post-tamoxifen injections. The number of peritoneal macrophages was determined in Tab1-deficient adult mice following LPS injection (Figure 6B). The frequency of CD11b+ F4/80+ macrophages in peritoneal fluid was lower in Tab1iKO mice compared to littermate controls following LPS stimulation. T-cells, B-cells and CD11b single positive cells were not significantly changed by Tab1 deficiency. Tab1 deficiency alone did not cause changes in macrophage population (Figure S2A). Importantly, the frequency of CD11b+ F4/80+ macrophages in peritoneal fluid was increased by LPS to around 30% from 10–15% under unstimulated conditions in control mice, but such increase was not observed in Tab1iKO mice. We noted that splenic CD11b+ F4/80+ macrophages were not significantly altered by LPS stimulation, and Tab1 deletion did not cause any alteration of splenic macrophages (Figure S2B), supporting the notion that participation of TAB1-dependent pathway in macrophage survival varies depending on macrophage type or activation state [48]. These suggest that TAB1 is important for activated macrophage maintenance in vivo.
Figure 6. LPS treated Tab1iKO macrophages are reduced in vivo.

(A) Tab1iKO or control including Tab1F+ Cre mice were intraperitoneally injected with 50 mg/kg tamoxifen for 3 consecutive days. After 3–5 weeks, peripheral blood was collected and leukocyte extracts were tested for Tab1 deletion by Western blotting. (B) Tab1iKO (n = 6) and control mice (n = 9) were intraperitoneally injected with 8 mg/kg LPS. Peritoneal leukocytes were collected at 72 hours and stained with fluorophore-conjugated antibodies. Shown is percent positive, excluding dead cells and debris, for the indicated markers. Percentages of CD11b+ F4/80+, CD11b+, CD3e+ or B220+ cells of total cells +/− SD is shown.
Discussion
Our current studies identify an essential role for TAK1 modulator proteins, TAB1 and TAB2, in LPS-activated macrophages, in BMDMs, and in a mouse model of inflammation (Figure 7). The roles of TAB1 and TAB2 in macrophages have not been well characterized in the published literature to date. We demonstrate that TAB1 and TAB2 are essential for LPS-activated macrophage survival but are dispensable for survival of naïve macrophages. Bacterial moieties including LPS are strong activators of macrophages, which polarize macrophages toward an inflammatory, M1 phenotype. M1 macrophages mediate acute inflammation by secreting cytokines and chemokines, which play a major role in initiation of inflammatory responses [49]. These classically activated macrophages are prone to cell death, which contributes to the termination of inflammatory responses [3]–[5]. Chronically activated macrophages are associated with many disease conditions including obesity, autoimmune diseases, atherosclerosis and asthma [50]. In contrast, naïve macrophages in tissues, so-called resident macrophages such as microglia, Kupffer cells and Langerhans cells, play indispensable roles during development and in tissue repair [51]–[53]. Resident unstimulated macrophages are also important for prompt responses to protect tissues from insults. Therefore, the presence of unstimulated resident macrophages is beneficial to maintain tissue integrity. Thus, limiting inflammatory conditions by controlling only an activated sub-set of macrophages could be a useful tool in treating disease. Our results demonstrate that deletion of either Tab1 or Tab2 effectively kills only LPS-activated macrophages in vitro, and that Tab1 deletion prevents increase of peritoneal macrophages upon LPS stimulation. These results indicate that TAB1 and TAB2 are potentially useful targets to selectively control the activated fraction of macrophages.
Figure 7. TAB1 and TAB2 are essential for LPS-activated macrophage survival.
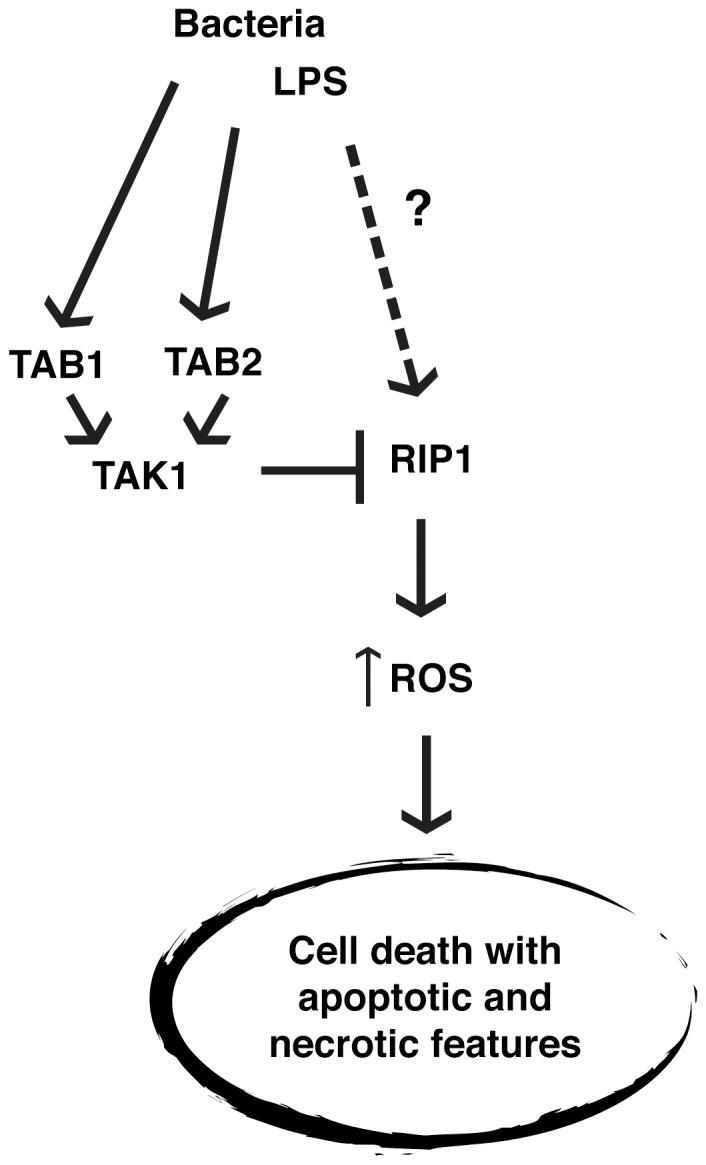
TAK1 binding proteins, TAB1 and TAB2 are essential for protecting BMDMs from LPS-induced cell death, which occurs downstream of RIP1, involves increased ROS, and shows features of both apoptosis and necrosis.
It is interesting that the Tab1 Tab2diKO shows diminished activation of IKK and p38 following LPS stimulation, which is similar to the Tak1iKO phenotype. When these genes are individually deleted, LPS-induced IKK and p38 activation are nearly normal and naïve macrophages persist. However, macrophages having deletion of either Tab1 or Tab2 could not withstand LPS-activation and Tab1iKO macrophages underwent necrosis after 3–4 days, while Tab2iKO die after 1 day, despite showing early LPS-induced TAK1 activation at or near WT levels (Figures 4B and C). In light of the nearly normal activation of NF-κB and p38 in Tab1iKO and Tab2iKO in response to LPS treatment, TAB1- and TAB2-dependent activated macrophage survival may occur through signaling pathway(s) independently of NF-κB and p38, which is in contrast to previous studies [4], [5].
Recapitulating our in vitro results, we found that the number of peritoneal macrophages was lesser in LPS-treated Tab1iKO mice when compared to control. Based on our in vitro data, we anticipate this disease model to produce a net increase in LPS-activated peritoneal macrophages in control but not Tab1iKO mice, however it cannot be conclusively excluded that the observed reduction is due to a defect in recruitment or expansion independent of cell death. Importantly, other hematopoietic cell types, including T cells, B cells and granulocytes, were found to be unaffected, suggesting TAB1-dependent survival signaling that is potentially unique to macrophages. We note here that liver was found to be undamaged in Tab1-deficient mice upon LPS injection under our experimental conditions, although higher doses of LPS are known to cause liver damage. Inferences based on our studies are limited by our mouse model, which has Tab1 deleted in all cells, such that one cannot rule out the effects from other cell types in vivo. Future studies analyzing macrophage-specific conditional knockout mice for Tab1 and Tab2 could bring insight to the roles of these genes in macrophage cell death. Further research focused on this important mechanism in macrophages could inform inflammatory disease models, particularly diseases in which microorganisms target macrophages. The data supporting that TAB1 is essential for macrophage survival in LPS-treated mice could be used to improve our understanding of the control of inflammation.
Supporting Information
Wild-type macrophages treated with LPS do not have reduced viability under experimental conditions. Viability of LPS-treated control macrophages. Tab2flox/flox BMDMs were cultured for 8 days with 0.3 µM 4-OHT followed by 3 days 1 µg/ml LPS, and viability was measured by Crystal Violet Assay. Shown are mean percentages of attached macrophages compared to 8 days treated with vehicle +/− SD of 3 independently performed experiments.
(TIF)
TAB1-dependent survival depends on type of macrophage. (A) Peritoneal leukocytes were collected from Tab1-deficient mice treated with vehicle (PBS) at 72 hours and stained with fluorophore-conjugated antibodies. Shown is percent positive of 2 control and 3 Tab1iKO for the indicated markers. Percentages of CD11b+ F4/80+, CD11b+, CD3e+ or B220+ cells of total cells ±SD is shown. (B) Tab1iKO and control mice were intraperitoneally injected with 8 mg/kg LPS. Splenocytes were collected and stained with fluorophore-conjugated antibodies. Shown is percent positive of 6 control and 4 Tab1iKO for CD11b+ F4/80+, CD11b+, CD3e+ or B220+ as a percentage of total cells ±SD.
(TIF)
Acknowledgments
We thank S. Akira for Tak1-floxed and Tab2-floxed mice, and S. Elliott, L. Hester, and J. Dow for support.
Funding Statement
This work was supported by “Keio Kanrinmaru Project,” the Promotion of Environmental Improvement for Independence of Young Researchers under the Special Coordination Funds for Promoting Science and Technology (JST, Japan) (to G. T.), and National Institutes of Health Grant GM068812 (to J. N-T.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496: 445–455 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weigert A, Johann AM, Knethen A von, Schmidt H, Geisslinger G, et al. (2006) Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 108: 1635–1642 10.1182/blood-2006-04-014852 [DOI] [PubMed] [Google Scholar]
- 3. He S, Liang Y, Shao F, Wang X (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A 108: 20054–20059 10.1073/pnas.1116302108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, et al. (2005) Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-kappaB as key regulators. Immunity 23: 319–329 10.1016/j.immuni.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 5. Ma Y, Temkin V, Liu H, Pope RM (2005) NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem 280: 41827–41834 10.1074/jbc.M510849200 [DOI] [PubMed] [Google Scholar]
- 6. Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132: 344–362 10.1016/j.cell.2008.01.020 [DOI] [PubMed] [Google Scholar]
- 7. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, et al. (1999) The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398: 252–256 10.1038/18465 [DOI] [PubMed] [Google Scholar]
- 8. Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, et al. (2003) TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol 326 105–115: 10.1016/S0022-2836(02)01404-3 [DOI] [PubMed] [Google Scholar]
- 9. Chen ZJ, Bhoj V, Seth RB (2006) Ubiquitin, TAK1 and IKK: is there a connection? Cell Death Differ 13: 687–692 10.1038/sj.cdd.4401869 [DOI] [PubMed] [Google Scholar]
- 10. Cheung PC, Nebreda AR, Cohen P (2004) TAB3, a new binding partner of the protein kinase TAK1. Biochem J 378: 27–34 10.1042/BJ20031794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, et al. (2003) Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J 22: 6277–6288 10.1093/emboj/cdg605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, et al. (2004) TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell 15: 535–548 10.1016/j.molcel.2004.08.008 [DOI] [PubMed] [Google Scholar]
- 13. Kishida S, Sanjo H, Akira S, Matsumoto K, Ninomiya-Tsuji J (2005) TAK1-binding protein 2 facilitates ubiquitination of TRAF6 and assembly of TRAF6 with IKK in the IL-1 signaling pathway. Genes Cells 10: 447–454 10.1111/j.1365-2443.2005.00852.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xia ZP, Sun L, Chen X, Pineda G, Jiang X, et al. (2009) Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461: 114–119 10.1038/nature08247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, et al. (2003) TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol 23: 1231–1238 10.1128/MCB.23.4.1231-1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ori D, Kato H, Sanjo H, Tartey S, Mino T, et al. (2013) Essential Roles of K63-Linked Polyubiquitin-Binding Proteins TAB2 and TAB3 in B Cell Activation via MAPKs. J Immunol 190: 4037–4045 10.4049/jimmunol.1300173 [DOI] [PubMed] [Google Scholar]
- 17. Ono K, Ohtomo T, Sato S, Sugamata Y, Suzuki M, et al. (2001) An evolutionarily conserved motif in the TAB1 C-terminal region is necessary for interaction with and activation of TAK1 MAPKKK. J Biol Chem 276: 24396–24400 10.1074/jbc.M102631200 [DOI] [PubMed] [Google Scholar]
- 18. Scholz R, Sidler CL, Thali RF, Winssinger N, Cheung PC, et al. (2010) Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process. J Biol Chem 285: 25753–25766 10.1074/jbc.M109.093468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inagaki M, Omori E, Kim JY, Komatsu Y, Scott G, et al. (2008) TAK1-binding protein 1, TAB1, mediates osmotic stress-induced TAK1 activation but is dispensable for TAK1-mediated cytokine signaling. J Biol Chem 283: 33080–33086 10.1074/jbc.M807574200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Omori E, Inagaki M, Mishina Y, Matsumoto K, Ninomiya-Tsuji J (2012) Epithelial transforming growth factor beta-activated kinase 1 (TAK1) is activated through two independent mechanisms and regulates reactive oxygen species. Proc Natl Acad Sci U S A 109: 3365–3370 10.1073/pnas.1116188109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu HH, Xie M, Schneider MD, Chen ZJ (2006) Essential role of TAK1 in thymocyte development and activation. Proc Natl Acad Sci U S A 103: 11677–11682 10.1073/pnas.0603089103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, et al. (2005) Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6: 1087–1095 10.1038/ni1255 [DOI] [PubMed] [Google Scholar]
- 23. Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, et al. (2006) TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int Immunol 18: 1405–1411 10.1093/intimm/dxl082 [DOI] [PubMed] [Google Scholar]
- 24. Wan YY, Chi H, Xie M, Schneider MD, Flavell RA (2006) The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol 7: 851–858 10.1038/ni1355 [DOI] [PubMed] [Google Scholar]
- 25. Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, et al. (2010) TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell 17: 481–496 10.1016/j.ccr.2010.03.021 [DOI] [PubMed] [Google Scholar]
- 26. Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, et al. (2010) Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A 107: 844–849 10.1073/pnas.0909781107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kajino-Sakamoto R, Inagaki M, Lippert E, Akira S, Robine S, et al.. (2008) Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J Immunol 181: 1143–1152. PMCID: PMC3065656, NIHMSID: NIHMS279618. [DOI] [PMC free article] [PubMed]
- 28. Kajino-Sakamoto R, Omori E, Nighot PK, Blikslager AT, Matsumoto K, et al. (2010) TGF-beta-activated kinase 1 signaling maintains intestinal integrity by preventing accumulation of reactive oxygen species in the intestinal epithelium. J Immunol 185: 4729–4737 10.4049/jimmunol.0903587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Omori E, Matsumoto K, Sanjo H, Sato S, Akira S, et al. (2006) TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J Biol Chem 281: 19610–19617 10.1074/jbc.M603384200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Omori E, Morioka S, Matsumoto K, Ninomiya-Tsuji J (2008) TAK1 regulates reactive oxygen species and cell death in keratinocytes, which is essential for skin integrity. J Biol Chem 283: 26161–26168 10.1074/jbc.M804513200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang M, Wei X, Guo Y, Breslin P, Zhang S, et al. (2008) TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J Exp Med 205: 1611–1619 10.1084/jem.20080297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ajibade AA, Wang Q, Cui J, Zou J, Xia X, et al. (2012) TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 36: 43–54 10.1016/j.immuni.2011.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eftychi C, Karagianni N, Alexiou M, Apostolaki M, Kollias G (2012) Myeloid TAKI [corrected] acts as a negative regulator of the LPS response and mediates resistance to endotoxemia. PloS One 7: e31550 10.1371/journal.pone.0031550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG (2013) TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol 33: 582–595 10.1128/MCB.01225-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inagaki M, Komatsu Y, Scott G, Yamada G, Ray M, et al. (2008) Generation of a conditional mutant allele for Tab1 in mouse. Genesis 46: 431–439 10.1002/dvg.20418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badea TC, Wang Y, Nathans J (2003) A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci 23: 2314–2322. PMID: 12657690. [DOI] [PMC free article] [PubMed]
- 37. Newton K, Sun X, Dixit VM (2004) Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24: 1464–1469 10.1128/MCB.24.4.1464-1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, et al. (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73: 457–467 10.1016/0092-8674(93)90134-C [DOI] [PubMed] [Google Scholar]
- 39. Lee TH, Shank J, Cusson N, Kelliher MA (2004) The Kinase Activity of Rip1 Is Not Required for Tumor Necrosis Factor-α-induced IκB Kinase or p38 MAP Kinase Activation or for the Ubiquitination of Rip1 by Traf2. J Biol Chem 279: 33185–33191 10.1074/jbc.M404206200 [DOI] [PubMed] [Google Scholar]
- 40. Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, et al. (2006) Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem 281: 39891–39896 10.1074/jbc.M608155200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H (2005) Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem 280: 7359–7368 10.1074/jbc.M407537200 [DOI] [PubMed] [Google Scholar]
- 42.Kishimoto K, Matsumoto K, Ninomiya-Tsuji J (2000) TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J Biol Chem 275: 7359–7364. PMID: 10702308 [DOI] [PubMed]
- 43. Jadrich JL, O'Connor MB, Coucouvanis E (2006) The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 133: 1529–1541 10.1242/dev.02333 [DOI] [PubMed] [Google Scholar]
- 44. Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, et al. (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 19: 2668–2681 10.1101/gad.1360605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komatsu Y, Shibuya H, Takeda N, Ninomiya-Tsuji J, Yasui T, et al. (2002) Targeted disruption of the Tab1 gene causes embryonic lethality and defects in cardiovascular and lung morphogenesis. Mech Dev 119: : 239–249. doi.org/10.1016/S0925-4773(02)00391-X. [DOI] [PubMed] [Google Scholar]
- 46. Higashi AY, Ikawa T, Muramatsu M, Economides AN, Niwa A, et al. (2009) Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J Immunol 182: 5633–5640 10.4049/jimmunol.0802413 [DOI] [PubMed] [Google Scholar]
- 47. Takaesu G, Inagaki M, Takubo K, Mishina Y, Hess PR, et al. (2012) TAK1 (MAP3K7) signaling regulates hematopoietic stem cells through TNF-dependent and -independent mechanisms. PloS One 7: e51073 10.1371/journal.pone.0051073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, et al. (2009) Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 457: 318–321 10.1038/nature07472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martinez FO, Sica A, Mantovani A, Locati M (2008) Macrophage activation and polarization. Front Biosci 13: 453–461 10.2741/2692 [DOI] [PubMed] [Google Scholar]
- 50. Soehnlein O, Lindbom L (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol 10: 427–439 10.1038/nri2779 [DOI] [PubMed] [Google Scholar]
- 51. Ji K, Miyauchi J, Tsirka SE (2013) Microglia: An Active Player in the Regulation of Synaptic Activity. Neural Plast 2013: 627325 10.1155/2013/627325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chorro L, Geissmann F (2010) Development and homeostasis of “resident” myeloid cells: the case of the Langerhans cell. Trends Immunol 31: 438–445 10.1016/j.it.2010.09.003 [DOI] [PubMed] [Google Scholar]
- 53. Jaeschke H (2011) Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol 26 Suppl 1 173–179 10.1111/j.1440-1746.2010.06592.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Wild-type macrophages treated with LPS do not have reduced viability under experimental conditions. Viability of LPS-treated control macrophages. Tab2flox/flox BMDMs were cultured for 8 days with 0.3 µM 4-OHT followed by 3 days 1 µg/ml LPS, and viability was measured by Crystal Violet Assay. Shown are mean percentages of attached macrophages compared to 8 days treated with vehicle +/− SD of 3 independently performed experiments.
(TIF)
TAB1-dependent survival depends on type of macrophage. (A) Peritoneal leukocytes were collected from Tab1-deficient mice treated with vehicle (PBS) at 72 hours and stained with fluorophore-conjugated antibodies. Shown is percent positive of 2 control and 3 Tab1iKO for the indicated markers. Percentages of CD11b+ F4/80+, CD11b+, CD3e+ or B220+ cells of total cells ±SD is shown. (B) Tab1iKO and control mice were intraperitoneally injected with 8 mg/kg LPS. Splenocytes were collected and stained with fluorophore-conjugated antibodies. Shown is percent positive of 6 control and 4 Tab1iKO for CD11b+ F4/80+, CD11b+, CD3e+ or B220+ as a percentage of total cells ±SD.
(TIF)