

Abstract
The effect of DHA on HO-1 expression in cancer cells has never been characterized. This study examines DHA-induced HO-1 expression in human cancer cell model systems. DHA enhanced HO-1 gene expression in a time- and concentration-dependent manner, with maximal induction at 21 hours of treatment. This induction of HO-1 expression was confirmed in vivo using a xenograft nude mouse model fed a fish oil-enriched diet. The increase in HO-1 gene transcription induced by DHA was significantly attenuated by the antioxidant N-Acetyl Cysteine (NAC), suggesting the involvement of oxidative stress. This was supported by direct measurement of lipid peroxide levels after DHA treatment. Using a human HO-1 gene promoter reporter construct, we identified two antioxidant response elements (AREs) that mediate the DHA-induced increase in HO-1 gene transcription. Knockdown of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) expression compromised the DHA-induced increase in HO-1 gene transcription, indicating the importance of the Nrf2 pathway in this event. However, the protein levels of Nrf2 remained unchanged upon DHA treatment. Further studies demonstrated that DHA reduces nuclear Bach1 protein expression by promoting its degradation and attenuates Bach1 binding to the AREs in the HO-1 gene promoter. In contrast, DHA enhanced Nrf2 binding to the AREs without affecting nuclear Nrf2 expression levels, indicating a new cellular mechanism that mediates DHA’s induction of HO-1 gene transcription. To our knowledge, this is the first characterization of DHA induced HO-1 expression in human malignant cells.
Keywords: Cancer, heme oxygenase-1, gene transcription regulation, n-3 PUFA, antioxidant response element
1. INTRODUCTION
Docosahexaenoic acid (DHA, 22:6, n-3) is a long chain n-3 polyunsaturated fatty acid (n-3 PUFA) that has been shown to have anticancer activity in various experimental model systems [1]. DHA induces apoptosis of cancer cells [2], inhibits cancer cell proliferation [3, 4], and suppresses tumor growth in vivo [2, 5]. The cellular and molecular mechanisms of long chain n-3 PUFA’s anticancer activity have been extensively investigated yet still remain elusive. Early studies revealed that DHA is incorporated into the cell membrane at the expense of arachidonic acid (AA) leading to reduction of AA-derived prostaglandins and inhibition of the COX2 pathway. Furthermore, DHA-derived lipid mediators such as resolvins and protectins may have anti-inflammatory and anticancer activity. DHA was also shown to regulate gene expression thereby promoting programmed cancer cell death, suppress cellular survival signaling pathways, and enhance cellular oxidative stress [6, 7]. Recent studies demonstrated that DHA induces autophagy [8] and endoplasmic reticulum (ER) stress [9] in cancer cells. Among the cellular mechanisms described, DHA-induced lipid peroxidation has been well recognized to mediate DHA’s anticancer activity. Upon incorporation of DHA into tumor cell membranes, their susceptibility to lipid peroxidation is increased [10] and accumulation of the lipid peroxidation by-products causes peroxidative damage, ultimately leading to cell death [11]. These observations are further supported by the fact that the killing of malignant cells by DHA can be accelerated by increased cellular oxidative stress [12]. Thus, the interplay between pro-oxidants and anti-oxidants is likely to contribute to DHA’s cytotoxicity toward cancer cells.
Heme oxygenase 1 (HO-1) is one of the rate-limiting enzymes in heme catabolism which catalyzes the stereospecific degradation of heme to biliverdin, with the concurrent release of iron and carbon monoxide (CO). Biliverdin is further converted to bilirubin by biliverdin reductase [13]. Because both bilirubin and CO have antioxidant activity, HO-1 is considered a cytoprotective antioxidant enzyme. In addition to free heme, many cellular stimuli, such as cytokines, heavy metals, physical stress, heat shock, and other oxidants can induce HO-1 expression [13]. Studies have demonstrated that most stimuli increase HO-1 transcription by targeting the keap1/Nrf2 signaling pathway [13]. The keap1 protein is normally bound to the Nrf2 transcription factor, thereby promoting Nrf2 protein degradation. During oxidative stress, the Nrf2 protein is released from keap1 and is translocated to the nucleus where it binds to AREs leading to the transcriptional activation of HO-1 and other antioxidant enzymes [14]. However, recent studies have pointed out that induction of HO-1 in eukaryotic cells is likely a compound-dependent event that requires further characterization [15].
Recent studies have shown that DHA induces HO-1 expression in non-cancerous model systems [16, 17], likely through multiple signaling mechanisms. However, the effect of DHA on HO-1 expression has never been thoroughly investigated in human malignant cells. The fact that cancer cells are more vulnerable to oxidative stress [18–20] and HO-1 is an established antioxidant enzyme intrigued us to investigate how DHA might regulate HO-1 expression in human cancer cells. As HO-1 has been proposed as a target for cancer therapy and for overcoming chemo-resistance [21, 22], a better understanding of whether and how DHA regulates HO-1 expression in cancer cells will provide novel strategies for further development of DHA as an effective anticancer agent.
In this study we have characterized the effects of DHA on HO-1 expression both in vitro, in A2780 (human ovarian cancer) cells, and in vivo, in a xenograft nude mouse model fed a fish oil diet. We demonstrate that treatment with DHA enhances HO-1 expression in our model systems. This increase in HO-1 expression by DHA was mediated at the transcriptional level and could be attenuated by the antioxidant NAC. Interestingly, we found that DHA increases HO-1 transcription by enhancing the degradation of the Bach1 protein, which led to increased binding of Nrf2 to the AREs in the HO-1 gene promoter, thus up-regulating HO-1 gene transcription.
2. MATERIALS AND METHODS
2.1 Materials
The pGL3/4.5-HO-1 luciferase reporter construct containing the 4.5 kilobase fragment of the human HO-1 gene promoter was kindly provided by Dr. Anupam Agarwal (University of Alabama at Birmingham, Birmingham, AL). The HO-1 3′-UTR and the glyceraldehydes-3-P-dehydrogenase (GAPDH) 3′-UTR reporter constructs were purchased from SwitchGear Genomics (Menlo Park, CA). The antibodies for Bach1 (sc-14700), Nrf2 (sc-722), PPARα (sc-9000) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The GAPDH antibody (Cat. 20035) was from ProMab Biotechnologies, Inc. (Albany, CA). The HO-1 antibody (SPA-896) was from Stressgen (Ann Arbor, MI). The Dual-Luciferase Reporter kit was from Promega (Madison, WI). The QuikChange Site-Directed Mutagenesis Kit was from Stratagene (La Jolla, CA). The Chromatin Immunoprecipitation (ChIP) Assay Kit was from Millipore (Temecula, CA). Bortezomib was from Selleck Chemicals (Houston, TX). Diphenyl-1-pyrenylphosphine (DPPP) and Prolong Gold Antifade were from Life Technologies Co. (Carlsbad, CA). MG132, α-eleostearic acid (α-ESA) and the Thiobarbituric Acid Reactive Substances (TBARS) assay kit were from Cayman Chemical (Ann Arbor, MI). The β-actin antibody (A5441), DHA, NAC and other chemical agents were analytic grade and purchased from Sigma-Aldrich (St. Louis, MO).
2.2 Cell culture
The human ovarian carcinoma cell line A2780 [23] was a kind gift from Dr. Stephen Howell (University of California, San Diego, CA). The EA.hy926 cell line [24] was kindly provided by Dr. Doris Benbrook (University of Oklahoma Health Sciences Center, Oklahoma City, OK). The human breast cancer line MCF7 was obtained from the American Type Culture Collection (Manassas, VA, USA). A2780 and MCF7 cells were cultivated in RPMI 1640 medium, and and EA.hy926 cells in DMEM medium, both being supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cells were routinely grown under a humid environment at 37°C, 5% CO2, and passaged twice a week. For DHA treatment, stock DHA was dissolved in ethanol at 30 mM and mixed with bovine serum albumin at 4:1 molar ratios in PBS before adding to cell culture. The final concentration of ethanol in the medium was below 0.5 percent. Control cells were treated with vehicle buffer.
2.3 Xenograft nude mouse study
Nude mice were purchased from Taconic Farms Inc. (Germantown, NY) and were used for in vivo evaluation of HO-1 induction by long chain n-3 polyunsaturated fatty acids, in accordance with the Institute Animal Care and Use Committee procedures and guidelines. A total of 5×106 A2780 cells were suspended in 100 μL PBS, mixed with 20% Matrigel, and injected s.c. into the flanks of 5-week old female nude mice. The mice were fed with either 7.5% (wt/wt) corn oil diet or 7.5% (wt/wt) fish oil diet containing enriched long chain n-3 PUFA such as DHA and eicosapentaenoic acid (EPA, Table 1). DHA and EPA account for 28% of the fat in the fish oil diet (Teklad Diets, Madison, WI). This type of diet has been frequently used for in vivo study of DHA’s anticancer activity [2, 25–29]. Tumor volume was determined three times a week as we previously described [30]. The tumor volume was calculated with following formula: v=l×w×0.5, where v is the tumor volume, l the length of tumor, and w the width of tumor. Animal weights were also recorded three times a week. For HO-1 expression analysis, animals were sacrificed four weeks after inoculation, the xenograft tissues were collected and immediately frozen using dry ice. Total RNA and proteins were extracted from the tissues and analyzed with reverse transcriptase-polymerase chain reaction (RT-PCR) using primers specific for HO-1 and GAPDH cDNA sequence, and Western blot using antibodies against HO-1 and GAPDH.
Table 1.
Composition of corn oil and fish oil diets (g/Kg).
| Diet ingredients | Fish oil | Corn oil |
|---|---|---|
|
| ||
| Casein | 200.0 | 200.0 |
| DL-Methionine | 3.0 | 3.0 |
| Sucrose | 475.0 | 475.0 |
| Corn Starch | 150.0 | 150.0 |
| Fish oil | 75.0 | 0.0 |
| Corn oil | 0.0 | 75.0 |
| Cellulose | 50.0 | 50.0 |
| Mineral Mix, AIN-76 | 35.0 | 35.0 |
| Vitamin Mix, AIN-76A | 10.0 | 10.0 |
| Choline Bitartrate | 2.0 | 2.0 |
| Ethoxyquin, antioxidant | 0.015 | 0.015 |
| Yellow food color | 0.1 | 0.1 |
2.4 Western blot analysis
Western blot was performed as described [30]. In brief, for cellular protein isolation, cells were lysed in buffer containing 50 mM Tris-HCl pH 7.8, 100 mM NaCl, 5 mM Na-EDTA, 0.1% SDS, 1 mM PMSF, 1% Triton-X-100, and 2.5% glycerol. The lysate was sonicated on ice for 3 strokes of 10 seconds each and centrifuged at 15,000×g for 15 minutes to remove insoluble material. For nuclear protein extraction, cells were detached by adding 2.5 ml wash buffer (for a 100-mm dish) containing 1 mM HEPES, pH 7.9, 150 μM MgCl2, 1 mM KCl, 50 μM Dithiothreitol (DTT), 100 μM PMSF, 200 ng/ml Aprotinin, 1μg/ml Leupeptin, 200 ng/ml Pepstatin A, and 0.01% NP-40. The lysate was centrifuged at 4,550×g for 2 minutes. Pellets were re-suspended in 50 μl suspension buffer containing 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 2 μg/ml aprotinin, 10 μg/ml leupeptin, 2 μg/ml pepstatin A. The suspension was incubated on ice for 30 minutes and centrifuged for 20 minutes at 15,000×g to remove insoluble material. Thirty to forty micrograms of protein from each sample was separated on a 10% SDS-PAGE gel, transferred to a PVDF membrane, and blotted with antibodies against HO-1, Nrf2, Bach1, PPARα, GAPDH, or β-actin. Protein expression of each specific band detected was semi-quantified by densitometry using Adobe Photoshop Elements 6.0 (San Jose, CA) and normalized to that of GAPDH or β-actin.
2.5 Transient transfection and luciferase activity assay
A2780 cells were seeded in 100-mm cell culture dishes and reached 70–80% confluence after 24 hours of plating. The cells were then transfected with the luciferase reporter constructs including the pGL3/4.5-HO-1 and its mutants, the HO-1-3′-UTR and GAPDH-3′-UTR using the Fugene HD transfection reagent (Roche, Mannheim, Germany) as previously described [31]. The next day, cells were lifted and plated into 24-well plates at a density of 2×105 per well. 48 hours after transfection, cells were treated with various reagents at indicated concentrations and durations. Cell lysates were prepared and luciferase activity was assayed using the Dual-Luciferase Reporter kit, as previously described [31]. The firefly luciferase activity was normalized to the amount of protein present in each sample. The data are expressed as percentages of luciferase activity detected in treated relative to untreated control cells.
2.6 siRNA knockdown of Nrf2 and PPARα
siRNA for Nrf2, PPARα were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Cells were co-transfected with 225 nM (final concentration) of Nrf2, PPARα, or scrambled non-specific siRNAs (as control) and 2 μg of PGL3/4.5-HO-1 into A2780 cells cultured in a 100-mm dish using the Fugene HD transfection reagent (Roche, Mannheim, Germany) following the manufacturer’s protocols. The next day, cells were lifted and plated into 24-well plates (for luciferase activity assay) and 100-mm dishes (for Western blot analysis). Forty-eight hours after the transfection, DHA was added to the medium at indicated concentrations for 21 hours. The cells were lysed and assayed for luciferase activity. The knockdown of Nrf2 and PPARα was confirmed by Western blot analysis.
2.7 Reverse transcriptase-polymerase chain reaction
Total RNA was isolated from xenograft tissues using the Trizol Reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. RNA samples were reverse-transcribed (RT) with the SuperScript II kit (Invitrogen, Carlsbad, CA) as previously described [32]. The cDNA was amplified by polymerase chain reaction (PCR) using the following transcript specific primers: HO-1, forward: 5′-GAG TCT AGA ATG CAG GCA TGC TGG CTC CC-3′, reverse: 5′-GAG TCT AGA CAA GCT ACT ATC AGA CAA TG C-3′; GAPDH, forward: 5′-TGGGGAAGGTGAAGGTCGG-3, reverse: 5′-GGGATCTCGCTCCTGGAAG-3′. The samples were initially denatured at 94 °C for 2 minutes prior to thermal cycling. The thermal cycle parameters for PCR were as follows: 94 °C for 1 minute, 53 °C (HO-1) or 48 °C (GAPDH) for 2 minutes, and 72 °C for 2 minutes, for a total of 31 cycles. The PCR products were separated on a 1% agarose gel containing ethidium bromide and visualized under ultraviolet light. The specific band visualized was semi-quantified by densitometry using Adobe Photoshop Elements 6.0 (San Jose, CA) and normalized to that of GAPDH.
2.8 Generation of PGL3/4.5-HO-1 mutants
The PGL3/4.5-HO-1 construct was used to generate mutants with deletion of the two AREs [33, 34] and two PPREs located within the promoter region [35]. This was achieved using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) as we described [36]. The mutations were confirmed by DNA sequencing.
2.9 Lipid Peroxidation Analysis
Lipid peroxidation was analyzed using both the DPPP staining and TBARS assay. For DPPP assay, A2780 cells were plated on sterile glass cover-slips in 6-well plates. After reaching 70–80% confluence, cells were treated with DHA or α-ESA for 21 hours, or pre-treated with NAC for 15 minutes followed by DHA treatment for 21 hours. The cells were then washed and fresh medium containing 50 μM DPPP was added. The cells were incubated in the dark at 37°C for 30 minutes, washed twice with PBS, and fixed with 4% para-formaldehyde at room temperature for 10 minutes. Cells were then washed twice with PBS and the cover-slips were lifted from the wells and placed face down onto a drop of AntiFade fixative on a glass slide. Slides were stored in the dark overnight prior to fluorescent microscope analysis. Cells were observed under a Nikon TE2000-E microscope with excitation at 351 nm and emission at 380 nm. All procedures prior to imaging were performed in the dark as DPPP is light-sensitive. The DPPP signaling detected was quantified using Adobe Photoshop Elements 6.0 (San Jose, CA), and is expressed as levels relative to untreated controls. For TBARS assay, A2780 cells were pre-treated with or without Vitamin E at indicated concentrations for 15 minutes followed by treatment with DHA for indicated durations. Cells were harvested according to the manufacture’s protocol. Briefly, cells were resuspended in PBS at a concentration of 2×107/ml and sonicated for 3×5 seconds on ice. The cellular lysate or malondialdehyde (MDA) standard were mixed with 100 μl of the SDS solution, and 4 ml of the color reagent. The reaction was stopped by boiling the samples for 1 hour. Samples were then incubated on ice for 10 minutes and centrifuged at 1,600×g for 10 min at 4°C. The reactions were then transferred to a black plate and the absorbance was recorded at 544 nm excitation, 590 nm emission.
2.10 Chromatin Immunoprecipitation Assay
The ChIP assay was performed following the manufacture’s protocol. In short, A2780 cells were treated with 100 μM DHA for 6 or 21 hours. Formaldehyde was then added directly to the medium (final concentration, 1%) to cross-link DNA and proteins at 37°C for 10 minutes. Cells were washed, removed from the dish, pelleted, and lysed on ice in 1% SDS buffer. The lysate was sonicated to shear the DNA into sizes ranging from 200 to 1000 bp and the DNA was pre-cleared for 30 min at 4°C with Protein A agarose/salmon sperm DNA (50% Slurry). The complex of protein and DNA was precipitated overnight at 4°C using Nrf2, Bach1 or PPARα antibody, and the complex was then incubated with Protein A agarose/salmon sperm DNA (50% Slurry) for 1 hour at 4°C, washed and eluted. The precipitants were incubated with NaCl at 65°C for 4 hours to reverse DNA cross-links. The precipitants were then digested with proteinase K, and DNA was extracted with phenol/chloroform and precipitated with ethanol. DNA was analyzed by PCR amplification using primers specific to the promoter regions of interest, and the PCR products were separated on a 1% agarose gel containing ethidium bromide and visualized under ultraviolet light. The primers used for DNA amplification of DNA fragment in the HO-1 gene promoter were as follows (HO-1 genomic sequence: NC_000022.10 (35777060-35790207)): ARE1 (-4074 to -4082) forward, 5′-CTG CCC AAA CCA CTT CTG TT-3′; reverse, 5′-GCA CTG GTG ACT CAG CAA AA -3′; ARE2 (-4010 to -4019) forward, 5′-GAC TCG CGG AAA CAA AGG-3′; reverse, 5′-GAA GCA TAA GAA GGC CTC-3′; PPRE (-1836 to -1881) forward, 5′-CTG GAG AGA GAA AGA GAC-3′; reverse, 5′-GTG GAG TCA AGA GGC TAC-3′.
2.11 Statistical Analysis
Statistical analysis was done with Graphpad Prism software (San Diego, CA). One-way ANOVA with Dunnett’s post-test was used to determine differences among control and experimental groups, with p < 0.05 or p < 0.01 as the level of statistical significance. The mouse survival curves were determined by the Kaplan-Meier method. The log-rank test was used to assess the differences in survival curves, with p < 0.05 as the level of statistical significance.
3. RESULTS
3.1 DHA enhances HO-1 expression through transcriptional regulation in human cancer cells
HO-1 was found to be barely detectable in A2780 cells as measured by Western blot (Figure 1). Treatment of A2780 cells with DHA enhanced HO-1 expression in a time-dependent manner (Figure 1A). A minimum of four hours of 100 μM DHA treatment was required to induce HO-1 expression, which was further enhanced at 21 hours. The up-regulation of HO-1 protein expression by DHA was also concentration-dependent. A detectable increase of HO-1 levels was seen with 50 μM DHA and a maximal induction with 200 μM DHA after 21 hours of treatment (Figure 1B). We then tested HO-1 induction by DHA in an A2780 mouse xenograft model system [30]. Nude mice were fed with either a corn oil-based diet (7.5%, wt/wt) containing enriched n-6 PUFAs (without DHA and EPA) or fish oil-based diet (7.5%, wt/wt) containing enriched n-3 PUFAs (mainly DHA and EPA at a 40/60 ratio). As shown in Figure 2A, tumor growth in mice fed a fish oil-based diet was significantly slower than that in mice fed a corn oil-based diet, indicating that a high DHA and EPA content suppresses tumor growth in vivo, results consistent with previous reports using various tumor model systems [2, 25–29]. More interestingly, HO-1 expression was higher at both the mRNA and protein levels in mice fed a fish oil-based diet versus mice fed a corn oil-based diet (Figure 2B, C). While body weight was similar between the two groups of mice, mice fed a fish oil-based diet survived significantly longer (Figure 2D).
Figure 1. Effects of DHA on HO-1 expression in cancer cells.
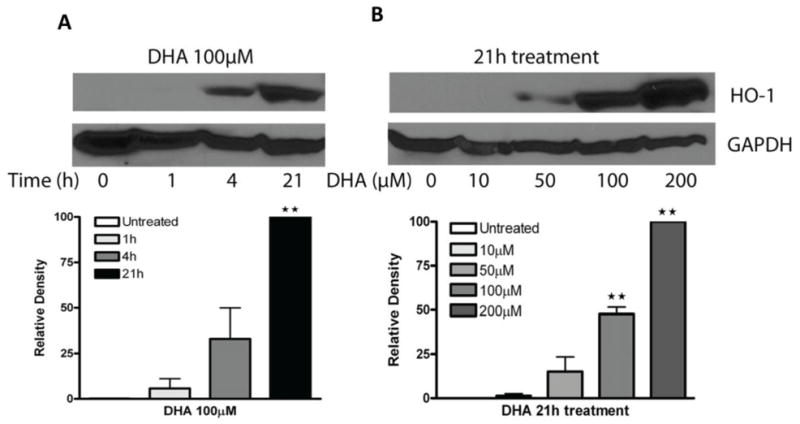
A2780 cells were treated with DHA at the indicated durations (A) or concentrations (B). Cell lysates were prepared and Western blot was performed using antibodies against HO-1 and GAPDH. Shown are representative images of three experiments. The relative density of the HO-1 band was quantified, normalized to that of GAPDH, and expressed as percentages relative to the highest expression level in DHA-treated cells. **, P < 0.01, compared to untreated cells using one-way ANOVA followed by Dunnett’s analysis.
Figure 2. Fish oil diet suppresses tumor growth and induces HO-1 expression in nude mice.
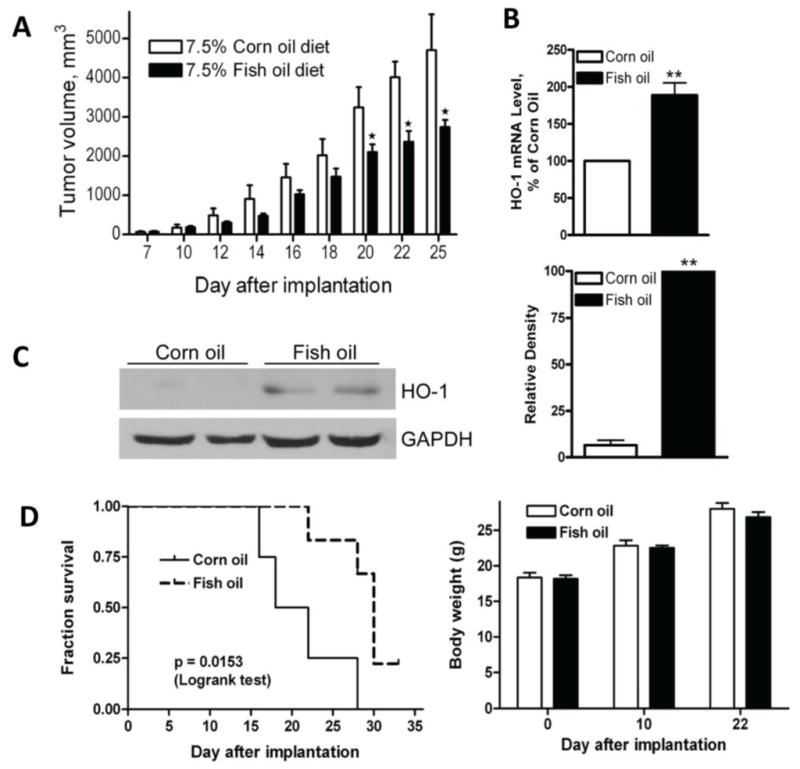
A. A2780 cells (5×106 in 100 l PBS mixed with 20% Matrigel) were injected s.c. into the flanks of 5-week-old female nude mice. Mice were on 7.5% fish oil diet or 7.5% corn oil diet, starting one week prior to implantation. Tumor volumes were determined as described in Materials and Methods. *, P<0.05, compared with mice fed with corn oil diet, using paired t-test (n=6). B. Xenograft tumor tissues were collected from nude mice. Total RNA were extracted, and analyzed with RT-PCR using primers specific for HO-1 and GAPDH. Shown is the HO-1 mRNA quantification of 4 individual animal samples from each group that is normalized to that of GAPDH, and expressed as percentages relative to the expression level in the tissues from mice fed corn oil diet. **, P < 0.01, compared to corn oil group using paired t-test. C. Xenograft tumor tissues were collected from nude mice. Total proteins were extracted, analyzed with Western blot analysis. Shown are representative images of 4 individual animal samples. The relative density of the HO-1 band was quantified, normalized to that of GAPDH, and expressed as percentages relative to the expression level in the tissues from mice fed fish oil diet. **, P < 0.01, compared to fish oil group using paired t-test. D. Survival curve and body weight of A2780-implanted mice.
To determine whether the enhanced expression of HO-1 by DHA is due to transcriptional or post-transcriptional regulation, we transfected A2780 cells with a PGL3/4.5-HO-1 promoter reporter construct or an HO-1-3′-UTR reporter construct and treated the cells with 50 or 100 μM DHA for 1, 4, and 21 hours. The luciferase activity levels indicated that HO-1 promoter activity is significantly induced by DHA, while the HO-1-3′-UTR-mediated luciferase activity remains unchanged after DHA treatment in A2780 cells (Figure 3). A reporter construct containing GAPDH-3′-UTR was included as a control. These results clearly indicate that the observed DHA-induced increase in HO-1 expression is mediated at the transcriptional level.
Figure 3. Effect of DHA on HO-1 gene transcription.
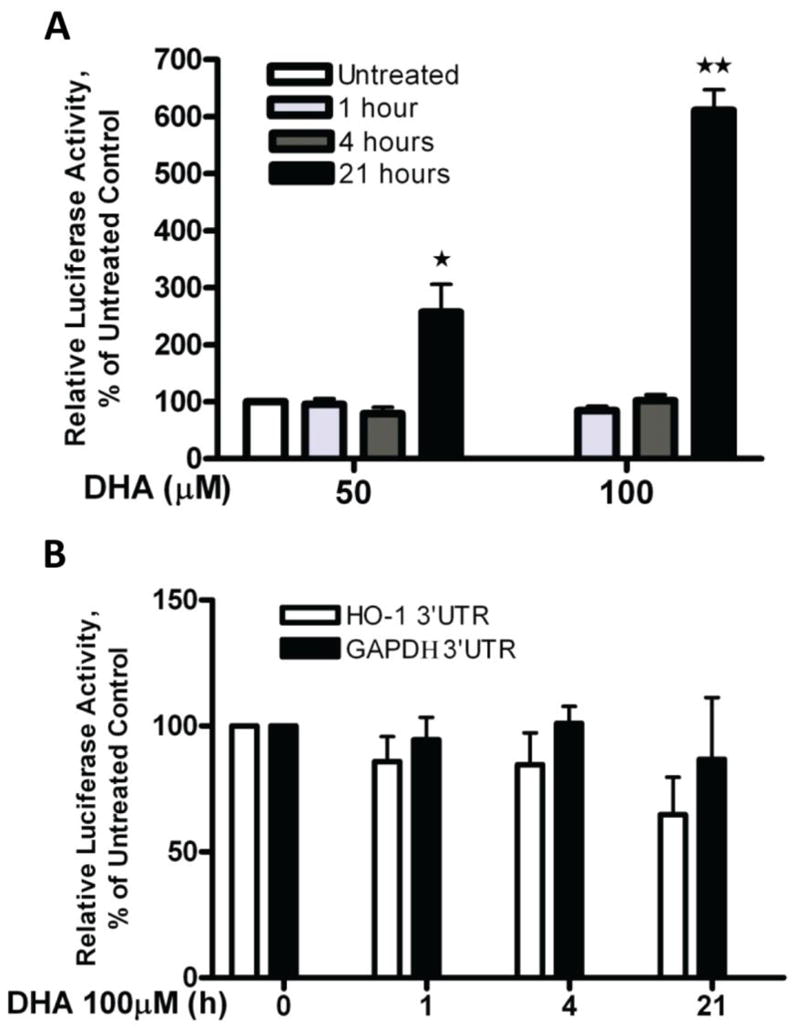
A. A2780 cells were transfected with the PGL3/4.5-HO-1 promoter reporter construct and treated with DHA at the indicated concentrations for 1, 4 or 21 hours. Cell lysates were prepared, and luciferase activity was assayed. Data are expressed as percentages of the value detected in untreated control cells (bars, SEM; n=3). **, P<0.01, *, P<0.05, compared to untreated control cells, using one-way ANOVA followed by Dunnett’s analysis. B. A2780 cells were transfected with the HO-1-3′-UTR or the GAPDH-3′-UTR reporter construct and treated with 100 μM DHA for 1, 4 or 21 hours. Cell lysates were prepared, and luciferase activity was assayed. Data are expressed as percentages of the value detected in untreated control cells (bars, SEM; n=3).
3.2 DHA is a more potent inducer of HO-1 expression than other fatty acids
To understand whether the induction of HO-1 is specific to DHA, A2780 cells transfected with a PGL3/4.5-HO-1 promoter reporter construct were treated with 100 μM of various fatty acids, including DHA (docosahexaenoic acid, 22:6, n-3), EPA (eicosapentaenoic acid, 20:5, n-3), AA (arachidonic acid, 20:5, n-6), α-LA (linolenic acid, 18:3, n-3), LA (linoleic acid, 18:3, n-6) or α-ESA (ethyl α-eleostearate, 20:3). Figure 4A shows that DHA dramatically enhanced HO-1 promoter activity after 21 hours of treatment (approximately 5-fold), whereas the other fatty acids had only a minor effect on HO-1 promoter activity (Figure 4A), demonstrating that DHA is the most potent inducer of HO-1 expression among the fatty acids tested. Because the fish oil diet contains DHA and EPA at a 40/60 ratio, we tested the combination of these two fatty acids on HO-1 gene promoter activity in A2780 cells. The combination of DHA (80 μM) and EPA (120 μM) induced HO-1 promoter activity to a level similar to that of DHA alone at 80 μM and much lower than that of DHA alone at 200 μM (Figure 4B). These results may explain the modest induction of HO-1 expression in vivo by fish oil diet (Figure 2). The induction of HO-1 transcription by DHA is likely due to its ability to initiate lipid peroxidation in our model system. The initiation of lipid peroxidation by DHA was confirmed using the DPPP probe, and the TBARS assay [32]. Treatment with DHA at 100 μM for 21 hours strongly enhanced the lipid peroxide levels as assayed by both assays in A2780 cells (Table 2). α-ESA and EPA both enhanced lipid peroxidation after 21 hours of treatment (Table 2) at levels significantly lower than that of DHA, consistent with their limited effect on HO-1 promoter activity. Pretreatment with NAC, a well-established antioxidant, dramatically reduced DHA-induced lipid peroxidation, and attenuated the DHA-induced increase in HO-1 promoter activity (Figure 4C), further indicating the involvement of oxidative stress in the DHA-induced increase in HO-1 gene transcription.
Figure 4. DHA-induced lipid peroxidation and HO-1 promoter activity.
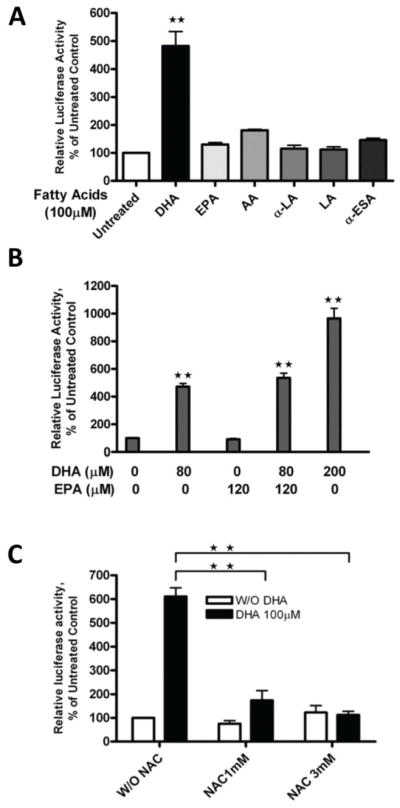
A. A2780 cells were transfected with the PGL3/4.5-HO-1 promoter reporter construct and treated with 100 μM of DHA (20:6, n-3), EPA (20:5, n-3), AA (20:5, n-6), alpha-LA (18:3, n-3), LA (18:3, n-6) or α-ESA (20:3, n-5) for 21 hours. B. A2780 cells were transfected with the PGL3/4.5-HO-1 promoter reporter construct and treated with DHA alone or in combination with EPA at indicated concentrations for 21 hours. C. A2780 cells were transfected with the PGL3/4.5-HO-1 promoter reporter construct and treated with 100 μM DHA or pre-treated with NAC at indicated concentrations and followed with 100 μM DHA for 21 hours. Cell lysates from A, B, C, were prepared, and luciferase activity was assayed. Data are expressed as percentages of the value detected in untreated control cells (bars, SEM; n=3). **, P<0.01, compared with untreated control cells, using one-way ANOVA followed by Dunnett’s analysis.
Table 2.
MDA and DPPP levels in A2780 cells after DHA treatment. A2780 cells were treated with 100 μM of DHA, EPA or ESA for 21 hours in the presence or absence of 200 μM of vitamin E (VitE) or 1 mM NAC. DPPP and TBARS assay were performed as described in Materials and Methods. Data are expressed as a percentage of the levels detected in untreated controls (n=3, SE).
| Groups | Untreated | DHA | VitE | DHA+VitE | EPA |
|---|---|---|---|---|---|
| TBARS | 100±0 | 1305±30a | 51±5 | 454±48b | 730±40b |
| Groups | Untreated | DHA | NAC | DHA+NAC | ESA |
| DPPP | 100±0 | 510±52a | 90±3 | 310±45b | 140±15b |
, p<0.01, compared with untreated control,
, p<0.01, compared with DHA-treated group, using one-way ANOVA followed by Dunnett analysis).
3.3 The involvement of the Nrf2 signaling pathway in mediating the DHA-induced increase in HO-1 expression
There are two potential signaling pathways that are likely involved in mediating the DHA-induced increase of HO-1 gene transcription. The first pathway is the Nrf2 signaling pathway, which is known to regulate HO-1 gene transcription by targeting AREs present in the HO-1 gene promoter in eukaryotic cells [14]. The second is the PPAR signaling pathway, which has been reported to mediate HO-1 gene transcription by targeting the PPRE elements in the HO-1 gene promoter [35, 37]. DHA is known to act as a nuclear ligand [38] to activate PPARα signaling in A2780 cells [39, 40]. To identify the DNA elements in the HO-1 promoter that mediate DHA’s induction of HO-1 gene transcription, we deleted two adjacent ARE elements and two adjacent PPRE elements in the PGL3/4.5-HO-1 promoter reporter construct (Figure 5A) [41]. As shown in Figure 5B, single deletion of the ARE1 (distal ARE) or the ARE2 compromised the DHA-induced increase in HO-1 promoter activity, with the ARE1 deletion being significantly more effective than the ARE2 deletion. Deletion of both ARE elements completely reversed the DHA-induced increase in HO-1 promoter activity (Figure 5C). In contrast, deletion of the PPRE elements, either individually or in combination, did not significantly alter the DHA-induced increase in HO-1 promoter activity (Figure 5B, C); strongly suggesting the involvement of the Nrf2 signaling pathway rather than the PPAR pathway in this event.
Figure 5. Identification of the DNA binding elements that mediate the DHA-induced increase in HO-1 promoter activity.
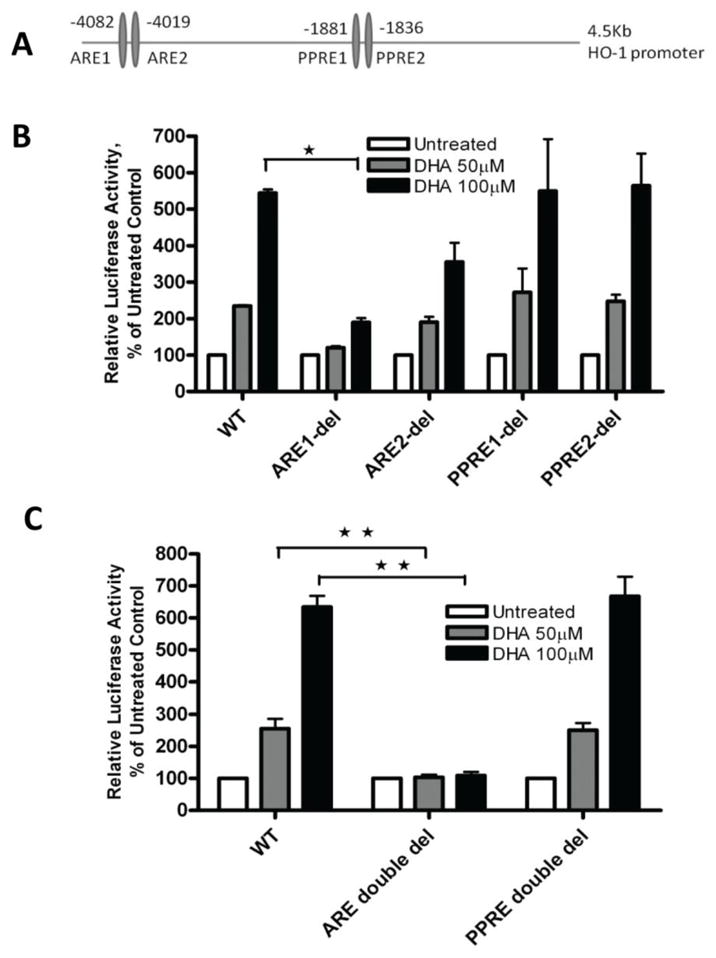
A. A diagram showing the relative locations of the AREs and PPREs present in the human HO-1 gene promoter. B. A2780 cells were transfected with the wild-type (WT) PGL3/4.5-HO-1 promoter reporter construct or a single deletion mutant and treated with DHA at the indicated concentrations for 21 hours at 48 hours post-transfection. Cell lysates were prepared and luciferase activity was assayed. C. A2780 cells were transfected with the wild-type PGL3/4.5-HO-1 promoter reporter construct or a double deletion mutant and treated with DHA at the indicated concentrations for 21 hours. Cell lysates from B and C were prepared and luciferase activity was assayed. Data are expressed as percentages of the value detected in untreated control cells (bar, SEM, n=3). *, P<0.05, **, P<0.01, compared with untreated control cells, using one-way ANOVA followed by Dunnett’s analysis.
To further confirm the involvement of the Nrf2 signaling pathway in the DHA-induced increase in HO-1 gene transcription, a sequence specific siRNA was applied to knockdown either Nrf2 or PPARα expression in A2780 cells (Figure 6A, B). Knockdown of Nrf2 dramatically attenuated the DHA-induced increase of HO-1 promoter activity, whereas knockdown of PPARα had no effect (Figure 6C); further supporting the conclusion that the Nrf2 signaling pathway mediates DHA-induced enhancement of HO-1 gene transcription.
Figure 6. The role of Nrf2 in DHA-induced increase in HO-1 promoter activity.
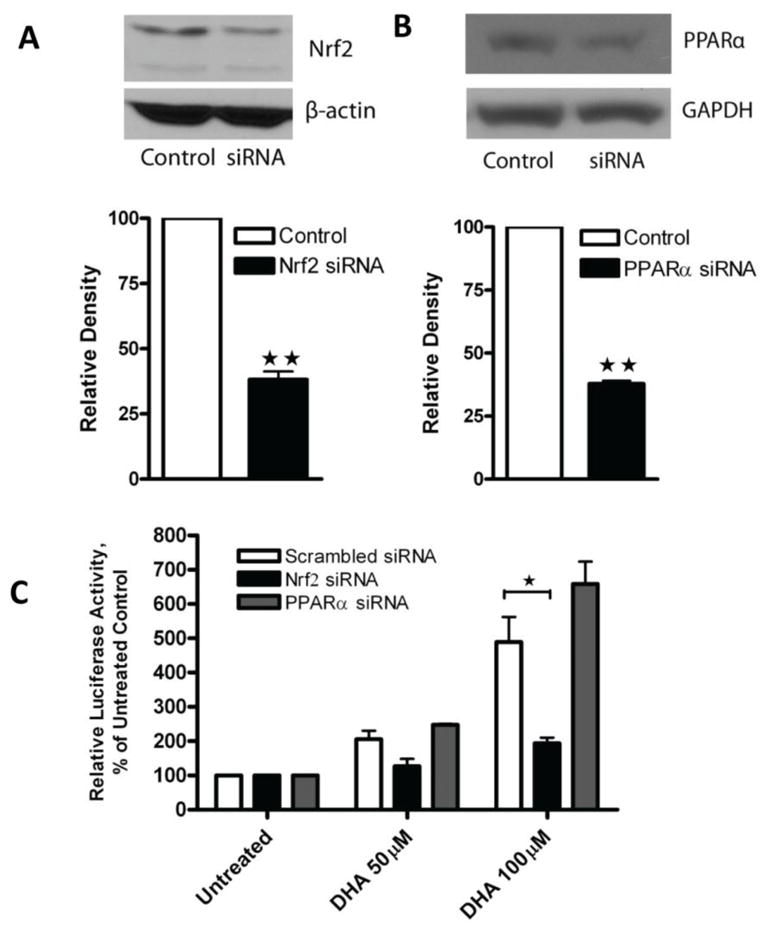
A. A2780 cells were transfected with 225 nM of scrambled siRNA or Nrf2 siRNA. After 48 hours whole cell lysates were prepared and Western blot was performed using antibodies against Nrf2 and β-actin. B. A2780 cells were transfected with 225 nM of scrambled siRNA, or PPARα siRNA. After 48 hours cell lysates were prepared and Western blot was performed using antibodies against PPARα and GAPDH. The relative density of the Nrf2 or PPARα band was quantified, normalized to that of β-actin or GAPDH, and expressed as percentages relative to the expression level in control cells. **, P < 0.01, compared to untreated cells using one-way ANOVA followed by Dunnett’s analysis. C. A2780 cells were co-transfected with the PGL3/4.5-HO-1 promoter reporter construct and scrambled siRNA, Nrf2 siRNA, or PPARα siRNA. Forty-eight hours after transfection, cells were treated with DHA at the indicated concentrations for 21 hours. Cell lysates were prepared and luciferase activity was assayed. Data are expressed as percentages of the value detected in untreated control cells (bar, SEM, n=3). *, P<0.05, compared to untreated control cells, using one-way ANOVA followed by Dunnett’s analysis.
3.4 DHA induces Bach1 degradation thereby enhancing the Nrf2-mediated HO-1 gene transcription
In order to obtain direct evidence of Nrf2 activation by DHA, we analyzed Nrf2 expression after DHA treatment in A2780 cells. To our surprise, treatment with 100 μM DHA for 6 or 21 hours (Figure 7A) (or 1–4 hours, data not shown) did not enhance nuclear Nrf2 expression levels in A2780 cells. On the other hand, treatment of the EA.hy926 cell line with 100 μM DHA for various times dramatically enhanced Nrf2 nuclear protein expression (Figure 7C), consistent with a previous report [42]. DHA-induced Nrf2 nuclear translocation and HO-1 gene transcription was also reported in human umbilical vein endothelial cells (HUVEC) [43].
Figure 7. Effects of DHA on Nrf2 nuclear protein expression and DNA binding.

A. A2780 cells were treated with 100 μM DHA for 6 or 21 hours. Cell lysates were prepared, nuclear proteins were extracted and Western blot was performed using antibodies against Nrf2, Bach1 and β-actin. B. A2780 cells were treated with 100 μM DHA for 6 or 21 hours. ChIP assay was performed as described in Material and Methods section. The amplified DNA fragments were separated on 1% agarose gel and visualized under ultraviolet light. Shown are representative gel images of 3 separate experiments. C. EA.hy926 cells were treated with 100 μM DHA for 0.5–3 hours. Nuclear proteins were extracted and Western blot was performed using antibodies against Nrf2, and β-actin. Shown are representative images of 2 separate experiments. The relative density of each detected bands in A, B, C were quantified, normalized to that of GAPDH, and expressed as percentages relative to the expression level in untreated control (A, B) or the highest expression level in DHA-treated cells (C). *, P<0.05, **, P<0.01, compared to untreated cells using one-way ANOVA followed by Dunnett’s analysis.
To further understand how Nrf2 mediates the DHA-induced increase of HO-1 gene transcription, ChIP analysis was applied to examine the DNA binding activity of Nrf2 in cells that had been treated with DHA. ChIP for Bach1 binding activity was also included because Bach1 is known to bind to AREs and may block Nrf2 binding activity [44, 45]. PPARα binding to PPRE analyzed by ChIP was included as a control. Interestingly, Nrf2 binding to the ARE1 was significantly enhanced after treatment with 100 μM DHA for 6–21 hours, and its binding to the ARE2 was also significantly elevated at 6 hours of DHA treatment (Figure 7B). In contrast, Bach1 binding to the ARE1 was significantly reduced after 6–21 hours of DHA treatment, consistent with the observation that nuclear Bach1 protein expression was reduced by DHA (Figure 7A). As expected, PPARα binding to the PPRE remains unchanged (Figure 7B). These results suggest that DHA enhances Nrf2 binding to the AREs by attenuating Bach1 protein expression and its DNA binding activity at the ARE sites. It is well known that Bach1 is degraded via the ubiquitin-proteasome pathway upon certain stimulation [14, 44, 46]. To determine if this is occurring upon DHA stimulation in our model system, we pre-treated A2780 cells with the proteasome inhibitors MG132 [47] or Bortezomib [48] to block protein degradation prior to DHA treatment. Figure 8A shows that the induction of HO-1 promoter activity by DHA is largely diminished by pre-treatment of the cells with proteasome inhibitors; suggesting that blocking the degradation of Bach1 reverses the DHA-induced increase in HO-1 gene transcription. Western blot analysis shows that pre-treatment of the cells with MG132 reversed DHA-induced degradation of Bach1 protein in A2780 cells (Figure 8B). The induction of HO-1 expression and the suppression of Bach1 expression was also evident in the breast cancer cell line MCF7 (Figure 9), suggesting that these DHA effects are not cell line dependent.
Figure 8. Effects of proteasome inhibitors on the DHA-induced increase in HO-1 promoter activity.

A. A2780 cells were transfected with the PGL3/4.5-HO-1 promoter reporter construct. Forty-eight hours post-transfection the cells were pre-treated with 10 μM MG132 or 1 μM bortezomib for 15 minutes followed by treatment with 100 μM DHA for 21 hours. Cell lysates were prepared and luciferase activity was assayed. Data are expressed as percentages of the value detected in untreated control cells (bar, SEM, n=3). **, P<0.01, compared to untreated control cells, using one-way ANOVA followed by Dunnett’s analysis. B. A2780 cells were treated with 10 μM MG132, 100 μM DHA, or pre-treated with 10 μM MG132 for 15 minutes followed by 100 μM DHA for 21 hours. Cell lysates were prepared and Western blot was performed using antibodies against Bach1 and β-actin. Shown are representative images of 3 separate experiments. The bands were quantified, normalized to β-actin and is expressed as percentages relative to the expression level in untreated controls. *, P<0.05, **, p<0.01, using one-way ANOVA followed by Dunnett’s analysis.
Figure 9. Effects of DHA on the expression of HO-1 and Bach1 in MCF7 cells.
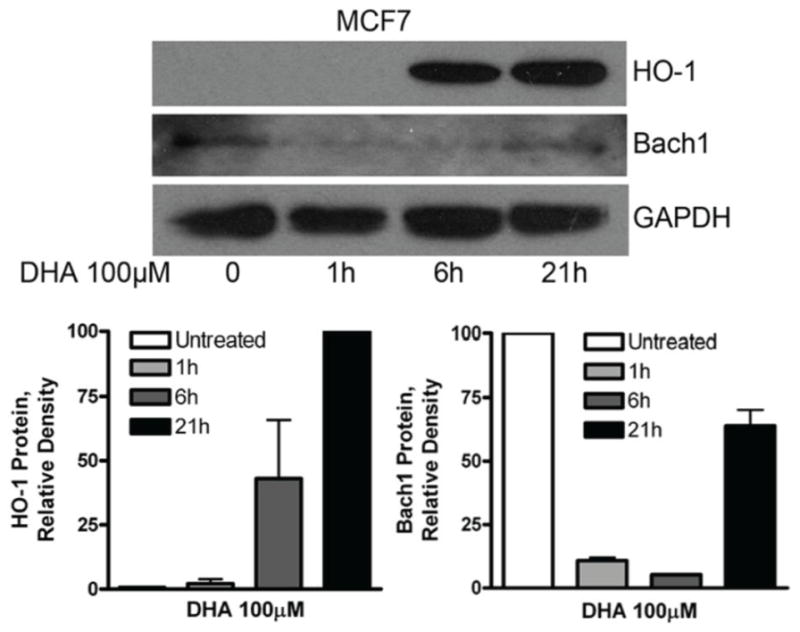
MCF7 cells were treated with 100 μM DHA for 6 or 21 hours. Cell lysates were prepared and Western blot was performed using antibodies against HO-1, Bach1 and GAPDH. Shown are representative images of two separate experiments. The bands were quantified, normalized to GAPDH and is expressed as percentages relative to the highest expression level detected.
4. DISCUSSION
In the present study, we have characterized the effects of DHA on HO-1 expression in a human cancer cell model system. Although HO-1 expression can be induced by many stimuli [14], to our knowledge, this is the first characterization of DHA-induced HO-1 expression in human malignant cells. The most interesting finding is that DHA enhances HO-1 gene transcription in cancer cells by promoting Bach1 degradation that results in enhanced binding of Nrf2 to the AREs present in the HO-1 gene promoter. In addition, we found that among the fatty acids tested, DHA is the most effective inducer of HO-1 expression in cancer cells. These results further support our previous claims that cellular antioxidant enzymes are responsive to DHA and could potentially mediate DHA’s anticancer activity [32, 39, 49].
The concentration of DHA used in the present study has been frequently used in vitro in cancer cell model systems [2, 39, 50–54] and the fish oil diet tested was also similar to previous reports [2, 25–29]. While the exact amount of long chain n-3 PUFAs in the plasma or tissues of the mice on fish oil diet may differ among different studies, there is a significant increase in DHA and EPA contents in the plasma and tissues compared with that of mice on standard diets [2, 29]. The plasma DHA contents could reach as high as nearly 400 μM in mice on a 5% fish oil diet for 6 weeks [2], and similar plasma and tumor tissue DHA concentrations were obtained when DHA was delivered to mice by gavage at 0.5 g/kg daily for 12 days [55]. In humans, to our knowledge, the highest orally tolerated dose of EPA plus DHA is 188 mg/kg/day [56], which is considerably higher than the usual adult intake, and there is no data available to indicate what concentrations of DHA are achieved in the plasma and tissues in this study. However a previous report indicates that daily consumption of 4.4g EPA plus DHA over 6 weeks resulted in a plasma DHA concentration at 51 g/mL (155 M) in humans, which is three times higher than the basal level [57]. Furthermore, it has been well described that plasma concentrations of n-3 PUFA reflect relative dietary n-3 PUFA intake in humans [58–60]. These previous reports suggest that the concentrations of DHA used in our study are physiologically relevant and achievable in vivo, and that the fish oil diet used is appropriate for the intended investigation.
DHA has been reported to induce HO-1 gene expression in human endothelial cells [42], mouse macrophages [17], mouse glial cells [16], and human cell lines [61–63]; although the mechanisms of induction seem to differ among the various model systems investigated. To understand how DHA induces HO-1 expression in our model system, we focused on the Nrf2 and PPAR signaling pathways. This was based on several considerations: First, DHA is known to initiate lipid peroxidation thereby enhancing cellular oxidative stress, which could potentially activate the Nrf2 pathway leading to up-regulation of HO-1 gene expression [13]. Second, DHA is an established PPAR ligand which can activate the PPAR signaling pathway thereby regulating expression of many target genes [64]. We have previously shown that DHA activates PPARα signaling in A2780 cells and PPARα mediates the suppression of superoxide dismutase 1 expression [39] and the cytotoxicity of DHA [40]. Third, there are well-established AREs and PPREs in the human HO-1 gene promoter and both DNA elements were previously described to regulate HO-1 gene transcription [14, 35, 37]. Our results clearly indicate that it is the Nrf2 signaling pathway, but not the PPAR signaling pathway, that mediates the DHA-induced increase in HO-1 gene transcription. The fact that deletion of both AREs completely reversed the DHA-induced increase in HO-1 gene transcription indicates that the Nfr2 pathway is solely responsible for this up-regulation. However, since vitamin E and NAC could not completely reverse DHA-induced lipid peroxidation or HO-1 gene promoter activity, and EPA, another long chain n-3 PUFA, did not significantly induce HO-1 gene transcription, other signaling pathways coupled by DHA may activate the Nrf2 pathway independent of oxidative stress. In this context, recent studies have shown that DHA targets autophagy [8], ER stress [9], as well as other cellular survival pathways in cancer cells [6, 7], and that the Nrf2 pathway can be activated by oxidative stress-independent stimuli [65, 66].
The Nrf2 protein is normally bound by keap1, which brings Nrf2 to the proteasome for degradation. When cells are subjected to various stresses, Nrf2 dissociates from keap1 and translocates to the nucleus where it binds to the AREs of various gene promoters and activates gene transcription [13, 14]. While Nrf2 is identified as an enhancer of HO-1 gene transcription, Bach1 is known to be a transcriptional repressor that may bind to the same DNA elements as that of Nrf2 [44, 45]. The balance of Nrf2 and Bach1 and their dynamic exchange in expression levels work in concert to regulate the expression of the HO-1 gene [44]. The results from the present study demonstrate that DHA treatment enhances HO-1 gene transcription by attenuating Bach1 protein expression in our model system, further supporting the concept that the relative expression levels of Nrf2 and Bach1 are critical in controlling HO-1 gene transcription. While knockdown of Nrf2, but not PPARα, attenuated the DHA-induced increase of HO-1 gene transcription and deletion of the ARE sites completely reversed the DHA-induced up-regulation of HO-1 promoter activity, DHA does not enhance Nrf2 protein expression levels. These interesting findings led us to focus on the binding of Nrf2 and Bach1 at the ARE sites in the HO-1 gene promoter. Whereas the nuclear protein level of Nrf2 was unchanged upon DHA treatment, Nrf2 binding to the AREs was significantly increased. In contrast, the nuclear protein level of Bach1 was significantly decreased, as was the binding of Bach1 to the AREs after DHA treatment. The use of proteasome inhibitors provided evidence indicating that DHA enhances Bach1 protein degradation thereby inducing Nrf2-mediated HO-1 gene transcription. These results along with previous reports [44, 45, 67] strongly support the conclusion that DHA enhances HO-1 gene transcription by promoting Bach1 degradation. Similar to our findings, a previous report described the induction of HO-1 gene transcription in human liver cells by cobalt protoporphyrin through suppression of Bach1 protein expression [67]. In a more general sense, these observations suggest that although HO-1 expression can be induced by many stimuli, the induction may be mediated through different mechanisms depending on the nature of the stress. Since there are limited reports available on Bach1 regulation of HO-1 induction in cancer cells, the details of how DHA promotes Bach1 degradation in our cancer cell model system and whether Bach1 mediates differential regulation of HO-1 expression in cancer cells versus normal cells merits further investigation.
DHA has been found to have anticancer activity in both in vitro and in vivo model systems [1, 68, 69], but the mechanisms of its action remain elusive. Lipid peroxidation initiated by DHA has been described as one of the mechanisms accounting for DHA’s cytotoxicity [52, 70–72]. If so, cellular antioxidant enzymes may play a critical role in DHA-induced cytotoxicity. We have previously demonstrated that DHA down-regulates expression of the SOD1 and GPx4 gene in a number of human cancer cell lines [49], leading to enhanced lipid peroxidation and cytotoxicity [32]. The results from the present study indicate that the antioxidant enzymes in cancer cells respond to DHA treatment in a more diverse manner, meaning that expression of some enzymes may be up-regulated and others down-regulated. The complex interplay of antioxidant enzymes after DHA treatment remains to be elucidated; however the up-regulation of HO-1 by DHA in cancer cells may provide therapeutic implications. While induction of HO-1 in normal cells is cytoprotective and anti-inflammatory [73, 74], expression of HO-1 in cancer cells may support malignant growth [75] and render cancer cells more resistant to chemotherapy [21]. In fact, HO-1 has been proposed as a potential cellular target for cancer therapy [22] and HO-1 inhibitors such as zinc protoporphyrin have been reported to suppress cancer growth in vitro and in vivo [76–78]. Therefore, the combination of DHA and HO-1 inhibitors could be an attractive approach to more effectively suppress cancer growth and future work to investigate this potential combinatorial approach is justified.
In summary, we have demonstrated in the present study that DHA induces HO-1 gene transcription in human cancer cells through the Nrf2 signaling pathway, targeting the ARE elements localized in the HO-1 gene promoter. Our results indicate that the Bach1 degradation is critical for this event, and DHA-initiated lipid peroxidation contributes to this induction. Given the cytoprotective role of HO-1 in eukaryotic cells, these findings suggest that the combination of DHA and HO-1 inhibitors could be an attractive approach to more effectively suppress cancer growth.
Acknowledgments
This work was supported in part by grants from the American Cancer Society (CNE-117557); the Susan G. Komen for the Cure Foundation (KG081083); the NIH OK-INBRE program (3P20RR016478-09S2); and the Oklahoma Center for the Advancement of Science and Technology (HR09-025).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jiang WG, Bryce RP, Horrobin DF. Essential fatty acids: molecular and cellular basis of their anti-cancer action and clinical implications. Crit Rev Oncol Hematol. 1998;27:179–209. doi: 10.1016/s1040-8428(98)00003-1. [DOI] [PubMed] [Google Scholar]
- 2.Kang KS, Wang P, Yamabe N, Fukui M, Jay T, Zhu BT. Docosahexaenoic acid induces apoptosis in MCF-7 cells in vitro and in vivo via reactive oxygen species formation and caspase 8 activation. PloS one. 2010;5:e10296. doi: 10.1371/journal.pone.0010296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardman WE, Barnes CJ, Knight CW, Cameron IL. Effects of iron supplementation and ET-18-OCH3 on MDA-MB 231 breast carcinomas in nude mice consuming a fish oil diet. Br J Cancer. 1997;76:347–54. doi: 10.1038/bjc.1997.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Begin ME, Ells G, Das UN, Horrobin DF. Differential killing of human carcinoma cells supplemented with n-3 and n-6 polyunsaturated fatty acids. J Natl Cancer Inst. 1986;77:1053–62. [PubMed] [Google Scholar]
- 5.Karmali RA, Marsh J, Fuchs C. Effect of omega-3 fatty acids on growth of a rat mammary tumor. J Natl Cancer Inst. 1984;73:457–61. doi: 10.1093/jnci/73.2.457. [DOI] [PubMed] [Google Scholar]
- 6.Gleissman H, Johnsen JI, Kogner P. Omega-3 fatty acids in cancer, the protectors of good and the killers of evil? Exp Cell Res. 2010;316:1365–73. doi: 10.1016/j.yexcr.2010.02.039. [DOI] [PubMed] [Google Scholar]
- 7.Stephenson JA, Al-Taan O, Arshad A, Morgan B, Metcalfe MS, Dennison AR. The multifaceted effects of omega-3 polyunsaturated Fatty acids on the hallmarks of cancer. Journal of lipids. 2013;2013:261247. doi: 10.1155/2013/261247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jing K, Song KS, Shin S, Kim N, Jeong S, Oh HR, et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy. 2011;7:1348–58. doi: 10.4161/auto.7.11.16658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakobsen CH, Storvold GL, Bremseth H, Follestad T, Sand K, Mack M, et al. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. J Lipid Res. 2008;49:2089–100. doi: 10.1194/jlr.M700389-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardman WE, Munoz J, Jr, Cameron IL. Role of lipid peroxidation and antioxidant enzymes in omega 3 fatty acids induced suppression of breast cancer xenograft growth in mice. Cancer Cell Int. 2002;2:10. doi: 10.1186/1475-2867-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez MJ. Fish oil, lipid peroxidation and mammary tumor growth. J Am Coll Nutr. 1995;14:325–35. doi: 10.1080/07315724.1995.10718517. [DOI] [PubMed] [Google Scholar]
- 12.Begin ME, Ells G, Horrobin DF. Polyunsaturated fatty acid-induced cytotoxicity against tumor cells and its relationship to lipid peroxidation. J Natl Cancer Inst. 1988;80:188–94. doi: 10.1093/jnci/80.3.188. [DOI] [PubMed] [Google Scholar]
- 13.Srisook K, Kim C, Cha YN. Molecular mechanisms involved in enhancing HO-1 expression: de-repression by heme and activation by Nrf2, the “one-two” punch. Antioxid Redox Signal. 2005;7:1674–87. doi: 10.1089/ars.2005.7.1674. [DOI] [PubMed] [Google Scholar]
- 14.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annual review of pharmacology and toxicology. 2010;50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 15.Wright MM, Schopfer FJ, Baker PR, Vidyasagar V, Powell P, Chumley P, et al. Fatty acid transduction of nitric oxide signaling: nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proc Natl Acad Sci U S A. 2006;103:4299–304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu DY, Tsao YY, Leung YM, Su KP. Docosahexaenoic acid suppresses neuroinflammatory responses and induces heme oxygenase-1 expression in BV-2 microglia: implications of antidepressant effects for omega-3 fatty acids. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2010;35:2238–48. doi: 10.1038/npp.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Khor TO, Saw CL, Lin W, Wu T, Huang Y, et al. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Molecular pharmaceutics. 2010;7:2185–93. doi: 10.1021/mp100199m. [DOI] [PubMed] [Google Scholar]
- 18.Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast cancer research : BCR. 2001;3:323–7. doi: 10.1186/bcr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 20.Schor NF, Kagan VE, Liang Y, Yan C, Tyurina Y, Tyurin V, et al. Exploiting oxidative stress and signaling in chemotherapy of resistant neoplasms. Biochemistry Biokhimiia. 2004;69:38–44. doi: 10.1023/b:biry.0000016349.75384.e6. [DOI] [PubMed] [Google Scholar]
- 21.Nuhn P, Kunzli BM, Hennig R, Mitkus T, Ramanauskas T, Nobiling R, et al. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol Cancer. 2009;8:37. doi: 10.1186/1476-4598-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gleixner KV, Mayerhofer M, Vales A, Gruze A, Hormann G, Cerny-Reiterer S, et al. Targeting of Hsp32 in solid tumors and leukemias: a novel approach to optimize anticancer therapy. Curr Cancer Drug Targets. 2009;9:675–89. doi: 10.2174/156800909789057024. [DOI] [PubMed] [Google Scholar]
- 23.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, et al. Radiation survival parameters of antineoplastic drug-sensitive and -resistant human ovarian cancer cell lines and their modification by buthionine sulfoximine. Cancer Res. 1985;45:2110–5. [PubMed] [Google Scholar]
- 24.van Oost BA, Edgell CJ, Hay CW, MacGillivray RT. Isolation of a human von Willebrand factor cDNA from the hybrid endothelial cell line EA.hy926. Biochemistry and cell biology = Biochimie et biologie cellulaire. 1986;64:699–705. doi: 10.1139/o86-096. [DOI] [PubMed] [Google Scholar]
- 25.Boudreau MD, Sohn KH, Rhee SH, Lee SW, Hunt JD, Hwang DH. Suppression of tumor cell growth both in nude mice and in culture by n-3 polyunsaturated fatty acids: mediation through cyclooxygenase-independent pathways. Cancer Res. 2001;61:1386–91. [PubMed] [Google Scholar]
- 26.Reddy BS, Maruyama H. Effect of dietary fish oil on azoxymethane-induced colon carcinogenesis in male F344 rats. Cancer Res. 1986;46:3367–70. [PubMed] [Google Scholar]
- 27.Rose DP, Connolly JM. Effects of dietary omega-3 fatty acids on human breast cancer growth and metastases in nude mice. J Natl Cancer Inst. 1993;85:1743–7. doi: 10.1093/jnci/85.21.1743. [DOI] [PubMed] [Google Scholar]
- 28.Kelavkar UP, Hutzley J, McHugh K, Allen KG, Parwani A. Prostate tumor growth can be modulated by dietarily targeting the 15-lipoxygenase-1 and cyclooxygenase-2 enzymes. Neoplasia. 2009;11:692–9. doi: 10.1593/neo.09334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akinsete JA, Ion G, Witte TR, Hardman WE. Consumption of high omega-3 fatty acid diet suppressed prostate tumorigenesis in C3(1) Tag mice. Carcinogenesis. 2012;33:140–8. doi: 10.1093/carcin/bgr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding WQ, Liu B, Vaught JL, Yamauchi H, Lind SE. Anticancer activity of the antibiotic clioquinol. Cancer Res. 2005;65:3389–95. doi: 10.1158/0008-5472.CAN-04-3577. [DOI] [PubMed] [Google Scholar]
- 31.Zhou J, Zhang S, Xue J, Avery J, Wu J, Lind SE, et al. Activation of Peroxisome Proliferator-activated Receptor alpha (PPARalpha) Suppresses Hypoxia-inducible Factor-1alpha (HIF-1alpha) Signaling in Cancer Cells. J Biol Chem. 2012;287:35161–9. doi: 10.1074/jbc.M112.367367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding WQ, Vaught JL, Yamauchi H, Lind SE. Differential sensitivity of cancer cells to docosahexaenoic acid-induced cytotoxicity: the potential importance of down-regulation of superoxide dismutase 1 expression. Mol Cancer Ther. 2004;3:1109–17. [PubMed] [Google Scholar]
- 33.Rushworth SA, Chen XL, Mackman N, Ogborne RM, O’Connell MA. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J Immunol. 2005;175:4408–15. doi: 10.4049/jimmunol.175.7.4408. [DOI] [PubMed] [Google Scholar]
- 34.Rushworth SA, MacEwan DJ. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood. 2008;111:3793–801. doi: 10.1182/blood-2007-07-104042. [DOI] [PubMed] [Google Scholar]
- 35.Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, et al. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007;27:1276–82. doi: 10.1161/ATVBAHA.107.142638. [DOI] [PubMed] [Google Scholar]
- 36.Xue J, Wang S, Wu J, Hannafon BN, Ding WQ. Zinc at sub-cytotoxic concentrations induces heme oxygenase-1 expression in human cancer cells. Cell Physiol Biochem. 2013;32:100–10. doi: 10.1159/000350128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen HH, Chen TW, Lin H. Pravastatin attenuates carboplatin-induced nephrotoxicity in rodents via peroxisome proliferator-activated receptor alpha-regulated heme oxygenase-1. Mol Pharmacol. 2010;78:36–45. doi: 10.1124/mol.109.061101. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Starodub O, McIntosh A, Kier AB, Schroeder F. Liver fatty acid-binding protein targets fatty acids to the nucleus. Real time confocal and multiphoton fluorescence imaging in living cells. J Biol Chem. 2002;277:29139–51. doi: 10.1074/jbc.M202923200. [DOI] [PubMed] [Google Scholar]
- 39.Tuller ER, Beavers CT, Lou JR, Ihnat MA, Benbrook DM, Ding WQ. Docosahexaenoic acid inhibits superoxide dismutase 1 gene transcription in human cancer cells: the involvement of peroxisome proliferator-activated receptor alpha and hypoxia-inducible factor-2alpha signaling. Mol Pharmacol. 2009;76:588–95. doi: 10.1124/mol.109.057430. [DOI] [PubMed] [Google Scholar]
- 40.Tuller ER, Brock AL, Yu H, Lou JR, Benbrook DM, Ding WQ. PPARalpha signaling mediates the synergistic cytotoxicity of clioquinol and docosahexaenoic acid in human cancer cells. Biochem Pharmacol. 2009;77:1480–6. doi: 10.1016/j.bcp.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Wright MM, Kim J, Hock TD, Leitinger N, Freeman BA, Agarwal A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem J. 2009;422:353–61. doi: 10.1042/BJ20090339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang YC, Lii CK, Wei YL, Li CC, Lu CY, Liu KL, et al. Docosahexaenoic acid inhibition of inflammation is partially via cross-talk between Nrf2/heme oxygenase 1 and IKK/NF-kappaB pathways. The Journal of nutritional biochemistry. 2012 doi: 10.1016/j.jnutbio.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 43.Ishikado A, Morino K, Nishio Y, Nakagawa F, Mukose A, Sono Y, et al. 4-Hydroxy hexenal derived from docosahexaenoic acid protects endothelial cells via Nrf2 activation. PloS one. 2013;8:e69415. doi: 10.1371/journal.pone.0069415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. Embo J. 2002;21:5216–24. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raval CM, Zhong JL, Mitchell SA, Tyrrell RM. The role of Bach1 in ultraviolet A-mediated human heme oxygenase 1 regulation in human skin fibroblasts. Free Radic Biol Med. 2012;52:227–36. doi: 10.1016/j.freeradbiomed.2011.10.494. [DOI] [PubMed] [Google Scholar]
- 46.Hou W, Shan Y, Zheng J, Lambrecht RW, Donohue SE, Bonkovsky HL. Zinc mesoporphyrin induces rapid and marked degradation of the transcription factor Bach1 and up-regulates HO-1. Biochim Biophys Acta. 2008;1779:195–203. doi: 10.1016/j.bbagrm.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nardinocchi L, Pantisano V, Puca R, Porru M, Aiello A, Grasselli A, et al. Zinc downregulates HIF-1alpha and inhibits its activity in tumor cells in vitro and in vivo. PloS one. 2010;5:e15048. doi: 10.1371/journal.pone.0015048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birle DC, Hedley DW. Suppression of the hypoxia-inducible factor-1 response in cervical carcinoma xenografts by proteasome inhibitors. Cancer Res. 2007;67:1735–43. doi: 10.1158/0008-5472.CAN-06-2722. [DOI] [PubMed] [Google Scholar]
- 49.Ding WQ, Lind SE. Phospholipid hydroperoxide glutathione peroxidase plays a role in protecting cancer cells from docosahexaenoic acid-induced cytotoxicity. Molecular cancer therapeutics. 2007;6:1467–74. doi: 10.1158/1535-7163.MCT-06-0608. [DOI] [PubMed] [Google Scholar]
- 50.Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, et al. The role of docosahexaenoic acid in mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis. 2005;26:1914–21. doi: 10.1093/carcin/bgi163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merendino N, Loppi B, D’Aquino M, Molinari R, Pessina G, Romano C, et al. Docosahexaenoic acid induces apoptosis in the human PaCa-44 pancreatic cancer cell line by active reduced glutathione extrusion and lipid peroxidation. Nutr Cancer. 2005;52:225–33. doi: 10.1207/s15327914nc5202_12. [DOI] [PubMed] [Google Scholar]
- 52.Leonardi F, Attorri L, Di Benedetto R, Di Biase A, Sanchez M, Nardini M, et al. Effect of arachidonic, eicosapentaenoic and docosahexaenoic acids on the oxidative status of C6 glioma cells. Free Radic Res. 2005;39:865–74. doi: 10.1080/10715760500145069. [DOI] [PubMed] [Google Scholar]
- 53.Collett ED, Davidson LA, Fan YY, Lupton JR, Chapkin RS. n-6 and n-3 polyunsaturated fatty acids differentially modulate oncogenic Ras activation in colonocytes. Am J Physiol Cell Physiol. 2001;280:C1066–75. doi: 10.1152/ajpcell.2001.280.5.C1066. [DOI] [PubMed] [Google Scholar]
- 54.Xue M, Wang Q, Zhao J, Dong L, Ge Y, Hou L, et al. Docosahexaenoic acid inhibited the Wnt/beta-Catenin pathway and suppressed breast cancer cells in vitro and in vivo. J Nutr Biochem. 2013 doi: 10.1016/j.jnutbio.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 55.Gleissman H, Segerstrom L, Hamberg M, Ponthan F, Lindskog M, Johnsen JI, et al. Omega-3 fatty acid supplementation delays the progression of neuroblastoma in vivo. Int J Cancer. 2011;128:1703–11. doi: 10.1002/ijc.25473. [DOI] [PubMed] [Google Scholar]
- 56.Burns CP, Halabi S, Clamon GH, Hars V, Wagner BA, Hohl RJ, et al. Phase I clinical study of fish oil fatty acid capsules for patients with cancer cachexia: cancer and leukemia group B study 9473. Clin Cancer Res. 1999;5:3942–7. [PubMed] [Google Scholar]
- 57.Garg ML, Leitch J, Blake RJ, Garg R. Long-chain n-3 polyunsaturated fatty acid incorporation into human atrium following fish oil supplementation. Lipids. 2006;41:1127–32. doi: 10.1007/s11745-006-5062-1. [DOI] [PubMed] [Google Scholar]
- 58.Kuriki K, Nagaya T, Tokudome Y, Imaeda N, Fujiwara N, Sato J, et al. Plasma concentrations of (n-3) highly unsaturated fatty acids are good biomarkers of relative dietary fatty acid intakes: a cross-sectional study. J Nutr. 2003;133:3643–50. doi: 10.1093/jn/133.11.3643. [DOI] [PubMed] [Google Scholar]
- 59.Thiebaut AC, Rotival M, Gauthier E, Lenoir GM, Boutron-Ruault MC, Joulin V, et al. Correlation between serum phospholipid fatty acids and dietary intakes assessed a few years earlier. Nutr Cancer. 2009;61:500–9. doi: 10.1080/01635580802710717. [DOI] [PubMed] [Google Scholar]
- 60.Brasky TM, Darke AK, Song X, Tangen CM, Goodman PJ, Thompson IM, et al. Plasma phospholipid fatty acids and prostate cancer risk in the SELECT trial. J Natl Cancer Inst. 2013;105:1132–41. doi: 10.1093/jnci/djt174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen HW, Chao CY, Lin LL, Lu CY, Liu KL, Lii CK, et al. Inhibition of matrix metalloproteinase-9 expression by docosahexaenoic acid mediated by heme oxygenase 1 in 12-O-tetradecanoylphorbol-13-acetate-induced MCF-7 human breast cancer cells. Archives of toxicology. 2013;87:857–69. doi: 10.1007/s00204-012-1003-3. [DOI] [PubMed] [Google Scholar]
- 62.Saw CL, Yang AY, Guo Y, Kong AN. Astaxanthin and omega-3 fatty acids individually and in combination protect against oxidative stress via the Nrf2-ARE pathway. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2013;62:869–75. doi: 10.1016/j.fct.2013.10.023. [DOI] [PubMed] [Google Scholar]
- 63.Wang S, Avery JE, Hannafon BN, Lind SE, Ding WQ. Zinc protoporphyrin suppresses cancer cell viability through a heme oxygenase-1-independent mechanism: the involvement of the Wnt/beta-catenin signaling pathway. Biochem Pharmacol. 2013 doi: 10.1016/j.bcp.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 64.Das UN. Essential Fatty acids - a review. Current pharmaceutical biotechnology. 2006;7:467–82. doi: 10.2174/138920106779116856. [DOI] [PubMed] [Google Scholar]
- 65.Merchant AA, Singh A, Matsui W, Biswal S. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood. 2011;118:6572–9. doi: 10.1182/blood-2011-05-355362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JM, Moehlenkamp JD, Hanson JM, Johnson JA. Nrf2-dependent activation of the antioxidant responsive element by tert-butylhydroquinone is independent of oxidative stress in IMR-32 human neuroblastoma cells. Biochem Biophys Res Commun. 2001;280:286–92. doi: 10.1006/bbrc.2000.4106. [DOI] [PubMed] [Google Scholar]
- 67.Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J. 2006;20:2651–3. doi: 10.1096/fj.06-6346fje. [DOI] [PubMed] [Google Scholar]
- 68.Bougnoux P. n-3 polyunsaturated fatty acids and cancer. Curr Opin Clin Nutr Metab Care. 1999;2:121–6. doi: 10.1097/00075197-199903000-00005. [DOI] [PubMed] [Google Scholar]
- 69.Hardman WE. (n-3) fatty acids and cancer therapy. J Nutr. 2004;134:3427S–30S. doi: 10.1093/jn/134.12.3427S. [DOI] [PubMed] [Google Scholar]
- 70.Maheo K, Vibet S, Steghens JP, Dartigeas C, Lehman M, Bougnoux P, et al. Differential sensitization of cancer cells to doxorubicin by DHA: a role for lipoperoxidation. Free Radic Biol Med. 2005;39:742–51. doi: 10.1016/j.freeradbiomed.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 71.Siddiqui RA, Harvey K, Stillwell W. Anticancer properties of oxidation products of docosahexaenoic acid. Chem Phys Lipids. 2008;153:47–56. doi: 10.1016/j.chemphyslip.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 72.Siddiqui RA, Harvey KA, Xu Z, Bammerlin EM, Walker C, Altenburg JD. Docosahexaenoic acid: a natural powerful adjuvant that improves efficacy for anticancer treatment with no adverse effects. Biofactors. 2011;37:399–412. doi: 10.1002/biof.181. [DOI] [PubMed] [Google Scholar]
- 73.Barbagallo I, Galvano F, Frigiola A, Cappello F, Riccioni G, Murabito P, et al. Potential therapeutic effects of natural heme oxygenase-1 inducers in cardiovascular diseases. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4360. [DOI] [PubMed] [Google Scholar]
- 74.Durante W. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Frontiers in bioscience : a journal and virtual library. 2011;16:2372–88. doi: 10.2741/3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Was H, Dulak J, Jozkowicz A. Heme oxygenase-1 in tumor biology and therapy. Current drug targets. 2010;11:1551–70. doi: 10.2174/1389450111009011551. [DOI] [PubMed] [Google Scholar]
- 76.Fang J, Sawa T, Akaike T, Akuta T, Sahoo SK, Khaled G, et al. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003;63:3567–74. [PubMed] [Google Scholar]
- 77.Hirai K, Sasahira T, Ohmori H, Fujii K, Kuniyasu H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int J Cancer. 2007;120:500–5. doi: 10.1002/ijc.22287. [DOI] [PubMed] [Google Scholar]
- 78.La P, Fernando AP, Wang Z, Salahudeen A, Yang G, Lin Q, et al. Zinc protoporphyrin regulates cyclin D1 expression independent of heme oxygenase inhibition. J Biol Chem. 2009;284:36302–11. doi: 10.1074/jbc.M109.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]