

Clustered, regularly interspaced, short palindromic repeat (CRISPR) RNA-guided nucleases (RGNs) are highly efficient genome editing tools.1–3 CRISPR-associated 9 (Cas9) RGNs are directed to genomic loci by guide RNAs (gRNAs) containing 20 nucleotides that are complementary to a target DNA sequence. However, RGNs can induce mutations at sites that differ by as many as five nucleotides from the intended target4–6. Here we report that truncated gRNAs, with shorter regions of target complementarity <20 nucleotides in length, can decrease undesired mutagenesis at off-target sites by as much as 5000-fold or more without sacrificing on-target genome editing efficiencies. In addition, combining truncated gRNAs with pairs of Cas9 variants that nick DNA (paired nickases) can lead to further reductions in off-target mutations. Our results delineate a simple, effective strategy to improve the specificities of Cas9 nucleases or paired nickases.
The Streptococcus pyogenes Cas9 nuclease (hereafter referred to as Cas9) can robustly induce insertion or deletion mutations (indels) or precise alterations via repair of Cas9-induced double-stranded breaks (DSBs) by non-homologous end-joining (NHEJ) or homology-directed repair (HDR), respectively. However, unwanted indel mutations can also be induced at off-target sites sharing sequence similarity to the on-target site4–6. Recently, several approaches to improve the specificity of RNA-guided Cas9 have been recently described including truncation of the 3' end of gRNA (which is derived from the tracrRNA domain that is believed to mediate interaction with Cas9) or addition of two G nucleotides to the 5' end of the gRNA (just before the 20 nt complementarity region); however, RGNs utilizing these altered gRNAs show decreased on-target activities6, 7. Alternatively, a “paired nicking” strategy (originally implemented with pairs of closely spaced zinc finger nickases8), in which two gRNAs targeted to adjacent sites on opposite DNA strands each recruit a Cas9 variant (Cas9-D10A) that nicks DNA instead of cutting both strands7, 9, 10, can reduce mutation frequencies at known off-target sites of single gRNA-guided Cas9 nuclease in human cells7, 10. Nevertheless, indel mutations can still be observed at some off-target sites10 and the addition of a second gRNA might introduce new off-target mutations because a single gRNA-directed Cas9 nickase can efficiently induce indels at some sites7, 9, 11. In addition, the need to express appropriately positioned and oriented paired gRNAs presents technical challenges if implemented with multiplex12–14 or genome-wide library-based applications of RGNs15, 16. Finally, the paired nickase strategy cannot be used to improve the specificities of catalytically inactive Cas9 (dCas9) fused to heterologous effectors, such as transcriptional activation domains17–19. Thus, the development of additional methods to improve the specificity of CRISPR-based systems remains an important priority.
We hypothesized that off-target effects of RGNs might be minimized by decreasing the length of the gRNA-DNA interface. Such an approach might seem counterintuitive, but we20 and others7, 10 have shown that lengthening the 5' end of the complementary region can reduce on-target editing efficiency, with some of these longer gRNAs processed back to standard length in human cells10. In contrast, certain gRNAs bearing either truncations or progressively greater numbers of mismatches at the 5' end of their complementarity targeting regions have been shown to retain robust Cas9-mediated on-target cleavage activities4, 9, 21. We hypothesized that these 5'-end nucleotides might not be necessary for full gRNA activity and that these nucleotides might normally compensate for mismatches at other positions along the gRNA-target DNA interface; therefore, we reasoned that shorter gRNAs might be more sensitive to mismatches and thus more specific (Supplementary Fig. 1).
To test our predictions, we constructed a series of progressively shorter gRNAs for a target site in the EGFP reporter gene containing 15, 17, 19, and 20 complementary nucleotides (Online Methods and Fig. 1a). We measured the abilities of these gRNAs to direct Cas9-induced indels at this target site in human U2OS.EGFP cells by quantifying mutation of a single integrated and constitutively expressed EGFP gene 4, 22 (Online Methods). gRNAs that have 17 or 19 nucleotides of target complementarity showed activities comparable to the full-length gRNA with 20 nucleotides of complementarity, whereas a gRNA containing 15 nucleotides of complementarity failed to show activity (Fig. 1b). To extend the generality of these findings, we assayed full-length gRNAs and matched gRNAs with 18, 17 and/or 16 nucleotides of complementarity to four additional EGFP reporter gene sites (EGFP sites #1, #2, #3 and #4; Fig. 1c). For all four target sites, gRNAs with 17 and/or 18 nucleotides of complementarity functioned as efficiently as (or, in one case, more efficiently than) their matched full-length counterparts (Fig. 1c). gRNAs with only 16 nucleotides of complementarity showed substantially decreased or undetectable activities at the two sites for which they could be made (Fig. 1c). Given these results, in this report we refer to truncated gRNAs with complementarity lengths of 17 or 18 nucleotides as “trugRNAs” and RGNs using these tru-gRNAs as “tru-RGNs”.
Figure 1. On-target genome editing activities of truncated gRNAs and Cas9 nuclease in human cells.
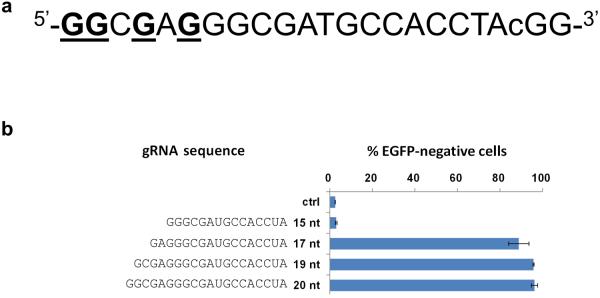



(a) Target site in the EGFP gene used to assess the activities of gRNAs that are truncated at their 5' end. Note that Gs near the 5' end (underlined) enable gRNAs with complementarity lengths of 15, 17, 19 and 20 nt to be expressed from a U6 promoter in human cells. The degenerate base of the protospacer adjacent motif (PAM) sequence (in this case, a C nucleotide) is shown in lowercase.
(b) Efficiencies of EGFP disruption in human cells mediated by Cas9 and gRNAs bearing variable length complementarity regions (shown in nts) for the target site of (a). Ctrl = control gRNA lacking a complementarity region. Error bars indicate standard errors of the mean (s.e.m.), n = 2.
(c) Efficiencies of EGFP disruption in human cells mediated by Cas9 and full-length or shortened gRNAs for four target sites in the EGFP reporter gene (sites #1 – #4). Lengths and sequences of the gRNA complementarity regions are shown. Ctrl, control gRNA lacking a complementarity region; nt, nucleotides. Error bars represent s.e.m., n = 2.
(d) Efficiencies of targeted indel mutations introduced at seven different human endogenous gene targets by matched standard and tru-RGNs. Lengths and sequences of gRNA complementarity regions are shown. Indel frequencies were measured by T7EI assay. Ctrl = control gRNA lacking a complementarity region. Three of the target sites used are named as previously described4: VEGFA site 1 (aka Target site 1), VEGFA Site 3 (aka Target Site 3), and EMX1 Site 1 (aka Target Site 4). VEGFA= vascular endothelial growth factor A, EMX1= empty spiracles homolog 1. Error bars represent s.e.m., n = 2.
(e) Efficiencies of precise alterations mediated by homology-directed repair (HDR) using single strand oligonucleotides (ssODN) at two endogenous human genes (VEGFA and EMX1) with either full-length RGNs or tru-RGNs. The percentage of alleles in which HDR had occurred (Percentage HDR) was measured using a BamHI restriction digest assay (Online Methods). Control gRNA = control gRNA lacking a complementarity region. Error bars represent s.e.m., n = 2.
To determine whether tru-RGNs could efficiently edit endogenous genes, we constructed trugRNAs for seven sites in three human genes (VEGFA, EMX1, and CLTA), including four sites targeted in previous studies4–6, 10(Fig. 1d). Five of these seven tru-gRNAs induced Cas9-mediated indel mutations with efficiencies comparable to those mediated by matched standard length RGNs (Fig. 1d) (Online Methods) with the two tru-gRNAs that showed lower activities than their full-length counterparts still exhibiting high absolute rates of mutagenesis (means of 13.3% and 16.6%) (Fig. 1d). Sanger sequencing confirmed that indels introduced by tru-RGNs originate at the expected site of cleavage and are essentially indistinguishable from those induced by their standard counterpart RGNs (Supplementary Fig. 2). We also found that tru-gRNAs containing a mismatched 5' G and 17 nucleotides of complementarity could efficiently direct Cas9-induced indels, whereas those bearing a mismatched 5' G and 16 nucleotides of complementarity showed lower or undetectable activities compared with matched full-length gRNAs (Supplementary Fig. 3), consistent with our results that a minimum of 17 nucleotide of complementarity is required for efficient RGN activity. Finally, we tested whether tru-RGNs could introduce a BamHI restriction site insertion via HDR with ssODNs and found that they did so for two sites in the VEGFA and EMX1 genes with efficiencies comparable to or higher than matched standard RGNs (Fig. 1e). We conclude that tru-RGNs can function efficiently to introduce on-target indels or HDR-mediated genome editing events in human cells.
To assess the specificities of tru-RGNs, we first tested whether they possess greater sensitivity to Watson-Crick mismatches at the gRNA-DNA interface by testing variants of the full-length and trugRNAs we had previously made for EGFP sites #1, #2 and #3 (Fig. 1c above). These variant gRNAs harbor single substitutions or adjacent double substitutions at each position within the complementarity region (except the 5' G) (Fig. 2a and 2b). Using the EGFP disruption assay in human U2OS.EGFP cells, we found that tru-RGNs were generally more sensitive to single and adjacent double mismatches than standard RGNs (compare bottom and top panels of Fig. 2a and 2b). Magnitudes of sensitivity to mismatches were site-dependent, with tru-RGNs to EGFP site #1 exhibiting less sensitivity to single and double mismatches than tru-RGNs to EGFP sites #2 and #3. Note that EGFP site #1 tru-gRNAs have 18 complementary nucleotides whereas the others have 17.
Figure 2. tru-gRNAs exhibit enhanced specificities and function efficiently with Cas9 nuclease and paired Cas9 nickases in human cells.




(a) Activities of RGNs targeted to three sites in EGFP using full-length (top) or tru-gRNAs (bottom) with single mismatches at each position (except at the 5'-most base which must remain a G for efficient expression from the U6 promoter). Grey boxes in the grids show the positions of Watson-Crick transversion mismatches. Empty gRNA control is a gRNA lacking a complementarity region. RGN activities were measured using the EGFP disruption assay and values shown represent the percentage of EGFP-negative observed relative to an RGN using a perfectly matched gRNA. Experiments were performed in duplicate and means with error bars representing s.e.m. are shown. Nucleotides present at each position of the gRNA complementarity region are shown at the bottom of each panel.
(b) Activities of RGNs targeted to three sites in EGFP using full-length (top) or tru-gRNAs (bottom) with adjacent double mismatches at each position (except at the 5'-most base which must remain a G for efficient expression from the U6 promoter). Data presented as in (a).
(c) The percentage of sequencing reads containing on- and off-target indel mutations induced by RGNs targeted to three different endogenous human gene sites as measured by deep sequencing. Indel frequencies are shown for the three target sites from cells in which targeted RGNs with a full-length gRNA, a tru-gRNA, or a control gRNA lacking a complementarity region were expressed. Sequences of the on-target and off-target (OT) sites for full-length and tru-gRNAs are shown in Table 1. The scale of the y-axis is the same for all three panels. Absolute counts of indel mutation data used to make these graphs can be found in Supplementary Table 2. On-target sites are named as previously described4 and as in Fig. 1d.
(d) Fold-improvements in off-target site specificities of three tru-RGNs compared to matched standard RGNs. Values shown represent the ratio of on-target to off-target activities for tru-RGNs divided by the ratio of on-target to off-target activities of standard RGNs for the off-target sites shown, calculated using the data from (c) and Supplementary Table 2. For the sites marked with an asterisk (*), no indels were observed with the tru-RGN and therefore the values shown represent conservative statistical estimates for the fold-improvements in specificities for these off-target sites (see Online Methods).
(e) Schematic illustrating locations of VEGFA sites 1 and 4 targeted by gRNAs and variant Cas9 nickases to generate paired double nicks. Target sites for the full-length gRNAs are underlined, with the first base in the PAM sequence shown in lowercase. Location of the BamHI restriction site inserted by homology-directed repair (HDR) with a ssODN donor is shown.
(f) Substitution of a full-length gRNA for VEGFA site 1 with a tru-gRNA does not reduce the efficiency of indel mutations observed with a paired full-length gRNA for VEGFA site 4 and Cas9-D10A nickases. Control gRNA used is one lacking a complementarity region. The frequency of indel mutations induced by the VEGFA site 4 gRNA alone with Cas9 nickase is highlighted in red.
(g) Substitution of a full-length gRNA for VEGFA site 1 with a tru-gRNA does not reduce the efficiency of BamHI sequence alterations introduced with a paired full-length gRNA for VEGFA site 4, Cas9-D10A nickase, and an ssODN donor template. Control gRNA used is one lacking a complementarity region.
We next examined whether tru-RGNs have reduced genomic off-target effects in human cells by using matched full-length and tru-gRNAs targeted to VEGFA site 1, VEGFA site 3,and EMX1 site 1 (Fig. 1d). We chose these target sites because previous studies had defined a total of 13 off-target sites for the full-length gRNAs targeted to these sequences4, 5. tru-RGNs exhibited substantially reduced mutagenesis activity relative to matched standard RGNs at all 13 previously identified off-target sites in human U2OS.EGFP cells (Table 1), with 11 sites having mutation frequencies below the reliable detection limit (2 – 5%) of the T7 Endonuclease I (T7EI) assay used for these experiments (Table 1; Online Methods). We also observed similar results in a different human cell line (FT-HEK293 cells) (Supplementary Table 1). To enable more sensitive detection of off-target mutations, we used high-throughput sequencing to assess 12 of the 13 off-target sites we had analyzed by T7EI assay (for technical reasons, we were unable to amplify the required shorter amplicon for one of the sites) as well as an additional, previously identified5 off-target site for EMX1 site 1 in U2OS.EGFP cells (Fig. 2c). These sequencing results showed that tru-RGNs induced substantially decreased mutagenesis frequencies at all 13 off-target sites relative to matched standard RGNs (Fig. 2c and Supplementary Table 2), with some sites showing decreases of ~5000-fold or more (Fig. 2d). No indel mutations were observed with RGNs for off-target sites (OT1–4 and OT1–11). Therefore, for these two sites, we conservatively estimated a likely upper boundary for the average indel frequencies and calculated a minimum improvement in tru-RGN specificities for these sites of >10,000 or more over standard RGNs (Online Methods; Fig. 2d).
Table 1.
Frequencies of indels induced at on- and off-target sites by tru-RGNs and matched standard RGNs in human U2OS.EGFP cells
| Target ID | Gene | Full-Length Target (20 nt) | Indel mutation frequency (%) | Truncated Target (17 or 18 nts) | Indel mutation frequency (%) |
|---|---|---|---|---|---|
| Target Site 1 | VEGFA | GGGTGGGGGGAGTTTGCTCCtGG | 23.69 [25.68, 21.70] | GTGGGGGGAGTTTGCTCCtGG | 23.93 [28.30, 19.55] |
| OT1–3 | IGDCC3 | GGATGGAGGGAGTTTGCTCCtGG | 17.25 [20.22, 14.28] | ATGGAGGGAGTTTGCTCCtGG | Not Detected |
| OT1–4 | LOC116437 | GGGAGGGTGGAGTTTGCTCCtGG | 6.23 [6.44, 6.03] | GAGGGTGGAGTTTGCTCCtGG | Not Detected |
| OT1–6 | CACNA2D | CGGGGGAGGGAGTTTGCTCCtGG | 3.73 [3.95, 3.50] | GGGGAGGGAGTTTGCTCCtGG | Not Detected |
| OT1–11 | GGGGAGGGGAAGTTTGCTCCtGG | 10.36 [11.02, 9.69] | GGAGGGGAAGTTTGCTCCtGG | Not Detected | |
|
| |||||
| Target Site 3 | VEGFA | GGTGAGTGAGTGTGTGCGTGtGG | 54.08 [55.10, 53.06] | GAGTGAGTGTGTGCGTGtGG | 50.49 [49.24, 51.74] |
| OT3–1 | (abParts) | GGTGAGTGAGTGTGTGTGTGaGG | 6.16 [6.71, 5.60] | GAGTGAGTGTGTGTGTGaGG | Not Detected |
| OT3–2 | MAX | AGTGAGTGAGTGTGTGTGTGgGG | 19.64 [18.58, 20.70] | GAGTGAGTGTGTGTGTGgGG | 5.52 [5.77, 5.27] |
| OT3–4 | GCTGAGTGAGTGTATGCGTGtGG | 7.95 [7.84, 8.06] | GAGTGAGTGTATGCGTGtGG | 1.69 [1.42, 1.95] | |
| OT3–9 | TPCN2 | GGTGAGTGAGTGCGTGCGGGtGG | Not Detected | GAGTGAGTGCGTGCGGGtGG | Not Detected |
| OT3–17 | SLIT1 | GTTGAGTGAATGTGTGCGTGaGG | 1.85 [1.77, 1.92] | GAGTGAATGTGTGCGTGaGG | Not Detected |
| OT3–18 | COMDA | TGTGGGTGAGTGTGTGCGTGaGG | 6.16 [6.72, 5.60] | GGGTGAGTGTGTGCGTGaGG | Not Detected |
| OT3–20 | AGAGAGTGAGTGTGTGCATGaGG | 10.47 [9.39, 11.55] | GAGTGAGTGTGTGCATGaGG | Not Detected | |
|
| |||||
| Target Site 4 | EMX1 | GAGTCCGAGCAGAAGAAGAAgGG | 41.56 [41.76, 41.37] | GTCCGAGCAGAAGAAGAAgGG | 43.01 [43.89, 42.15] |
| OT4–1 | HCN1 | GAGTTAGAGCAGAAGAAGAAaGG | 19.26 [18.54, 19.99] | GTTAGAGCAGAAGAAGAAaGG | Not Detected |
| OT4_53* | MFAP1 | GAGTCTAAGCAGAAGAAGAAgAG | 4.37 [3.79, 4.96] | GTCTAAGCAGAAGAAGAAgAG | Not Detected |
Mutation frequencies were assessed by T7EI assay with means (bold text) of duplicate measurements (in brackets) shown.
Off-target site OT4_53 is the same as EMX1 target 3 OT31 from ref. 5.
We then sought to assess whether tru-RGNs might induce additional off-target mutations in the human genome beyond those previously identified for matched full-length RGNs. Based on the results of our EGFP disruption assay, we reasoned that genomic sites with the fewest mismatches compared to the on-target site would be the most likely to be mutated. Therefore, we computationally identified sites in the human genome with one, two or three mismatches relative to tru-gRNAs targeted to VEGFA site 1, VEFGA site 3 and EMX1 site 1 (Supplementary Table 3a), excluding known off-target sites that had already been examined by deep sequencing above (Supplementary Table 3b). We used the T7EI assay to examine 97 potential off-target sites including all with one mismatch and either all or some with two or three mismatches (Supplementary Table 3c and Supplementary Table 4). Only one of these 97 sites showed detectable levels of indel mutations by T7EI assay in human U2OS.EGFP cells and none showed detectable indels in human FT-HEK293 cells (Supplementary Table 4). The one site for which indels were observed was also mutated by the corresponding standard full-length RGN (Supplementary Results and Supplementary Fig. 4), demonstrating that this off-target site is not unique to the tru-RGN. We also used deep sequencing to examine 30 of the most closely matched potential off-target sites (including all sites with one mismatch for all three RGNs and nearly all sites with two mismatches for the RGNs targeted to VEGFA Site #1 and EMX1 Site #1 (Supplementary Table 4d)) and found undetectable or very low rates of indel mutations (Supplementary Table 5), comparable to those induced by tru-RGNs for other previously known off-target sites (Supplementary Table 2). Of note, the percentage of sites with mutation rates above 0.1% decreases with increasing numbers of mismatches (Supplementary Table 4e), validating our focus on sites with fewest mismatches. tru-RGNs generally appear to induce either very low or undetectable levels of mutations at potential off-target sites that differ by one or two mismatches; by contrast, our previous study using standard RGNs showed high levels of mutations at numerous off-target sites bearing up to four or five mismatches4.
Because neither tru-gRNAs nor paired nickases completely eliminate off-target effects, we explored whether combining these strategies might further reduce such mutations. First, we used a pair of gRNAs targeted to sites in the human VEGFA gene (VEGFA site 1 and VEGFA site 4; Fig. 2e) previously shown to work with the paired Cas9-D10A nickase approach10. Substitution of the full-length gRNA for VEGFA Site #1 with a tru-gRNA did not adversely affect the induction of indels (Fig. 2f) or incorporation of a restriction site sequence by HDR (Fig. 2g). We next used deep sequencing to examine mutation frequencies at four previously validated off-target sites of the VEGFA site 1 gRNA and found that mutation rates dropped below the detection limit of the sequencing assay at all four of these off-target sites when using paired nickases with one full-length gRNA and one tru-gRNA (Supplementary Table 6). By contrast, both a single tru-RGN (Supplementary Table 2) and paired nickases with full-length gRNAs (Supplementary Table 6) still induced off-target mutations at one of these four off-target sites (OT1–3). Although we substituted a tru-gRNA for only one of the two full-length gRNAs in our VEGFA paired nickase experiments, we have shown at another locus (in the EMX1 gene) that use of two tru-gRNAs with Cas9 nickase can robustly induce indels at an endogenous human gene locus (Supplementary Fig. 5). Taken together, we conclude that tru-gRNAs can further reduce the off-target effects of paired Cas9 nickases (and vice versa) without compromising the efficiency of on-target genome editing.
Our results show that tru-RGNs can generally introduce mutations via NHEJ or HDR at on-target sites with high efficiencies and show reduced mutagenic effects at closely matched off-target sites. One potential model to explain our overall results is that standard RGNs with full-length gRNAs might possess more affinity for their target sites than is required (perhaps not unsurprisingly because it might be beneficial for naturally occurring CRISPR systems to tolerate the introduction of alterations in the target sequence) and that truncation of the gRNA might poise the tru-RGN/DNA complex to be more sensitive to mismatches, perhaps by reducing binding energy at the gRNA/DNA interface. The concept of excess affinity affecting the specificity of DNA binding domains has previously been suggested by others for engineered zinc finger proteins23, 24.
The use of tru-gRNAs for improving CRISPR specificity offers important advantages over the paired Cas9 nickase strategy. tru-gRNAs should be technically simpler to implement with applications involving multiplex12–14 or genome-wide libraries of gRNAs15, 16 and, unlike the paired nickase strategy, can also be used to improve the specificities of dCas925 or dCas9 fusions to heterologous effector domains such as transcriptional regulatory domains17–19. In this regard, we note that we have found that tru-gRNAs can efficiently recruit dCas9-VP64 transcriptional activators to an endogenous human gene (Y.F., V.M.C., and J.K.J., unpublished data). tru-RGNs also avoid the need to use a second gRNA that on its own can potentially induce additional unwanted mutations with a Cas9 nickase. For example, consistent with previously published results7, 9, 11, we found that a single gRNA to VEGFA site 4 was capable of efficiently inducing indel mutations with Cas9 nickases (red bar, Fig. 2f).
Our findings have several important implications for how to choose potential RGN target sites. The use of shorter gRNAs does not decrease the targeting range of the platform because target sites with 17 or 18 nts of complementarity will each occur in random DNA with frequencies equal to those with 20 nts of complementarity. Our results show that tru-gRGNs generally appear to induce very low or undetectable levels of mutagenesis at off-target sites with as few as one or two mismatches and suggest that sites with three or more mismatches will not have mutations at high frequencies, if at all. Thus, a reasonable strategy for choosing target sites to minimize off-target effects might be to choose tru-gRNA target sites that are unique in the genome and that have the smallest possible number of potential off-target sites with 1 or 2 mistmatches. We have modified our web-based ZiFiT Targeter program26, 27 so that it can identify tru-gRNA sites with 17 or 18 nts of complementarity and provide the user with information about potential off-target sites that differ by 0, 1 or 2 positions in the published genomes of fruit flies, roundworm, zebrafish, mice, rats, humans using the Bowtie program28. This modified version of ZiFiT is currently accessible at http://zifit.partners.org.
Our results raise several important questions to be addressed in future experiments. Although we found that tru-gRNAs with 17 or 18 nts of complementarity generally function efficiently at the intended target site and have improved specificities, it is possible that certain gRNAs with shorter and longer complementarity lengths might also possess such properties. Testing of greater numbers of truncated gRNAs in future experiments should help to determine this and also whether certain characteristics such as GC content might predict activity levels. In addition, we presume our tru-gRNAs are generally being expressed at the same levels as their full-length counterparts because they show comparable activities and because titration experiments performed for EGFP sites #1, #2, and #3 in which the amounts of gRNA and Cas9 expression vectors are varied demonstrate that shortened gRNAs give activity curves similar to their full-length counterparts (Supplementary Fig. 6); however, more direct, quantitative assessments of tru-gRNA expression levels will be required to definitively establish this and to better understand why and how tru-gRNAs function with high efficiencies and specificities.
In summary, tru-gRNAs provide a simple and flexible approach to minimize the off-target effects of individual Cas9 nucleases, paired Cas9 nickases and, potentially, dCas9 fusion proteins in human cells. However, we note that definitive assessment of the relative efficacies of any platform designed to improve specificities will require the development of an unbiased approach for globally assessing off-target effects in human cells. Continued efforts to develop methods for assessing and improving specificity will further accelerate the use of CRISPR-based reagents for research and therapeutic applications.
Online Methods
Plasmid construction
All gRNA expression plasmids were assembled by designing, synthesizing, annealing, and cloning pairs of oligonucleotides (IDT) harboring the complementarity region into plasmid pMLM3636 (available from Addgene; http://www.addgene.org/crispr-cas) as previously described4. The resulting gRNA expression vectors encode a ~100 nt gRNA whose expression is driven by a human U6 promoter. The sequences of all oligonucleotides used to construct gRNA expression vectors are shown in Supplementary Table 7. The Cas9 D10A nickase expression plasmid (pJDS271) bearing a mutation in the RuvC endonuclease domain was generated by mutating plasmid pJDS246 using a QuikChange kit (Agilent Technologies) with the following primers: Cas9 D10A sense primer 5'-tggataaaaagtattctattggtttagccatcggcactaattccg-3'; Cas9 D10A antisense primer 5'-cggaattagtgccgatggctaaaccaatagaatactttttatcca-3'. All the targeted gRNA plasmids and the Cas9 nickase plasmids used in this study will be made available through the non-profit plasmid distribution service Addgene.
Human cell-based EGFP disruption assay
U2OS.EGFP cells harboring a single-copy, integrated EGFP-PEST gene reporter have been previously described22. These cells were maintained in Advanced DMEM (Life Technologies) supplemented with 10% FBS, 2 mM GlutaMax (Life Technologies), penicillin/streptomycin and 400 μg/ml G418. To assay for disruption of EGFP expression, 2 × 105 U2OS.EGFP cells were transfected in duplicate with gRNA expression plasmid or an empty U6 promoter plasmid as a negative control, Cas9 expression plasmid (pJDS246)4, and 10 ng of td-Tomato expression plasmid (to control for transfection efficiency) using a LONZA 4D-Nucleofector™, with SE solution and DN100 program according to the manufacturer's instructions. We used 25 ng/250 ng, 250 ng/750 ng, 200 ng/750 ng, and 250 ng/750 ng of gRNA expression plasmid/Cas9 expression plasmid for experiments with EGFP site #1, #2, #3, and #4, respectively. Two days following transfection, cells were trypsinized and resuspended in Dulbecco's modified Eagle medium (DMEM, Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and analyzed on a BD LSRII flow cytometer. For each sample, transfections and flow cytometry measurements were performed in duplicate.
Transfection of human cells and isolation of genomic DNA
To assess the on-target and off-target indel mutations induced by RGNs targeted to endogenous human genes, we transfected plasmids into U2OS.EGFP or FT-HEK293 (Life Technologies) cells using the following conditions: U2OS.EGFP cells were transfected using the same conditions as for the EGFP disruption assay described above. FT-HEK293 cells were transfected by seeding them at a density of 1.65 × 105 cells per well in 24 well plates in Advanced DMEM (Life Technologies) supplemented with 10% FBS and 2 mM GlutaMax (Life Technologies) at 37°C in a CO2 incubator. After 22 – 24 hours of incubation, cells were transfected with 125 ng of gRNA expression plasmid or an empty U6 promoter plasmid (as a negative control), 375 ng of Cas9 expression plasmid (pJDS246)4, and 10 ng of a td-Tomato expression plasmid, using Lipofectamine LTX reagent according to the manufacturer's instructions (Life Technologies). Medium was changed 16 hours after transfection. For both types of cells, genomic DNA was harvested two days post-transfection using an Agencourt DNAdvance genomic DNA isolation kit (Beckman) according to the manufacturer's instructions. For each RGN sample to be assayed, we performed 12 individual 4D transfection replicates, isolated genomic DNA from each of these 12 transfections, and then combined these samples to create two “duplicate” pools each consisting of six pooled genomic DNA samples. We then assessed indel mutations at on-target and off-target sites from these duplicate samples by T7EI assay, Sanger sequencing, and/or deep sequencing as described below.
To assess frequencies of precise alterations introduced by HDR with ssODN donor templates, 2×105 U2OS.EGFP cells were transfected 250 ng of gRNA expression plasmid or an empty U6 promoter plasmid (as a negative control), 750 ng Cas9 expression plasmid (pJDS246), 50 pmol of ssODN donor (or no ssODN for controls), and 10 ng of td-Tomato expression plasmid (as the transfection control). Genomic DNA was purified three days after transfection using Agencourt DNAdvance and assayed for the introduction of a BamHI site at the locus of interest as described below. All of these transfections were performed in duplicate.
For experiments involving Cas9 nickases, 2 × 105 U2OS.EGFP cells were transfected with 125 ng of each gRNA expression plasmid (if using paired gRNAs) or 250 ng of gRNA expression plasmid (if using a single gRNA), 750 ng of Cas9-D10A nickase expression plasmid (pJDS271), 10 ng of td-Tomato plasmid, and (if performing HDR) 50 pmol of ssODN donor template (encoding the BamHI site). All transfections were performed in duplicate. Genomic DNA harvested two days after transfection (if assaying for indel mutations) or three days after transfection (if assaying for HDR/ssODN-mediated alterations) using the Agencourt DNAdvance genomic DNA isolation kit (Beckman).
U2OS.EGFP and FT-HEK293 cell lines used in this study were tested for mycoplasma contamination every two weeks.
T7EI assays for quantifying frequencies of indel mutations
T7EI assays were performed as previously described4. In brief, PCR reactions to amplify specific on-target or off-target sites were performed with Phusion high-fidelity DNA polymerase (New England Biolabs) using one of the two following programs: (1) Touchdown PCR program [(98°C, 10 s; 72–62°C, −1 °C/cycle, 15 s; 72°C, 30 s) × 10 cycles, (98°C, 10 s; 62°C, 15 s; 72°C, 30 s) × 25 cycles] or (2) Constant Tm PCR program [(98°C, 10 s; 68°C or 72°C, 15 s; 72°C, 30 s) × 35 cycles], with 3% DMSO or 1 M betaine if necessary. All primers used for these amplifications are listed in Supplementary Table 8. Resulting PCR products ranged in size from 300 to 800 bps and were purified by Ampure XP beads (Agencourt) according to the manufacturer's instructions. 200ng of purified PCR products were hybridized in 1 × NEB buffer 2 in a total volume of 19 μl and denatured to form heteroduplexes using the following conditions: 95 °C, 5 minutes; 95 to 85 °C, −2 °C/s; 85 to 25 °C, −0.1 °C/s; hold at 4 °C. 1 μl of T7 Endonuclease I (New England Biolabs, 10 units/μl) was added to the hybridized PCR products and incubated at 37°C for 15 minutes. The T7EI reaction was stopped by adding 2 μl of 0.25 M EDTA solution and the reaction products were purified using AMPure XP beads (Agencourt) with elution in 20 μl 0.1× EB buffer (QIAgen). Reactions products were then analyzed on a QIAXCEL capillary electrophoresis system and the frequencies of indel mutations were calculated using the same formula as previously described22. A more detailed protocol for the T7EI assay and examples of sample capillary electrophoresis traces has been previously described22.
Sanger sequencing for quantifying frequencies of indel mutations
Purified PCR products used for T7EI assay were ligated into a Zero Blunt TOPO vector (Life Technologies) and transformed into chemically competent Top 10 bacterial cells. Plasmid DNAs were isolated and sequenced by the Massachusetts General Hospital (MGH) DNA Automation Core, using an M13 forward primer (5'-GTAAAACGACGGCCAG-3').
Restriction digest assay for quantifying specific alterations induced by HDR with ssODNs
PCR reactions of specific on-target sites were performed using Phusion high-fidelity DNA polymerase (New England Biolabs). The VEGFA and EMX1 loci were amplified using a touchdown PCR program ((98 °C, 10 s; 72–62 °C, −1 °C/cycle, 15 s; 72 °C, 30 s) × 10 cycles, (98 °C, 10 s; 62 °C, 15 s; 72 °C, 30 s) × 25 cycles), with 3% DMSO. The primers used for these PCR reactions are listed in Supplementary Table 8. PCR products were purified by Ampure XP beads (Agencourt) according to the manufacturer's instructions. For detection of the BamHI restriction site encoded by the ssODN donor template, 200 ng of purified PCR products were digested with BamHI at 37 °C for 45 minutes. The digested products were purified by Ampure XP beads (Agencourt), eluted in 20ul 0.1×EB buffer and analyzed and quantified using a QIAXCEL capillary electrophoresis system.
TruSeq library generation and sequencing data analysis
Locus-specific primers were designed to flank on-target and potential and verified off-target sites to produce PCR products ~300bp to 400 bps in length. Genomic DNAs from the pooled duplicate samples described above were used as templates for PCR. All PCR products were purified by Ampure XP beads (Agencourt) per the manufacturer's instructions. Purified PCR products were quantified on a QIAXCEL capillary electrophoresis system. PCR products for each locus were amplified from each of the pooled duplicate samples (described above), purified, quantified, and then pooled together in equal quantities for deep sequencing. Pooled amplicons were ligated with dual-indexed Illumina TruSeq adaptors as previously described29. The libraries were purified and run on a QIAXCEL capillary electrophoresis system to verify change in size following adaptor ligation. The adapter-ligated libraries were quantified by qPCR and then sequenced using Illumina MiSeq 250 bp paired-end reads performed by the Dana-Farber Cancer Institute Molecular Biology Core Facilities. We analyzed between 75,000 and 1,270,000 (average ~422,000) reads for each sample. The TruSeq reads were analyzed for rates of indel mutagenesis as previously described30. Specificity ratios were calculated as the ratio of observed mutagenesis at an on-target locus to that of a particular off-target locus as determined by deep sequencing. Fold-improvements in specificity with tru-RGNs for individual off-target sites were calculated as the specificity ratio observed with tru-gRNAs to the specificity ratio for that same target with the matched full-length gRNA. As mentioned in the text, for two of the known off-target sites, no indel mutations were detected with tru-gRNAs. For these particular sites, it was difficult to quantify the on-target to off-target ratios for tru-RGNs and, therefore, also the magnitude of improved specificity for tru-RGNs to standard RGNs. For example, we did not observe any tru-RGN-induced indels at sites OT1–4 and OT1–11 and thus the ratio of on-target to off-target rates would calculate to be infinite in these cases. For these sites, we reasoned it was possible that the true rates could be below the detection limit of our assay. Using the following equation, we calculated that we would have a 95% probability of observing 1 or more mutagenesis events if the mean number of mutagenesis events was 3:
Therefore, we used 3 as a conservative estimate of the number of events (instead of the 0 observed) as the numerator to estimate a lower bound for the fold-improvement of the tru-gRNA in place of infinite improvement suggested by observing 0 events. All sequencing data has been deposited with National Center for Biotechnology Information Sequence Read Archive (NCBI SRA), accession number SRP033215.
Supplementary Material
Acknowledgments
We thank Morgan Maeder, Shengdar Tsai, and James Angstman for helpful discussions and comments on the manuscript and Jennifer Foden for technical assistance. This work was funded by a National Institutes of Health (NIH) Director's Pioneer Award (DP1 GM105378), NIH R01 GM088040, NIH P50 HG005550, and the Jim and Ann Orr Massachusetts General Hospital (MGH) Research Scholar Award. This material is based upon work supported by, or in part by, the U. S. Army Research Laboratory and the U. S. Army Research Office under grant number W911NF-11-2-0056. J.K.J. has financial interests in Editas Medicine and Transposagen Biopharmaceuticals. J.K.J.'s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.K.J. and J.D.S. have filed a patent application on the tru-gRNA/tru-RGN technology. J.K.J. and J.D.S. are consultants for Editas Medicine, a company focused on developing genome-editing therapeutics.
Footnotes
Author Contributions Y.F., J.D.S., D.R., and J.K.J. conceived of and designed experiments. Y.F., J.D.S., and V.M.C. performed experiments. D.R. developed the updated version of ZiFiT software. Y.F., J.D.S., and J.K.J. wrote the paper.
References
- 1.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 2.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charpentier E, Doudna JA. Biotechnology: Rewriting a genome. Nature. 2013;495:50–51. doi: 10.1038/495050a. [DOI] [PubMed] [Google Scholar]
- 4.Fu Y, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pattanayak V, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho SW, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2013 doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim E, et al. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012;22:1327–1333. doi: 10.1101/gr.138792.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jao LE, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci U S A. 2013;110:13904–13909. doi: 10.1073/pnas.1308335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbert LA, et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang WY, et al. Heritable and Precise Zebrafish Genome Editing Using a CRISPR-Cas System. PLoS One. 2013;8:e68708. doi: 10.1371/journal.pone.0068708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reyon D, et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotech. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beerli RR, Dreier B, Barbas CF., 3rd Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci U S A. 2000;97:1495–1500. doi: 10.1073/pnas.040552697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sander JD, et al. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38:W462–468. doi: 10.1093/nar/gkq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D. Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res. 2007;35:W599–605. doi: 10.1093/nar/gkm349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher S, et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 2011;12:R1. doi: 10.1186/gb-2011-12-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sander JD, et al. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.