

Abstract
Pancreatic cancer is highly chemoresistant. A major contributing factor is the characteristic extensive stromal or fibrotic reaction, which comprises up to 90% of the tumor volume. Over the last decade there has been intensive research into the role of the pro-fibrogenic pancreatic stellate cells (PSCs) and their interaction with pancreatic cancer cells. As a result of the significant alterations in the tumor microenvironment following activation of PSCs, tumor progression, and chemoresistance is enhanced. This review will discuss how PSCs contribute to chemoresistance in pancreatic cancer.
Keywords: pancreatic cancer, chemoresistance, pancreatic stellate cells, stroma, fibrosis, hypoxia
Introduction
Pancreatic cancer is a highly aggressive malignancy with a notoriously dismal prognosis. Contributing to ~227,000 annual deaths worldwide, this insidious disease is the fourth leading cause of cancer-related death in developed countries (Raimondi et al., 2009; Hidalgo, 2010; Vincent et al., 2011). Remarkably, 80–85% of patients present with unresectable and incurable tumors, putting the median survival period at <6 months and the overall 5-years survival rate at <5% (Hidalgo, 2010; Vincent et al., 2011).
A major contributor to this poor clinical outcome is pancreatic cancer's prominent chemoresistance (Zalatnai and Molnar, 2007; Wang et al., 2011). In fact, the best treatments only prolong life by ~8–16 weeks (Wolfgang et al., 2013). Until recently, most research efforts focused solely on cancer cells. However, significant stakeholders in this chemoresistance are key pro-fibrogenic cells of the pancreas known as pancreatic stellate cells (PSCs), which when co-opted and activated by cancer cells, orchestrate the strong desmoplasia that characterizes pancreatic cancer (Apte et al., 2004; Bachem et al., 2005). The resultant stromal landscaping yields an exclusive microenvironment where cross-talk between cancer-associated human PSCs (CA-hPSCs) and cancer cells promotes local tumor progression, metastasis and chemoresistance (Apte et al., 2004; Bachem et al., 2005; Hwang et al., 2008; Vonlaufen et al., 2008a,b; Xu et al., 2010; Erkan et al., 2012; Phillips, 2012).
CA-hPSCs establish fibrosis via excessive extracellular matrix (ECM) deposition, which compresses and distorts intratumoural vasculature, causing hypoxia (Olive et al., 2009; Phillips, 2012; Stylianopoulos et al., 2012; Jacobetz et al., 2013). Hypoxia stimulates the epithelial-mesenchymal transition (EMT) of cancer cells, which is a more chemoresistant phenotype (Arumugam et al., 2009; Kikuta et al., 2010; Wang et al., 2011). Furthermore, the fibrosis sequesters chemotherapeutics in the stromal compartment, impairing successful drug delivery to cancer cells (Olive et al., 2009; Provenzano et al., 2012; Jacobetz et al., 2013). Despite being in a hostile microenvironment replete with cytotoxic drugs and hypoxia, CA-hPSCs not only survive, but actually thrive and proliferate causing the tumor microenvironment to occupy up to 90% of the tumor volume (Li et al., 2010; Neesse et al., 2011; Michl and Gress, 2013).
A major limitation to our understanding of the role of CA-hPSCs in chemoresistance is their survival mechanisms in this noxious microenvironment. However, mounting evidence suggests CA-hPSCs are both direct and indirect drivers of pancreatic cancer chemoresistance and spread, and thus their inhibition may potentiate current chemotherapy and intercept tumor-facilitatory bidirectional interactions.
A growth permissive microenvironment
The histopathological hallmark of pancreatic cancer that underlies its aggressiveness is the severe desmoplastic and fibroinflammatory reaction which generates a high stromal-to-epithelial ratio (Li et al., 2010). In fact, pancreatic ductal adenocarcinoma (PDAC), which comprises >85% of all pancreatic cancer subtypes, is one of the most stroma-rich malignancies (Bardeesy and Depinho, 2002; Feig et al., 2012). Despite PDAC demonstrating similar chemosensitivity as other cancers in vitro, PDAC patients are less responsive to chemotherapeutics than other cancers (Li et al., 2010), implicating the unique microenvironment in PDAC's chemoresistance. Such desmoplasia facilitates a mechanopathology known as growth-induced solid stress (GISS), resulting in collapsed or compressed intratumoural blood vessels or lymphatics, which respectively lead to increased hypoxia and interstitial fluid pressure (IFP); both attenuate chemosensitivity (Stylianopoulos et al., 2012). Non-invasive quantification of physiological parameters in human pancreatic tumors had confirmed that blood flow is reduced and metabolic activity is increased relative to normal pancreas (Komar et al., 2009). This recapitulates the significant consequences of GISS, as deficient vasculature reduces blood flow, yielding hypoxia (Stylianopoulos et al., 2012). Subsequently, stromal and cancer cells undergo aerobic glycolysis—“The Warburg Effect”—leading to their increased metabolic activity, as a means of surviving in this hypoxic microenvironment (Tod et al., 2013).
The stroma in PDAC is composed of abundant ECM proteins, such as collagen and hyaluronan, along with nerves, blood, and lymphatics, and a versatile cellular population including inflammatory cells and activated PSCs (Erkan et al., 2010; Neesse et al., 2011). Several paracrine and autocrine factors induce stroma production (Li et al., 2010; Neesse et al., 2011; Phillips, 2012) (Figure 1).
Figure 1.

Mechanisms of pancreatic stellate cell (PSC) activation. A central feature of PSCs is the transformation from a quiescent (fat storing phenotype) to an activated (myofibroblast-like phenotype) state. Growth factors and pro-inflammatory cytokines released by neighboring cells (pancreatic cancer cells, injured acinar or ductal cells, inflammatory cells, and endothelial cells) all induce PSC activation. Activated PSCs can then perpetuate this activation state via autocrine stimuli, leading to increased proliferation, migration, and excessive ECM production. In pancreatic cancer, activation of PSCs leads to the production of extensive fibrosis which in turn contributes to disease progression, metastases and chemoresistance. CTGF, connective tissue growth factor; COX-2, cyclooxygenase-2; ECM, extracellular matrix; EMMPRIN, extracellular matrix metalloproteinase inducer; ET-1, endothelin 1; IL, interleukin; PDGF, platelet derived growth factor; TGF-β, transforming growth factor β; TNFα, tumor necrosis factor α; TRAIL, TNF-related apoptosis-inducing ligand.
Pancreatic stellate cells—key fibrogenic cells in pancreatic cancer
In health, PSCs are quiescent, represent ~4% of the cell population, express desmin, and glial fibrillary acidic protein and store cytoplasmic vitamin A-containing lipid droplets (Apte et al., 1998, 2006). The latter markers are PSC-specific, distinguishing them from normal pancreatic fibroblasts. Disease onset heralds the accumulation of reactive oxygen species, cytokines and growth factors secreted by injured cells, which can all activate PSCs (Apte et al., 1999; Mews et al., 2002; Gao and Brigstock, 2005; Kordes et al., 2005; Vonlaufen et al., 2010) (Figure 1).
Such paracrine stimuli induce quiescent PSCs to transdifferentiate into a myofibroblast-like phenotype, gaining expression of α-smooth muscle actin (α-SMA) and losing the anti-fibrogenic lipid droplets (Apte et al., 1998). These morphological changes are accompanied by functional changes (Figure 1), such as: (1) increased proliferation and migration (Apte et al., 1999; Schneider et al., 2001; Mews et al., 2002; Phillips et al., 2003b; Omary et al., 2007); (2) excessive synthesis of ECM proteins as well as matrix metalloproteinases (MMPs) and their inhibitors (Apte et al., 1999, 2004; Schneider et al., 2001; Phillips et al., 2003a; Bachem et al., 2005); and (3) secretion of growth factors and cytokines involved in autocrine loops that perpetuate PSC activation (Shek et al., 2002; Ohnishi et al., 2003; Aoki et al., 2006a,b; Omary et al., 2007; Jiang et al., 2009).
Bidirectional interactions between pancreatic stellate cells and cancer cells
Substantial evidence corroborates the notion that bidirectional interplay occurs between cancer cells and CA-hPSCs, commensally facilitating tumor progression (Apte et al., 2004; Bachem et al., 2005; Hwang et al., 2008; Vonlaufen et al., 2008a; Xu et al., 2010). Cancer cells recruit PSCs via mitogenic and fibrogenic factors which promote PSC activation, proliferation, migration and ECM remodeling capability. For instance, supernatants from PDAC cell lines stimulated PSC proliferation and ECM synthesis in a dose-dependent manner, with these effects abrogated by neutralizing antibodies against platelet-derived growth factor (PDGF), fibroblast growth factor, and transforming growth factor-β1 (TGF-β1) (Bachem et al., 2005). Also, PSC synthesis of MMP-2, a protein crucial for basement membrane degradation, is increased by ECM metalloproteinase inducer (EMMPRIN), which is secreted by cancer cells (Schneiderhan et al., 2007). Such modulation of the ECM is critical for degradation of basement membrane and may influence cancer progression.
PSC recruitment is consequential for cancer cell behavior, as stellate or cancer cell-derived growth factors, cytokines and ECM components are sequestered in a fortified niche. For example, Bachem et al. (2005) originally demonstrated that subcutaneous co-injection of PSCs and cancer cells into nude mice accelerated tumor growth relative to solely injecting cancer cells. This was substantiated in an orthotopic mouse model which employed intra-pancreatic co-injections of PSCs and cancer cells, replicating the aforementioned augmented tumor growth and showing enhanced local and distant metastases relative to solely injecting cancer cells (Vonlaufen et al., 2008a). Xu et al. (2010) also demonstrated that CA-hPSCs co-migrate with metastasizing cancer cells, which is likely to aid cancer cell seeding and growth. Furthermore, PSC-conditioned medium increased proliferation, migration, invasion, and chemoresistance of cancer cells and reduced their apoptosis in vitro (Hwang et al., 2008; Vonlaufen et al., 2008a; Gao et al., 2010), while orthotopic co-injection of PSCs and cancer cells increased primary tumor incidence and size in vivo (Hwang et al., 2008). Moreover, co-culturing with PSCs promoted EMT of PDAC cells (Kikuta et al., 2010). Evidence by Watanabe et al. (2003) also demonstrated that extensive intratumoural fibroblastic cell proliferation correlates with a poorer disease outcome in pancreatic cancer patients. This significant evidence suggests that abolishing CA-hPSCs or their activity may reduce PDAC's aggressiveness, necessitating an improved understanding of CA-hPSC survival mechanisms.
Pancreatic stellate cells and chemoresistance of pancreatic cancer
PDAC is highly refractory to chemotherapeutics (Wolfgang et al., 2013). While the cause of chemoresistance is multifactorial, three major processes have been distinguished: (1) reduced drug uptake; (2) increased energy-dependent drug efflux; and (3) alterations in cellular capabilities affecting drug cytotoxicity, such as reduced apoptosis and dysregulated drug metabolism (Szakacs et al., 2006; Zalatnai and Molnar, 2007). However, another major determinant of pancreatic cancer chemoresistance is the extensive fibrosis produced by PSCs, which results in significant intratumoural hypoxia and a self-perpetuating hypoxia-fibrosis cycle (Figure 2) (Koong et al., 2000; Evans and Koch, 2003; Erkan et al., 2009). CA-hPSCs extend this chemoresistant profile via the hypoxia-fibrosis cycle (Masamune et al., 2008; Phillips, 2012). This impairs drug delivery to cancer cells and stimulates their EMT and genetic instability, yielding a more chemoresistant phenotype (Arumugam et al., 2009; Kikuta et al., 2010; Wang et al., 2011).
Figure 2.

Growth-induced solid stress (GISS), the hypoxia-fibrosis cycle and their contribution to chemoresistance. Continuous PSC activation results in excessive ECM deposition, particularly of tensile-resistant fibrillar collagen as well as compression-resistant hyaluronan. This eventually leads to prominent fibrosis which, along with deformation caused by the number of proliferating stromal cells and cancer cells, results in GISS. Consequently, this reduces the caliber of intratumoral lymphatics and blood vessels. The former leads to increased IFP, which can impair drug perfusion, while the latter reduces blood flow, consequently leading to intrastromal and intratumoral hypoxia. This loops back onto PSCs, driving their activation and hence generating more fibrosis, which creates a hypoxia–fibrosis cycle. The cycle indirectly contributes to chemoresistance by impairing drug delivery to cancer cells. Moreover, hypoxia is capable of increasing the genetic instability and EMT of pancreatic cancer cells, directly fuelling their chemoresistance. EMT, epithelial-mesenchymal transition.
Seminal proof-of-principle studies have substantiated the crucial influence of the fibrotic hypovascular stroma on GISS and chemoresistance in PDAC using genetically engineered mouse models. One study interrupted hedgehog signaling in the stroma (Olive et al., 2009), while two others enzymatically ablated hyaluronan, the main ECM determinant of the fibrotic stroma (Provenzano et al., 2012; Jacobetz et al., 2013). In all studies, the stroma was dramatically depleted leading to increased intratumoral vascular density and normalization of IFP. Following stromal depletion, administering chemotherapeutics such as gemcitabine or doxorubicin resulted in enhanced intratumoral drug perfusion, rendering tumors vulnerable to cytotoxicity, and hence inhibiting tumor growth and prolonging overall survival (Olive et al., 2009; Provenzano et al., 2012; Jacobetz et al., 2013). However, a Phase II clinical trial following the Olive et al. (2009) study ceased due to increased mortality in the treatment arm. This may have resulted from removal of the tumor-containing fibrotic barrier, encouraging the escape of aggressive clones which underwent metastatic evolution in response to the microenvironment's high selection pressure, however this needs further investigation. Regardless, these landmark studies elegantly showed that tumor-associated stromal collapse in mice is therapeutically effective. Hence, what if we deplete the stroma-producing CA-hPSCs rather than the stroma itself? Since stromal and cancer cells have been shown to contribute to GISS, their depletion can relieve it (Stylianopoulos et al., 2012).
In addition to the hypoxia-induced chemoresistance, PSCs can directly impact the response of cancer cells to chemotherapy. hPSC secretions have been shown to confer a chemoresistant cancer cell phenotype by (i) suppressing H2O2-induced apoptosis and increased survival of pancreatic cancer cells (Vonlaufen et al., 2008a); and (ii) decreased pancreatic cancer cell sensitivity to gemcitabine and radiation therapy (Hwang et al., 2008). These results were supported by Muerkoster et al. (2004), who demonstrated that pancreatic cancer cells co-cultured with PSCs are less sensitive to etoposide. In addition, pancreatic cancer cells cultured with ECM proteins produced by PSCs promoted resistance to 5-fluorouracil (5-FU), cisplatin, and doxorubicin (Miyamoto et al., 2004). However, a limitation in the field is that no studies have examined the influence of PSCs on proteins which protect tumor cells against chemotherapy agents (for example multi-drug resistant drug transporters).
It is important to note that in addition to the impact which stellate cells have on pancreatic cancer chemoresistance, it is now well established that the immune cells also impact on tumor progression and chemoresistance (reviewed in Evans and Costello, 2012; Hamada et al., 2013). Immune cells within the tumor microenvironment also activate PSCs (Figure 1), which may further potentiate the effect of stellate cells on chemoresistance.
Hypoxia and chemoresistance
As mentioned above, hypoxia is a cancer driver that induces EMT, a phenotypic change associated with increased chemoresistance and invasiveness of cancer cells (Castellanos et al., 2013). In addition, hypoxia increases genomic instability by elevating intracellular levels of mutagenic reactive oxygen species and suppressing DNA repair (Bristow and Hill, 2008; Chan and Bristow, 2010). This microenvironment selects for cancer cell clones carrying mutations and phenotypic alterations that improve their survival, increase chemoresistance, and enhance migration out of the inhospitable microenvironment to metastatic sites (Bao et al., 2012).
The cellular effects of hypoxia are primarily initiated by hypoxia-induced factor-1 alpha (HIF-1α), a well-established marker for cells under hypoxic stress (Ke and Costa, 2006). HIF-1α contains an oxygen-responsive degradation domain that is hydroxylated under normoxic conditions by prolyl hydroxylases, leading to ubiquitination and rapid proteasomal degradation (Srinivas et al., 1999; Masson et al., 2001). Low oxygen levels inactivate these prolyl hydroxylases, stabilizing HIF-1α, allowing it to heterodimerize with HIF-1β, forming HIF-1, a transcription factor that regulates numerous genes responsible for the cellular response to hypoxia (Greijer et al., 2005; Semenza et al., 2006). HIF-1α has been detected in both pancreatic cancer cells and surrounding stromal cells, in pancreatic cancer tissue specimens (Shibaji et al., 2003; Ide et al., 2007; Sun et al., 2007; Hoffmann et al., 2008; Schwartz et al., 2010). Positive staining correlated with increased lymph node metastases, decreased apoptotic index, increased intratumoral microvessel density, advanced tumor stage and poorer overall survival (Shibaji et al., 2003; Ide et al., 2007; Sun et al., 2007; Hoffmann et al., 2008; Schwartz et al., 2010).
The influence of hypoxia on pancreatic cancer growth and metastatic spread has been more directly investigated in mouse models of the disease. Buchler et al. (2004) used an orthotopic mouse model of pancreatic cancer that develops distant metastases and measured primary tumor oxygenation using the Eppendorf histograph. The group found a significant correlation between lower tumor oxygenation and increased metastatic score (Buchler et al., 2004). Chang et al. (2011) later reiterated these results using orthotopic implants of pancreatectomy samples from pancreatic cancer patients. The group measured intratumoral hypoxia by immunohistochemistry using the hypoxia marker EF5, and observed a correlation of higher EF5 staining with rapid tumor growth, increased proliferation and increased metastases (Chang et al., 2011). Ide et al. (2007) suggested a potential molecular pathway for the increased invasiveness by associating increased levels of HIF1α with elevated paracrine signaling proteins hepatocyte growth factor and c-met in pancreatic cancer tissue specimens. Importantly, Salnikov et al. (2009) showed that while hypoxia drives EMT in both cancer stem cells and cancer cells, it is the stem cells that gain the more invasive phenotype.
Hypoxia in pancreatic cancer also selects for cancer cell clones with phenotypic changes that confer a survival advantage and consequently enhance their aggressiveness and chemoresistance. Erkan et al. (2005) nicely demonstrated this influence in their investigation of BNIP3, a hypoxia-inducible pro-apoptotic gene, in pancreatic cancer cells and pancreatic cancer patient samples. They firstly observed that downregulation of BNIP3 in patient samples correlated with poorer patient survival. Upon silencing BNIP3 in pancreatic cancer lines in vitro, the group observed a marked increase in chemoresistance to 5-FU and gemcitabine (Erkan et al., 2005). The results suggested that the hypoxic microenvironment imposes selective pressure that favors PC cells with the ability to bypass cell death mechanisms, and as a consequence, that can better resist chemotherapeutics. In addition, Arumugam et al. (2009) have demonstrated the link between hypoxia-induced EMT in pancreatic cancer cells and resistance to 5-FU, gemcitabine and cisplatin in vitro. Tumor cells under hypoxia are also driven to switch their metabolism from an oxygen-consuming pathway to a glycolytic pathway of ATP production (Chen et al., 2009). Such an environment selects for cells with enhanced glycolytic activity, as evidenced by increased levels of glycolytic enzymes in pancreatic cancer tissues and the correlation of glycolytic enzyme polymorphisms with poorer overall survival and enhanced tumor growth (Mikuriya et al., 2007; Dong et al., 2011).
PSCs are also influenced by hypoxia, leading to phenotypic changes that further stimulate pancreatic cancer cells and that facilitate the self-perpetuation of hypoxia. Masamune et al. (2008) investigated the effect of hypoxia on human CA-PSCs in vitro. When cultured under hypoxic conditions, CA-PSCs exhibited increased migration and type I collagen production, allowing perpetuation of fibrosis and hypoxia. Conditioned media from these cells also induced endothelial cell proliferation, migration and angiogenesis via increased PSC-mediated VEGF production, both in vitro and in vivo (Masamune et al., 2008). Spivak-Kroizman et al. (2013) showed that hypoxia also indirectly induces collagen secretion in PSCs, by increasing PC cell sonic hedgehog secretion. Erkan et al. (2009) reconciled the apparent contradictory role of PSCs in perpetuating hypoxia via induction of fibrosis, and in reducing it via pro-angiogenic signals. Using co-culture of PSCs and PC cells in vitro, the group showed that although hypoxia alone drives a proangiogenic PSC phenotype, activated PSCs induce PC cells to increase anti-angiogenic endostatin production, thus perpetuating hypoxia by inhibiting angiogenesis. More recently, Eguchi et al. (2013) demonstrated that hypoxia indirectly increases the invasiveness of PC cells in vitro, by inducing secretion of connective tissue growth factor from PSCs. It is therefore not surprising that many anti-angiogenic therapies result in increased metastases, as they create a hypoxic microenvironment that favors this phenotype (Ebos et al., 2009; Paez-Ribes et al., 2009). The major role of PSCs in establishing and perpetuating the hypoxic microenvironment makes them ideal therapeutic targets in pancreatic cancer. Given PSCs regulate ECM turnover, such an approach would ideally include co-administration of an anti-fibrotic reagent such as pirfenidone, to ensure fibrolysis occurs after targeting PSCs.
Potential stromal targeting agents in the clinic
One reason drug trials have failed in pancreatic cancer is due to inclusion of all patients in a particular trial, regardless of the biological characteristics of an individual's tumor and no consideration of the stroma, which based on the evidence provided above is likely to influence a patient's response to chemotherapy. Understanding how stromal proteins influence drug resistance, drug delivery and patient survival has the potential to help clinicians make better use of available treatments to improve the outcome of patients with pancreatic cancer. Importantly, protein expression profiles of the stroma are now considered strong predictors of patient outcome (Conklin and Keely, 2012). For example, high stromal activity (αSMA positive PSCs) correlates with poor prognosis in pancreatic cancer patients (Erkan et al., 2008). Below we include discussion on a few of the promising stromal targeting therapies currently being investigated in the clinic.
At the 2013 ASCO meeting, a phase 1b study of gemcitabine plus PEGPH20 (PEGylated recombinant human hyularonidase) in patients with stage IV previously untreated pancreatic cancer was presented by Hingorani et al. (2013). PEGPH20 works by enzymatically depleting hyaluronic acid (a glycosaminoglycan), which is extremely abundant in pancreatic cancers and thought to contribute to the high interstitial fluid pressure (Thompson et al., 2010). The study separated out the nine patients with high levels of stromal hyaluronan and five of these patients showed a partial response to PEGPH20 treatment (response rate 56%). In addition, the PEGPH20 appeared to be well tolerated. The scientific and clinical community eagerly await the results of Phase II studies.
A Phase 3 trial is currently testing gemcitabine in combination with TH-302 in patients with locally advanced unresectable or metastatic pancreatic adenocarcinoma. As outlined above, pancreatic cancer is highly hypoxic and TH-302 is a chemotherapeutic agent which is selectively activated in a hypoxic tumor microenvironment (Sun et al., 2012).
Angiotensin II type I receptor inhibitors have also been explored as potential therapeutics to inhibit PSC activity. For example, Yamada et al. (2003) demonstrated that oral administration of candesartan (a widely used angiotensin II type-I receptor inhibitor) decreased ECM production and αSMA expression (activated PSC marker) in a rat chronic pancreatitis model. Furthermore, a retrospective clinical study suggested that pancreatic cancer patients treated with angiotensin II type 1 receptor inhibitors in combination with gemcitabine may have improved clinical outcome (Nakai et al., 2010). To expand on these findings the same authors recently completed a multicenter phase II clinical trial (35 patients with advanced pancreatic cancer) to examine whether patients treated with candesartan in combination with gemcitabine would have improved survival. The treatment regime was tolerated using moderate doses of candesartan with gemcitabine, but failed to demonstrate any significant clinical activity (Nakai et al., 2013).
More recently, in an elegant and comprehensive study by Chauhan et al. (2013), the angiotensin II Type 1 receptor inhibitor losartan was found to reduce stromal collagen and hyaluronan production, as well as decrease the number of αSMA positive PSCs in orthotopic pancreatic tumors. As a result, the treatment of tumors with losartan reduced GISS and increased vascular perfusion. The overall outcome was reduced hypoxia and increased sensitivity to chemotherapy agents. This promising pre-clinical study has now moved forward to a clinical trial in pancreatic cancer (NCT01821729). Notably, losartan has been shown to have a higher tumor penetration when compared to other angiotensin II Type 1 receptor inhibitors including candesartan. Indeed, inadequate tumor penetration of candesartan may in part explain its modest effect on chemosensitivity in the recent clinical trial (Nakai et al., 2013).
A recent article by Kozono et al. (2013), demonstrated that an anti-fibrotic agent pirfenidone decreased tumor growth/metastases and increased drug sensitivity in a mouse orthotopic model of pancreatic cancer (co-injection of PSCs and tumor cells into the pancreas). Pirfenidone is currently approved for the treatment of idiopathic pulmonary fibrosis in Europe and based on evidence by Kozono et al. (2013), it may be a promising anti-fibrotic agent which can be repurposed to reduce the activity of PSCs in pancreatic cancer.
Finally, Nab-paclitaxel or Abraxane® (Abraxis Bioscience) has generated great interest as a novel therapeutic for pancreatic cancer. A recent phase III MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) trial demonstrated that the addition of Nab-paclitaxel with gemcitabine was able to significantly improve the median survival of metastatic pancreatic cancer patients (8.5 months) when compared to gemcitabine treated only arm (6.7 months) and reduce toxicities (neuropathy and neutropenia) commonly associated with the cremaphor formulation used to dissolve paclitaxel thereby, allowing for a higher paclitaxel dose to be delivered to the patient (Von Hoff et al., 2013). Interestingly, several studies have also shown that Nab-paclitaxel alone or in combination with gemcitabine depletes PCSs and desmoplastic stroma (Von Hoff et al., 2011; Alvarez et al., 2013). Moreover, it is hypothesized that the albumin-bound Nab-paclitaxel may selectively accumulate in the pancreatic stroma via its binding to secreted protein acidic and rich in cysteine (SPARC) matricellular glycoprotein which binds albumin and is overexpressed in tumor stroma. Indeed, high SPARC expression has been correlated to poor survival outcome and has been suggested as a possible predictive biomarker for Nab-paclitaxel (Von Hoff et al., 2011; Alvarez et al., 2013). However, a recent study by Neesse et al. (2013) showed that the effects of Nab-paclitaxel were largely dose-dependent and that SPARC expression in the tumor stroma did not influence drug accumulation in a pancreatic cancer mouse model. However, they did report increased Nab-paclitaxel concentrations in plasma suggesting a potential interaction with circulating SPARC. Therefore, it is possible that high circulating SPARC may aid in increased drug retention and tissue delivery. Future studies will be needed to evaluate tissue and plasma SPARC expression as a predictive biomarker for Nab-paclitaxel.
Conclusions
As outlined above, a significant body of work corroborates the notion that CA-hPSCs are not relegated to a bystander role in PDAC chemoresistance, but instead, they are critical in driving it via both direct and indirect mechanisms. This occurs by way of PSC generated fibrosis creating a hypoxic microenvironment that directly fuels the chemoresistance of cancer cells. Concurrently, fibrosis impairs effective drug perfusion to cancer cells, which indirectly augments chemoresistance. Furthermore, there is mounting evidence that not only is stromal depletion therapeutically possible in mice with pancreatic tumors, but it also facilitates the delivery of chemotherapeutics to pancreatic tumors. Notably, stromal depletion entails a reversion of fibrosis as well as a decline in the number of cells generating fibrosis because reducing the number of activated PSCs in turn would diminish collagen production. Thus, in light of their major contribution to fibrosis in pancreatic cancer as well as their highly proliferative capacity, cancer-associated PSCs are ideal candidates for the identification of stromal-related therapeutic targets that may act as additional targets for potentiating existing chemotherapeutics (Figure 3).
Figure 3.
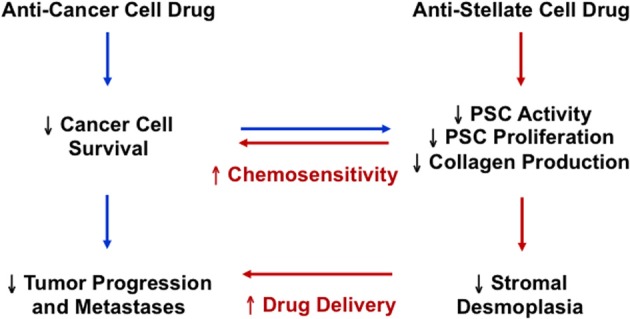
The potential therapeutic implication of targeting both pancreatic cancer cells and pancreatic stellate cells in pancreatic cancer.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Phoebe A. Phillips, Joshua A. McCarroll, Maria Kavallaris, and David Goldstein are supported by grants from the National Health and Medical Research Council (NHMRC), Cancer Council New South Wales (Maria Kavallaris), Cure Cancer Australia Foundation Grant (Phoebe A. Phillips), Cancer Institute New South Wales Career Development Fellowship (Joshua A. McCarroll, George Sharbeen), NHMRC Career Development Fellowship (Phoebe A. Phillips) and NHMRC Senior Research Fellowship (Maria Kavallaris). Phoebe A. Phillips, Joshua A. McCarroll, and David Goldstein are also supported by a University of New South Wales Goldstar Award.
Glossary
Abbreviations
- CA-hPSC
cancer-associated human pancreatic stellate cell
- COX-2
cyclooxygenase-2
- CTGF
connective tissue growth factor
- PSC
pancreatic stellate cell
- ECM
extracellular matrix
- EMMPRIN
extracellular matrix metalloproteinase inducer
- EMT
epithelial-mesenchymal transition
- ET-1
endothelin 1
- IFP
interstitial fluid pressure
- IL
interleukin
- PDAC
pancreatic ductal adenocarcinoma
- GISS
growth-induced solid stress
- MMPs
matrix metalloproteinases
- PDGF
platelet-derived growth factor
- TGFβ
transforming growth factor β
- TNFα
tumor necrosis factor α
- TRAIL
TNF-related apoptosis-inducing ligand HIF-1α, hypoxia-induced factor-1 alpha.
References
- Alvarez R., Musteanu M., Garcia-Garcia E., Lopez-Casas P. P., Megias D., Guerra C., et al. (2013). Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 109, 926–933 10.1038/bjc.2013.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki H., Ohnishi H., Hama K., Ishijima T., Satoh Y., Hanatsuka K., et al. (2006a). Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am. J. Physiol. Cell Physiol. 290, C1100–C1108 10.1152/ajpcell.00465.2005 [DOI] [PubMed] [Google Scholar]
- Aoki H., Ohnishi H., Hama K., Shinozaki S., Kita H., Yamamoto H., et al. (2006b). Existence of autocrine loop between interleukin-6 and transforming growth factor-beta1 in activated rat pancreatic stellate cells. J. Cell. Biochem. 99, 221–228 10.1002/jcb.20906 [DOI] [PubMed] [Google Scholar]
- Apte M. V., Haber P. S., Applegate T. L., Norton I. D., McCaughan G. W., Korsten M. A., et al. (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 43, 128–133 10.1136/gut.43.1.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte M. V., Haber P. S., Darby S. J., Rodgers S. C., McCaughan G. W., Korsten M. A., et al. (1999). Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 44, 534–541 10.1136/gut.44.4.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte M. V., Park S., Phillips P. A., Santucci N., Goldstein D., Kumar R. K., et al. (2004). Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 29, 179–187 10.1097/00006676-200410000-00002 [DOI] [PubMed] [Google Scholar]
- Apte M. V., Pirola R. C., Wilson J. S. (2006). Battle-scarred pancreas: role of alcohol and pancreatic stellate cells in pancreatic fibrosis. J. Gastroenterol. Hepatol. 21(Suppl. 3), S97–S101 10.1111/j.1440-1746.2006.04587.x [DOI] [PubMed] [Google Scholar]
- Arumugam T., Ramachandran V., Fournier K. F., Wang H., Marquis L., Abbruzzese J. L., et al. (2009). Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 69, 5820–5828 10.1158/0008-5472.can-08-2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem M. G., Schunemann M., Ramadani M., Siech M., Beger H., Buck A., et al. (2005). Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921 10.1053/j.gastro.2004.12.036 [DOI] [PubMed] [Google Scholar]
- Bao B., Azmi A. S., Ali S., Ahmad A., Li Y., Banerjee S., et al. (2012). The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim. Biophys. Acta. 1826, 272–296 10.1016/j.bbcan.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N., Depinho R. A. (2002). Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2, 897–909 10.1038/nrc949 [DOI] [PubMed] [Google Scholar]
- Bristow R. G., Hill R. P. (2008). Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8, 180–192 10.1038/nrc2344 [DOI] [PubMed] [Google Scholar]
- Buchler P., Reber H. A., Lavey R. S., Tomlinson J., Buchler M. W., Friess H., et al. (2004). Tumor hypoxia correlates with metastatic tumor growth of pancreatic cancer in an orthotopic murine model. J. Surg. Res. 120, 295–303 10.1016/j.jss.2004.02.014 [DOI] [PubMed] [Google Scholar]
- Castellanos J. A., Merchant N. B., Nagathihalli N. S. (2013). Emerging targets in pancreatic cancer: epithelial-mesenchymal transition and cancer stem cells. Onco. Targets. Ther. 6, 1261–1267 10.2147/ott.s34670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan N., Bristow R. G. (2010). “Contextual” synthetic lethality and/or loss of heterozygosity: tumor hypoxia and modification of DNA repair. Clin. Cancer Res. 16, 4553–4560 10.1158/1078-0432.ccr-10-0527 [DOI] [PubMed] [Google Scholar]
- Chang Q., Jurisica I., Do T., Hedley D. W. (2011). Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 71, 3110–3120 10.1158/0008-5472.can-10-4049 [DOI] [PubMed] [Google Scholar]
- Chauhan V. P., Martin J. D., Liu H., Lacorre D. A., Jain S. R., Kozin S. V., et al. (2013). Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 4, 2516 10.1038/ncomms3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Cairns R., Papandreou I., Koong A., Denko N. C. (2009). Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS ONE 4:e7033 10.1371/journal.pone.0007033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin M. W., Keely P. J. (2012). Why the stroma matters in breast cancer: insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adh. Migr. 6, 249–260 10.4161/cam.20567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Tang H., Hess K. R., Abbruzzese J. L., Li D. (2011). Glucose metabolism gene polymorphisms and clinical outcome in pancreatic cancer. Cancer 117, 480–491 10.1002/cncr.25612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos J. M., Lee C. R., Cruz-Munoz W., Bjarnason G. A., Christensen J. G., Kerbel R. S. (2009). Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 10.1016/j.ccr.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi D., Ikenaga N., Ohuchida K., Kozono S., Cui L., Fujiwara K., et al. (2013). Hypoxia enhances the interaction between pancreatic stellate cells and cancer cells via increased secretion of connective tissue growth factor. J. Surg. Res. 181, 225–233 10.1016/j.jss.2012.06.051 [DOI] [PubMed] [Google Scholar]
- Erkan M., Adler G., Apte M. V., Bachem M. G., Buchholz M., Detlefsen S., et al. (2012). StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 61, 172–178 10.1136/gutjnl-2011-301220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan M., Kleeff J., Esposito I., Giese T., Ketterer K., Buchler M. W., et al. (2005). Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 24, 4421–4432 10.1038/sj.onc.1208642 [DOI] [PubMed] [Google Scholar]
- Erkan M., Michalski C. W., Rieder S., Reiser-Erkan C., Abiatari I., Kolb A., et al. (2008). The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 6, 1155–1161 10.1016/j.cgh.2008.05.006 [DOI] [PubMed] [Google Scholar]
- Erkan M., Reiser-Erkan C., Michalski C. W., Deucker S., Sauliunaite D., Streit S., et al. (2009). Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 11, 497–508 10.1593/neo.81618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan M., Reiser-Erkan C., Michalski C. W., Kleeff J. (2010). Tumor microenvironment and progression of pancreatic cancer. Exp. Oncol. 32, 128–131 [PubMed] [Google Scholar]
- Evans A., Costello E. (2012). The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 3:270 10.3389/fphys.2012.00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S. M., Koch C. J. (2003). Prognostic significance of tumor oxygenation in humans. Cancer Lett. 195, 1–16 10.1016/S0304-3835(03)00012-0 [DOI] [PubMed] [Google Scholar]
- Feig C., Gopinathan A., Neesse A., Chan D. S., Cook N., Tuveson D. A. (2012). The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276 10.1158/1078-0432.ccr-11-3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R., Brigstock D. R. (2005). Connective tissue growth factor (CCN2) in rat pancreatic stellate cell function: integrin alpha5beta1 as a novel CCN2 receptor. Gastroenterology 129, 1019–1030 10.1053/j.gastro.2005.06.067 [DOI] [PubMed] [Google Scholar]
- Gao Z., Wang X., Wu K., Zhao Y., Hu G. (2010). Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology 10, 186–193 10.1159/000236012 [DOI] [PubMed] [Google Scholar]
- Greijer A. E., Van Der Groep P., Kemming D., Shvarts A., Semenza G. L., Meijer G. A., et al. (2005). Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J. Pathol. 206, 291–304 10.1002/path.1778 [DOI] [PubMed] [Google Scholar]
- Hamada S., Masamune A., Shimosegawa T. (2013). Alteration of pancreatic cancer cell functions by tumor-stromal cell interaction. Front. Physiol. 4:318 10.3389/fphys.2013.00318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M. (2010). Pancreatic cancer. N. Engl. J. Med. 362, 1605–1617 10.1056/NEJMra0901557 [DOI] [PubMed] [Google Scholar]
- Hingorani S. R., Harris W. P., Beck J. T., Berdov B. A., Wagner S. A., Pshevlotsky E. M. (2013). A phase Ib study of gemcitabine plus PEGPH20 (pegylated recombinant human hyaluronidase) in patients with stage IV previously untreated pancreatic cancer. J. Clin. Oncol. 31:4010 [Google Scholar]
- Hoffmann A. C., Mori R., Vallbohmer D., Brabender J., Klein E., Drebber U., et al. (2008). High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 10, 674–679 10.1593/neo08292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang R. F., Moore T., Arumugam T., Ramachandran V., Amos K. D., Rivera A., et al. (2008). Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926 10.1158/0008-5472.can-07-5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T., Kitajima Y., Miyoshi A., Ohtsuka T., Mitsuno M., Ohtaka K., et al. (2007). The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signaling. Ann. Surg. Oncol. 14, 2600–2607 10.1245/s10434-007-9435-3 [DOI] [PubMed] [Google Scholar]
- Jacobetz M. A., Chan D. S., Neesse A., Bapiro T. E., Cook N., Frese K. K., et al. (2013). Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 62, 112–120 10.1136/gutjnl-2012-302529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Abiatari I., Kong B., Erkan M., De Oliveira T., Giese N. A., et al. (2009). Pancreatic islet and stellate cells are the main sources of endocrine gland-derived vascular endothelial growth factor/prokineticin-1 in pancreatic cancer. Pancreatology 9, 165–172 10.1159/000178888 [DOI] [PubMed] [Google Scholar]
- Ke Q., Costa M. (2006). Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 70, 1469–1480 10.1124/mol.106.027029 [DOI] [PubMed] [Google Scholar]
- Kikuta K., Masamune A., Watanabe T., Ariga H., Itoh H., Hamada S., et al. (2010). Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 403, 380–384 10.1016/j.bbrc.2010.11.040 [DOI] [PubMed] [Google Scholar]
- Komar G., Kauhanen S., Liukko K., Seppanen M., Kajander S., Ovaska J., et al. (2009). Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin. Cancer Res. 15, 5511–5517 10.1158/1078-0432.ccr-09-0414 [DOI] [PubMed] [Google Scholar]
- Koong A. C., Mehta V. K., Le Q. T., Fisher G. A., Terris D. J., Brown J. M., et al. (2000). Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 48, 919–922 10.1016/S0360-3016(00)00803-8 [DOI] [PubMed] [Google Scholar]
- Kordes C., Brookmann S., Haussinger D., Klonowski-Stumpe H. (2005). Differential and synergistic effects of platelet-derived growth factor-BB and transforming growth factor-beta1 on activated pancreatic stellate cells. Pancreas 31, 156–167 10.1097/01.mpa.0000168222.05591.a0 [DOI] [PubMed] [Google Scholar]
- Kozono S., Ohuchida K., Eguchi D., Ikenaga N., Fujiwara K., Cui L., et al. (2013). Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 73, 2345–2356 10.1158/0008-5472.can-12-3180 [DOI] [PubMed] [Google Scholar]
- Li J., Wientjes M. G., Au J. L. (2010). Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J. 12, 223–232 10.1208/s12248-010-9181-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamune A., Kikuta K., Watanabe T., Satoh K., Hirota M., Shimosegawa T. (2008). Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G709–G717 10.1152/ajpgi.90356.2008 [DOI] [PubMed] [Google Scholar]
- Masson N., Willam C., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001). Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 20, 5197–5206 10.1093/emboj/20.18.5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P., Phillips P., Fahmy R., Korsten M., Pirola R., Wilson J., et al. (2002). Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. 50, 535–541 10.1136/gut.50.4.535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michl P., Gress T. M. (2013). Current concepts and novel targets in advanced pancreatic cancer. Gut. 62, 317–326 10.1136/gutjnl-2012-303588 [DOI] [PubMed] [Google Scholar]
- Mikuriya K., Kuramitsu Y., Ryozawa S., Fujimoto M., Mori S., Oka M., et al. (2007). Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int. J. Oncol. 30, 849–855 10.3892/ijo.30.4.849 [DOI] [PubMed] [Google Scholar]
- Miyamoto H., Murakami T., Tsuchida K., Sugino H., Miyake H., Tashiro S. (2004). Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 28, 38–44 10.1097/00006676-200401000-00006 [DOI] [PubMed] [Google Scholar]
- Muerkoster S., Wegehenkel K., Arlt A., Witt M., Sipos B., Kruse M. L., et al. (2004). Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 64, 1331–1337 10.1158/0008-5472.CAN-03-1860 [DOI] [PubMed] [Google Scholar]
- Nakai Y., Isayama H., Ijichi H., Sasaki T., Sasahira N., Hirano K., et al. (2010). Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br. J. Cancer 103, 1644–1648 10.1038/sj.bjc.6605955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y., Isayama H., Ijichi H., Sasaki T., Takahara N., Ito Y., et al. (2013). A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Invest. New Drugs 31, 1294–1299 10.1007/s10637-013-9972-5 [DOI] [PubMed] [Google Scholar]
- Neesse A., Frese K. K., Chan D. S., Bapiro T. E., Howat W. J., Richards F. M., et al. (2013). SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut. [Epub ahead of print]. 10.1136/gutjnl-2013-305559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neesse A., Michl P., Frese K. K., Feig C., Cook N., Jacobetz M. A., et al. (2011). Stromal biology and therapy in pancreatic cancer. Gut. 60, 861–868 10.1136/gut.2010.226092 [DOI] [PubMed] [Google Scholar]
- Ohnishi N., Miyata T., Ohnishi H., Yasuda H., Tamada K., Ueda N., et al. (2003). Activin A is an autocrine activator of rat pancreatic stellate cells: potential therapeutic role of follistatin for pancreatic fibrosis. Gut. 52, 1487–1493 10.1136/gut.52.10.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., McIntyre D., Honess D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 10.1126/science.1171362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary M. B., Lugea A., Lowe A. W., Pandol S. J. (2007). The pancreatic stellate cell: a star on the rise in pancreatic diseases. J. Clin. Invest. 117, 50–59 10.1172/jci30082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Vinals F., et al. (2009). Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231 10.1016/j.ccr.2009.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P. (2012). Pancreatic stellate cells and fibrosis, in Pancreatic Cancer and Tumor Microenvironment, eds Grippo P. J., Munshi H. G. (Trivandrum: Transworld Research Network; ). [PubMed] [Google Scholar]
- Phillips P. A., McCarroll J. A., Park S., Wu M. J., Pirola R., Korsten M., et al. (2003a). Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut. 52, 275–282 10.1136/gut.52.2.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P. A., Wu M. J., Kumar R. K., Doherty E., McCarroll J. A., Park S., et al. (2003b). Cell migration: a novel aspect of pancreatic stellate cell biology. Gut. 52, 677–682 10.1136/gut.52.5.677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P. P., Cuevas C., Chang A. E., Goel V. K., Von Hoff D. D., Hingorani S. R. (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429 10.1016/j.ccr.2012.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi S., Maisonneuve P., Lowenfels A. B. (2009). Epidemiology of pancreatic cancer: an overview. Nat. Rev. Gastroenterol. Hepatol. 6, 699–708 10.1038/nrgastro.2009.177 [DOI] [PubMed] [Google Scholar]
- Salnikov A. V., Kusumawidjaja G., Rausch V., Bruns H., Gross W., Khamidjanov A., et al. (2009). Cancer stem cell marker expression in hepatocellular carcinoma and liver metastases is not sufficient as single prognostic parameter. Cancer Lett. 275, 185–193 10.1016/j.canlet.2008.10.015 [DOI] [PubMed] [Google Scholar]
- Schneider E., Schmid-Kotsas A., Zhao J., Weidenbach H., Schmid R. M., Menke A., et al. (2001). Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am. J. Physiol. Cell Physiol. 281, C532–C543 [DOI] [PubMed] [Google Scholar]
- Schneiderhan W., Diaz F., Fundel M., Zhou S., Siech M., Hasel C., et al. (2007). Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J. Cell Sci. 120, 512–519 10.1242/jcs.03347 [DOI] [PubMed] [Google Scholar]
- Schwartz D. L., Bankson J. A., Lemos R., Jr., Lai S. Y., Thittai A. K., He Y., et al. (2010). Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 9, 2057–2067 10.1158/1535-7163.mct-09-0768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L., Shimoda L. A., Prabhakar N. R. (2006). Regulation of gene expression by HIF-1. Novartis Found. Symp. 272, 2–8 discussion: 8–14, 33–16. 10.1002/9780470035009.ch2 [DOI] [PubMed] [Google Scholar]
- Shek F. W., Benyon R. C., Walker F. M., McCrudden P. R., Pender S. L., Williams E. J., et al. (2002). Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 160, 1787–1798 10.1016/S0002-9440(10)61125-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibaji T., Nagao M., Ikeda N., Kanehiro H., Hisanaga M., Ko S., et al. (2003). Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 23, 4721–4727 [PubMed] [Google Scholar]
- Spivak-Kroizman T. R., Hostetter G., Posner R., Aziz M., Hu C., Demeure M. J., et al. (2013). Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 73, 3235–3247 10.1158/0008-5472.can-11-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas V., Zhang L. P., Zhu X. H., Caro J. (1999). Characterization of an oxygen/redox-dependent degradation domain of hypoxia-inducible factor alpha (HIF-alpha) proteins. Biochem. Biophys. Res. Commun. 260, 557–561 10.1006/bbrc.1999.0878 [DOI] [PubMed] [Google Scholar]
- Stylianopoulos T., Martin J. D., Chauhan V. P., Jain S. R., Diop-Frimpong B., Bardeesy N., et al. (2012). Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 15101–15108 10.1073/pnas.1213353109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H. C., Qiu Z. J., Liu J., Sun J., Jiang T., Huang K. J., et al. (2007). Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int. J. Oncol. 30, 1359–1367 10.3892/ijo.30.6.1359 [DOI] [PubMed] [Google Scholar]
- Sun J. D., Liu Q., Wang J., Ahluwalia D., Ferraro D., Wang Y., et al. (2012). Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin. Cancer Res. 18, 758–770 10.1158/1078-0432.ccr-11-1980 [DOI] [PubMed] [Google Scholar]
- Szakacs G., Paterson J. K., Ludwig J. A., Booth-Genthe C., Gottesman M. M. (2006). Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219–234 10.1038/nrd1984 [DOI] [PubMed] [Google Scholar]
- Thompson C. B., Shepard H. M., O'Cconnor P. M., Kadhim S., Jiang P., Osgood R. J., et al. (2010). Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 9, 3052–3064 10.1158/1535-7163.mct-10-0470 [DOI] [PubMed] [Google Scholar]
- Tod J., Jenei V., Thomas G., Fine D. (2013). Tumor-stromal interactions in pancreatic cancer. Pancreatology 13, 1–7 10.1016/j.pan.2012.11.311 [DOI] [PubMed] [Google Scholar]
- Vincent A., Herman J., Schulick R., Hruban R. H., Goggins M. (2011). Pancreatic cancer. Lancet 378, 607–620 10.1016/s0140-6736(10)62307-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D. D., Ervin T., Arena F. P., Chiorean E. G., Infante J., Moore M., et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 10.1056/NEJMoa1304369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D. D., Ramanathan R. K., Borad M. J., Laheru D. A., Smith L. S., Wood T. E., et al. (2011). Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J. Clin. Oncol. 29, 4548–4554 10.1200/jco.2011.36.5742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonlaufen A., Joshi S., Qu C., Phillips P. A., Xu Z., Parker N. R., et al. (2008a). Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093 10.1158/0008-5472.can-07-2477 [DOI] [PubMed] [Google Scholar]
- Vonlaufen A., Phillips P. A., Xu Z., Goldstein D., Pirola R. C., Wilson J. S., et al. (2008b). Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 68, 7707–7710 10.1158/0008-5472.can-08-1132 [DOI] [PubMed] [Google Scholar]
- Vonlaufen A., Phillips P. A., Yang L., Xu Z., Fiala-Beer E., Zhang X., et al. (2010). Isolation of quiescent human pancreatic stellate cells: a promising in vitro tool for studies of human pancreatic stellate cell biology. Pancreatology 10, 434–443 10.1159/000260900 [DOI] [PubMed] [Google Scholar]
- Wang Z., Li Y., Ahmad A., Banerjee S., Azmi A. S., Kong D., et al. (2011). Pancreatic cancer: understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 8, 27–33 10.1038/nrgastro.2010.188 [DOI] [PubMed] [Google Scholar]
- Watanabe I., Hasebe T., Sasaki S., Konishi M., Inoue K., Nakagohri T., et al. (2003). Advanced pancreatic ductal cancer: fibrotic focus and beta-catenin expression correlate with outcome. Pancreas 26, 326–333 10.1097/00006676-200305000-00003 [DOI] [PubMed] [Google Scholar]
- Wolfgang C. L., Herman J. M., Laheru D. A., Klein A. P., Erdek M. A., Fishman E. K., et al. (2013). Recent progress in pancreatic cancer. CA Cancer J. Clin. 63, 318–348 10.3322/caac.21190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Vonlaufen A., Phillips P. A., Fiala-Beer E., Zhang X., Yang L., et al. (2010). Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 177, 2585–2596 10.2353/ajpath.2010.090899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T., Kuno A., Masuda K., Ogawa K., Sogawa M., Nakamura S., et al. (2003). Candesartan, an angiotensin II receptor antagonist, suppresses pancreatic inflammation and fibrosis in rats. J. Pharmacol. Exp. Ther. 307, 17–23 10.1124/jpet.103.053322 [DOI] [PubMed] [Google Scholar]
- Zalatnai A., Molnar J. (2007). Review. Molecular background of chemoresistance in pancreatic cancer. In Vivo 21, 339–347 [PubMed] [Google Scholar]