

Abstract
Reducing the enduring vulnerability to relapse is a therapeutic goal in treating drug addiction. Studies with animal models of drug addiction show a marked increase in extrasynaptic glutamate in the core subcompartment of the nucleus accumbens (NAcore) during reinstated drug seeking. However, the synaptic mechanisms linking drug-induced changes in extrasynaptic glutamate to relapse are poorly understood. Here, we discovered impaired glutamate elimination in rats extinguished from heroin self-administration that leads to spillover of synaptically released glutamate into the nonsynaptic extracellular space in NAcore and investigated whether restoration of glutamate transport prevented reinstated heroin seeking. Through multiple functional assays of glutamate uptake and analyzing NMDA receptor-mediated currents, we show that heroin self-administration produced long-lasting downregulation of glutamate uptake and surface expression of the transporter GLT-1. This downregulation was associated with spillover of synaptic glutamate to extrasynaptic NMDA receptors within the NAcore. Ceftriaxone restored glutamate uptake and prevented synaptic glutamate spillover and cue-induced heroin seeking. Ceftriaxone-induced inhibition of reinstated heroin seeking was blocked by morpholino-antisense targeting GLT-1 synthesis. These data reveal that the synaptic glutamate spillover in the NAcore results from reduced glutamate transport and is a critical pathophysiological mechanism underling reinstated drug seeking in rats extinguished from heroin self-administration.
Keywords: extrasynaptic glutamate receptor, glutamate spillover, glutamate uptake, heroin self-administration, nucleus accumbens, relapse
Introduction
Relapse to using addictive drugs is linked to activating prefrontal cortex projections to the nucleus accumbens (Goldstein and Volkow, 2002). Neuroimaging studies reveal that presenting drug-associated cues to drug-dependent humans induces metabolic activation of anterior cingulate, orbital cortex, and nucleus accumbens that is often correlated with the intensity of reported desire to relapse to drug use (Goldstein and Volkow, 2002). Similarly, animal models of relapse show that activation of prelimbic prefrontal cortex (PL) glutamatergic projections into the core subcompartment of the accumbens (NAcore) is associated with cue-, stress-, or drug-induced reinstatement of drug seeking (Kalivas, 2009). Importantly, evidence supporting a role for glutamate transmission in the accumbens has accrued in drug-dependent humans and in animal models regardless of the class of addictive drug.
Given the role of glutamate transmission in the nucleus accumbens, much research into the neurobiology of drug relapse has focused on measuring glutamate release and cellular neuroadaptations at excitatory synapses in the accumbens (Kalivas, 2009; Wolf, 2010; Lüscher and Malenka, 2011). Using microdialysis or biosensor measurement of extracellular glutamate in the NAcore, a common finding is that reinstated drug seeking in animal models is associated with a marked increase in extracellular glutamate, largely derived from PL afferents (McFarland et al., 2003; McFarland et al., 2004; LaLumiere and Kalivas, 2008; Gass et al., 2011; Gipson et al., 2013b). This includes studies of reinstated alcohol, cocaine, heroin, and nicotine seeking induced by Pavlovian cues, stress, or acute, noncontingent drug administration. Further supporting a pathogenic role for increasing extracellular glutamate during relapse, the increased extracellular glutamate is not measured in the reinstated seeking of natural reinforcer (McFarland et al., 2003).
The access of synaptic glutamate to the extrasynaptic space is gated by glial glutamate transporters that are often strategically expressed adjacent to the synaptic cleft (Minelli et al., 2001). This tripartite synaptic architecture (i.e., astrocytic process adjacent to the synapse plus the presynaptic and postsynaptic neurons; see Fig. 5) minimizes glutamate spillover, thereby permitting efficient synaptic signaling by rapidly eliminating released glutamate and localizing synaptic glutamate transmission to the synaptic site of release (Perea et al., 2009; Rusakov et al., 2011). The glial excitatory amino acid transporter 2 (GLT-1) is particularly efficient at reducing synaptic glutamate spillover (Danbolt, 2001). Notably, compounds that increase expression of the GLT-1 (i.e., N-acetylcysteine, ceftriaxone, and propentofylline) can inhibit the reinstatement of drug seeking (Knackstedt et al., 2009; Sari et al., 2009; Knackstedt et al., 2010; Fischer-Smith et al., 2012; Trantham-Davidson et al., 2012; Fischer et al., 2013; Reissner et al., 2014).
Figure 5.

Model of how downregulated GLT-1 promotes reinstated heroin seeking due to increase synaptic glutamate spillover to extrasynaptic glutamate receptors. The size of the symbol indicates increased or reduced stimulation by glutamate (mGluR5 or GluN2B) or glutamate transport (GLT-1). The dashed line and smaller arrowhead signify reduced glutamate relative to the solid line and larger arrowhead in response to the same amount of synaptically released glutamate.
Here, we used in vivo and in vitro assays of glutamate uptake to show that heroin use induces an enduring downregulation of GLT-1 in the NAcore that leads to spillover of synaptically released glutamate that may be pathogenic for cue-induced reinstatement of heroin seeking.
Materials and Methods
Animals and surgery
All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Assessment and Accreditation of Laboratory Animal Care. Male Sprague Dawley rats (250 g on arrival; Charles River Laboratories) were individually housed in a temperature- and humidity-controlled environment with a 12 h dark/light cycle (lights on 6:00 P.M.). Experiments were conducted during the rats' dark cycle. Rats received food ad libitum until 1 d before behavioral training, after which food restriction procedures (20 g/d of rat chow) were implemented and maintained throughout the duration of the experiment. Rats were allowed 1 week to acclimate to the vivarium before inducing anesthesia and implanting jugular cannula. The rats used for no-net-flux had microdialysis guide cannulas implanted into the NAcore immediately after catheter implantation into the jugular vein. The surgical details have been described in a previous study (Shen et al., 2011).
Heroin self-administration procedures
All self-administration experiments occurred in standard operant chambers with two retractable levers, a house light, and a cue light and tone generator (Med Associates). During 3 h sessions on 14 consecutive days, rats were trained to press the active lever on a fixed ratio 1 schedule with 20 s timeout for an infusion of heroin-hydrochloride (100 μg/infusion for days 1–2, 50 μg/4 s infusion for days 3–4, 25 μg/infusion for days 5–14). The descending dosing protocol was used to facilitate the acquisition of heroin self-administration and to increase the number of reinstated lever presses (Fuchs and See, 2002). Heroin was kindly provided by the National Institute on Drug Abuse. Saline control rats were yoked to heroin self-administering rats and thereby received noncontingent infusions of saline in the same temporal pattern as heroin infusions were self-administered. Concurrent with the drug infusion, a cue tone (2900 Hz) and cue light immediately above the active lever turned on. After 14 d of self-administration, rats began extinction training. Active lever presses produced no drug infusion or light/tone cues. For cue-induced reinstatement, no injection was given, but the session program was identical to the self-administration session program (i.e., cue tone and light turned on with an active lever press). Rats did not receive intravenous drug infusions during either mode of reinstatement trial.
Systemic treatment of ceftriaxone and intra-NAcore delivery of GLT-1 morphilinos
Ceftriaxone (Nova Plus) was dissolved in saline and administered in a dose of 200 mg/kg (Knackstedt et al., 2010; Trantham-Davidson et al., 2012). Heroin-trained or yoked saline controls were administered daily treatment with ceftriaxone starting on day 8 of extinction training and continuing for 7 d, after which time rats entered a cue-induced reinstatement session. Twenty-four hours after reinstatement testing, these animals were killed for uptake assay or whole-cell recordings. Knock down of GLT-1 in the NAcore was performed using a vivo-morpholino strategy that we showed previously to yield a 56 ± 10% (n = 7) reduction in GLT-1 expression (Reissner et al., 2012). Two morpholino constructs (Gene Tools) were used: GLT-1 specific morpholino (5′-TGTTGGCACCCTCGGTTGATGCCAT-3′) and a random sequence as a negative control. Morpholino stock solutions were diluted 50-fold in sterile PBS (10 mm Na2HPO4, 2 mm KH2PO4, 137 mm NaCl, 2.7 mm KCl) and injected into the NAcore through 26 ga cannulae (Plastics One) implanted bilaterally into NAcore at +1.5 A/P, +1.8 M/L, −5.5 D/V relative to bregma (Paxinos and Watson, 2005). A sham microinjection was performed on day 4 of extinction training. On days 5, 6, and 7 of extinction training, 1.0 μl of morpholino solution was infused at 0.5 μl/min using 33 ga microinjectors (Plastics One) that extended into the NAcore 2 mm below the base of the cannula. Microinjectors were left in place for 60 s to allow for diffusion. Our previous study showed that this protocol reduced GLT-1 expression in the dissected NAcore subregion at 7 d after the last microinjection (Reissner et al., 2012), but it also remains possible that diffusion of the morpholino altered protein expression in tissue adjacent to the NAcore.
Whole-cell recordings in acute brain slices
Rats were anesthetized with ketamine HCl (87.5 mg/kg, i.p.) and brains were rapidly removed into ice-cold aCSF. Coronal slices (230 μm thick) containing the NAcore were cut in ice-cold aCSF with a vibratome and incubated in 32°C aCSF containing the following (in mm): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 25 NaHCO3, and 11 glucose for at least 45 min before returning to room temperature. Picrotoxin (50 μm) was added to block GABAA receptor-mediated IPSCs during recording. Cells were visualized using infrared-differential interference contrast optics. To assess excitatory synaptic transmission, neurons were voltage clamped at −80 mV using a Multiclamp 700A amplifier (Molecular Devices). Patch electrodes (1.5–2 MΩ) were filled with internal solution containing the following (in mm): 124 cesium methanesulfonate, 10 NaCl, 1 MgCl2, 10 HEPES, 1 EGTA, 1 QX-314, 2 MgATP, and 0.3 MgGTP, 275–285 mOsm, pH 7.2–7.4. Series resistance (<20 MΩ) and input resistance were monitored online with a 5 mV depolarizing step (100 ms) given with each stimulus. Afferents were stimulated at 0.05 Hz by a concentric bipolarectrode placed ∼150 μm dorsomedial to the recorded neurons. The intensity of stimulation was chosen to evoke a 500–600 pA EPSC at −80 mV clamping voltage. Glutamatergic afferents were electrically stimulated and the NMDA currents were pharmacologically isolated by bath applying the AMPAR antagonist CNQX (10 μM) at +40 mV clamping voltage. Data were filtered at 2 kHz, digitized at 5 kHz, and collected and analyzed using AxoGraph X software (AxoGraph Scientific). NMDA current decay was quantified by the time elapsed from the EPSC peak to 50% peak amplitude using AxoGraph X.
[3H]-glutamate uptake assay
Crude membrane fractionation was used for determining the Na+-dependent glutamate uptake (Salvatore et al., 2012). Briefly, rats underwent live decapitation and the NAcore was rapidly dissected. Briefly, fresh NAcore tissue was homogenized by tissue grinders in cold 0.32 m sucrose buffer containing 10 mm HEPES and 1 mm EDTA, pH 7.4, and then centrifuged at 1000 × g for 10 min to remove nuclei and large debris. The resulting supernatants were concentrated at 15,000 × g for 20 min to pellet the crude membrane fraction. The pellet was subsequently suspended with Kreb's-Ringer's solution phosphate buffer (KRP) containing the following (in mm): 140 NaCl, 1.2 CaCl2, KH2PO4, 5 HEPES, 10 glucose, and 1 MgCl2. For Na+-independent glutamate uptake, NaCl in KRP buffer was replaced by 140 mm choline. Glutamate uptake measurements were initiated by adding [3H] glutamate (0.24 μm, 50 Ci/mmol; PerkinElmer) in the presence of unlabeled glutamate (10 μm) in a final volume of 250 μl of KRP buffer. After incubation at 37°C for 15 min, the uptake was terminated by washing the slices with ice-cold nonradioactive choline-containing buffer. Crude membranes were then solubilized using 1% SDS and the level of radioactivity was determined using a liquid scintillation counter. Protein content in each sample was measured using the BCA assay. Uptake of [3H] glutamate that occurred in choline-containing KRP buffer was subtracted from that measured in the presence of NaCl to determine Na+-dependent uptake component. Counts per minute were converted to uptake/milligram of protein/15 min.
No-net-flux microdialysis
For the NAcore dialysis experiments, cannulas (20 ga; Plastics One) were implanted bilaterally into NAcore at +1.7 mm A/P, +2.5 mm M/L at 6° angle, −5.5 mm D/V relative to bregma (Paxinos and Watson, 2005) simultaneously with insertion of the indwelling jugular catheter. On the night before testing, the microdialysis probes were inserted into the NAcore, with the internal cannula extending 2 mm beyond the end of the guide cannula. To estimate the basal level of extracellular glutamate, the no-net-flux procedure was conducted by changing the perfusate to dialysis buffer containing 2.5, 5, or 10 μm glutamate. Three samples were collected at 20 min intervals for each concentration. The net flux of the analyte diffusing into or out of the probe was determined by subtracting the concentration of the analyte added to the buffer from the concentration of the analyte in the samples. A plot of the analyte flux at each concentration added to the buffer yields an estimate of basal levels (y = 0; or the point at which there is no-net flux of analyte into or out of the probe) and elimination (slope, or clearance of the analyte from the probe). The details of the microdialysis procedure and quantification of glutamate have been described in a previous study (Baker et al., 2003).
Biotinylation assay
The NAcore was dissected and sliced into prism-shaped sections (200 μm) with a McIlwain tissue chopper (Vibratome). The tissue was incubated for 30 min in 300 μl of PBS containing 1 mg/ml Sulfo-NHS-Biotin (Pierce) at 4°C with gentle shaking. Reaction was quenched by adding ice-cold 100 mm glycine in PBS. The tissues were washed twice with ice-cold 100 mm glycine in PBS and then homogenized by sonication in 300 μl of 1% SDS in RIPA lysis buffer (Pierce) containing protease inhibitor mixture (Roche). After incubating for 30–60 min on ice, the protein-containing solution was centrifuged at 10,000 × g for 10 min at 4°C to remove insoluble material. The supernatant was added with 100 μl of NeutrAvidin agarose resin and incubated for 2 h at 4°C with gentle rotation. After washing with ice-cold PBS twice, the biotinylated protein was eluted by 50 μl of loading buffer (1% SDS and 50 mm DTT) with heating at 80°C for 5 min. The proteins of interest (GLT-1 and xCT) in the biotinylated fraction could be detected with immunoblotting. Antibodies used for immunoblotting include mouse monoclonal antibodies against the GLT-1 (BD Biosciences), α-tubulin (Santa Cruz Biotechnology), and rabbit polyclonal antibodies against xCT (Szumlinski et al., 2004).
Enzyme-based glutamate biosensor
R2 ceramic-based microelectrode arrays (MEAs) were prepared for in vivo recordings as described previously (Burmeister et al., 2002; Quintero et al., 2011; Onifer et al., 2012). Briefly, platinum sites were electroplated with an m-phenylenediamine dihydrochloride size exclusion layer (Acros Organics) to block potential interfering analytes such as ascorbic acid (AA), catecholamines, and indoleamines (Burmeister et al., 2002; Hascup et al., 2007; Zhang et al., 2008). After a 24 h incubation, recoding sites were coated with a glutamate oxidase (GluOx) enzyme solution (U.S. Biological) containing a final concentration of 1% BSA (Sigma-Aldrich), 0.125% glutaraldehyde (Sigma-Aldrich), and 1% GluOx. The GluOx enzyme is required for measurement of glutamate because it metabolizes glutamate to α-ketoglutarate, which is then converted to the reporter molecule H2O2. When a potential of +0.7 V versus a silver/silver chloride reference electrode is applied to the MEA, H2O2 is oxidized, resulting in the transfer of two electrons to the platinum recording surface. The resulting change in current was amplified and digitized by fast analytical sensing technology (FAST16 MKIII recording system; Quanteon).
Electrode calibration.
MEAs were calibrated to determine their sensitivity to glutamate and selectivity against AA using constant potential amperometry with a FAST16 MKIII system, as described previously (Burmeister et al., 2002; Quintero et al., 2011; Onifer et al., 2012). Briefly, the MEA was submerged into 40 ml of a continuously stirred solution of 0.05 m PBS titrated to pH 7.4 and allowed to reach a stable baseline for ∼30 min before calibrating. Phosphate buffer temperature was maintained at 37°C using a circulating water bath (Gaymar Industries). Aliquots of freshly made 20 mm AA and 20 mm glutamate were used to obtain final concentrations of 250 μm AA and 20, 40, and 60 μm Glu for all calibrations. Selectivity ratios for glutamate over AA were calculated in addition to the limit of detection (LOD) and linearity (R2) for all glutamate MEAs. Reported as mean ± SEM, the MEAs had average selectivity ratios of 50 ± 22 to 1, limit of detections of 0.9 ± 0.64 μm, and R2 values of 0.9952 ± 0.003 (n = 7 MEAs). The MEAs were also tested to compare the recording capability among the platinum recording sites using H2O2 (8.8 μm, final concentration) as a test substance. Electrodes that did not respond to H2O2, had R2 values <0.99, or had LODs >5 μm were excluded. After calibration, MEAs were fitted with single-barrel glass capillaries bumped to an inner tip diameter of ∼10 μm. Pulled pipettes were embedded in modeling clay fixed to the printed circuit board above the MEA ceramic tip. Molten sticky wax was applied to the embedded pipette to stabilize the assembly and to prevent movement during the recording. Attachment of the pipette was performed under a dissection scope to place the tip of the pipette above the glutamate sensitive sites 50–100 μm from the surface of the MEA (Burmeister et al., 2002; Michael and Borland, 2007). After attachment to the MEA, the pipette was filled with freshly made KCl solution containing 70 mm KCl, 79 mm NaCl, and 2.5 mm Ca2Cl, pH 7.4, attached to the head stage, and connected to a Picospritzer III (Parker Instruments).
Electrode implantation.
Rats were anesthetized with 30% urethane at 5 mg/kg and placed in a stereotaxic apparatus (David Kopf Instruments) fitted with a 37°C with a heating pad. After removal of the scalp tissue, rats underwent a 4 mm × 4 mm craniotomy to remove the skull above the NAcore on both the right and left side, leaving bregma intact. Rats were then implanted with a glutamate-selective MEA-KCl pipette assembly into either the left or right NAcore (AP, +1.8 mm; ML, ±1.5 mm; DV, −7 mm vs bregma) based on the coordinates from Paxinos and Watson (2005). A small hole was drilled at the back of the skull above the cerebellum and used for placement of the silver/silver chloride reference electrode.
Stimulation protocol.
Once the glutamate MEA was lowered to the NAcore, rats underwent a minimum 30 min acclimation period to establish a stable baseline recording. After the equilibration/baseline period, pressure ejections of 70 mm KCl solution were given using the Picospritzer III microinjection dispensing system. Approximately 150 nl of KCl solution was pressure ejected adjacent to the microelectrode to induce depolarization and subsequent release of glutamate in the NAcore. KCl ejection resulted in reproducible glutamate peaks detected by MEAs. After each KCl ejection, recordings were allowed to return to baseline for at least 5 min before repeating the procedure. This procedure was repeated 5 times for each recording site. The location of each recording was verified after each experiment using cresyl violet staining.
Data analysis.
The FAST16 MKIII recording system saved amperometric data, time, and experimenter-mediated ejection marks. All data traces from microelectrode arrays were analyzed with FAST Analysis software (Jason Burmeister Consulting), a program written and compiled in MATLAB (The MathWorks). The FAST Analysis software was used to determine first uptake rate (μm/s) and Tc (T20–T60; mm/s). Given that glutamate uptake follows a first order decay rate, the decay of the glutamate signal can be fitted to the slope of the linear regression of the natural log transformation of the data over time; when this value is multiplied by the maximum amplitude of the signal, glutamate uptake rate in micromoles per second is obtained (Michael and Borland, 2007). Tc (T20–T60) is defined as the change in amplitude between T20 and T60 divided by the change in time from T20 to T60. This rate has units of millimoles per second and the parameters T20 and T60 are the seconds between the maximum amplitude and the time when the level has decreased to 20% and 60% from maximum, respectively, as follows: Tc = (C20 − C60/T20 − T60) All data were passed through a low stringency wavelet low-pass filter using the Daubechies wavelet to remove high-frequency noise.
Statistics
The data were statistically analyzed using a two-tailed unpaired t test when two groups were compared. For comparing multiple measurements in the same experiment, the data were analyzed using a two-way ANOVA. Bonferroni's or Tukey's tests were applied for multiple comparisons and p < 0.05 was considered statistically significant. All statistical tests are conducted using the SPSS or Prism (GraphPad) software.
Results
Heroin self-administration reduces glutamate uptake via GLT-1
We trained rats to self-administer a descending dose of intravenous heroin for 14 d, followed by a 14 d period of daily extinction training (Fig. 1A). Figure 1B shows the rates of active lever pressing for the animals trained to self-administer heroin and the yoked saline controls that were used for the biochemical, electrophysiological, and behavioral data described in our study. Figure 1B also shows the mean number of heroin infusions. In the first experiment, rats were killed and the NAcore was examined for changes in expression and function of the glial glutamate transporter EAAT2 (GLT-1; responsible for eliminating >90% of synaptically released glutamate; Danbolt, 2001). Using a biotinylation strategy to quantify surface expression, we found that GLT-1 surface protein was reduced by ∼25% (t(25) = 2.13, p = 0.043; normalized to saline optical density; Fig. 1C) and was accompanied by an ∼20% reduction (t(10) = 2.59, p = 0.027) in the in vitro Na+-dependent uptake of 3H-glutamate into NAcore tissue slices in heroin (4582 ± 152 cpm/mg protein/min) compared with yoked-saline animals (5387 ± 197 cpm/mg protein/min; Fig. 1D). Because previous studies with rats trained to self-administer cocaine showed parallel reductions in GLT-1 and the activity of the cystine-glutamate exchanger (Kau et al., 2008; Knackstedt et al., 2010), we also blotted for surface expression of the catalytic subunit of the exchanger (xCT) and quantified Na+-independent uptake of 3H-glutamate into NAcore tissue slices, which largely results from transport by the cystine-glutamate exchanger (Bridges et al., 2012). Figure 1C shows that, in contrast to cocaine, extinction from heroin self-administration was associated with an increase in surface expression of xCT (t(18) = 2.32, p = 0.032), although this increase may not reflect upregulated activity of the cystine-glutamate exchanger because there was no difference in Na+-independent 3H-glutamate uptake (t(10) = 0.24, p = 0.814) between heroin (199 ± 8 cpm/mg protein/min) and yoked-saline animals (201 ± 4 cpm/mg protein/min; Fig. 1D). Whole-cell lysate values for GLT-1 and xCT were not significantly different in heroin compared with yoked saline animals (t(12) = 1.99, p = 0.070 for GLT-1; t(11) = 1.41, p = 0.1869 for xCT; Fig. 1C).
Figure 1.

Heroin self-administration induces an enduring decrease in glutamate uptake. A, Training protocol for heroin self-administration and yoked-saline control. All measurements were performed after 2 weeks of extinction training. B, Self-administration and extinction active lever pressing of all yoked-saline and heroin-trained rats used in all data shown in Figures 1, 2, 3, and 4. C, Decreased surface expression of GLT-1 (normalized to saline optical density) and increased surface expression of xCT without changes in total protein from whole-cell lysate. Representative Western blots for GLT-1 and xCT are shown on the right. S, Yoked-saline; H, heroin-trained. Open arrowhead refers to the band quantified. D, Na+-dependent [3H]-glutamate uptake (normalized to saline CPM) in NAcore tissue slices is reduced in heroin compared with yoked-saline animals, whereas Na+-independent 3H-glutamate uptake did not differ between the two treatment groups. The number of determinations is shown in the bar. *p < 0.05 comparing heroin with yoked saline.
Heroin self-administration reduces glutamate uptake in vivo
To assess more directly glutamate in the nonsynaptic extracellular space and to measure effects of heroin self-administration/extinction on glutamate clearance in vivo, we inserted glutamate biosensors into the NAcore and measured the rate of elimination of extrasynaptic glutamate released by a depolarizing pulse of K+. The release of glutamate by K+ is calcium dependent and thought to reflect largely synaptic release, although calcium-dependent release by glia cannot be excluded (Cousin et al., 1993; Longuemare and Swanson, 1997). The decay time required for glutamate to return to basal levels is controlled largely by the elimination of extracellular glutamate by glutamate transport (Day et al., 2006). Figure 2A shows that the in vivo rate of elimination of glutamate released by applying K+ (70 mm; 150 nl) adjacent to a biosensor in the NAcore was markedly reduced in heroin compared with yoked-saline rats (t(9) = 2.39, p = 0.041). In a second in vivo experiment, a dialysis probe was inserted into the NAcore and no-net-flux microdialysis of glutamate was used to estimate the concentration and elimination of extracellular glutamate. We found that, whereas basal glutamate was not altered, heroin rats showed evidence of altered elimination of glutamate, as indicated by an increased slope in heroin compared with yoked-saline subjects (t(11) = 2.90, p = 0.015; Fig. 2B).
Figure 2.
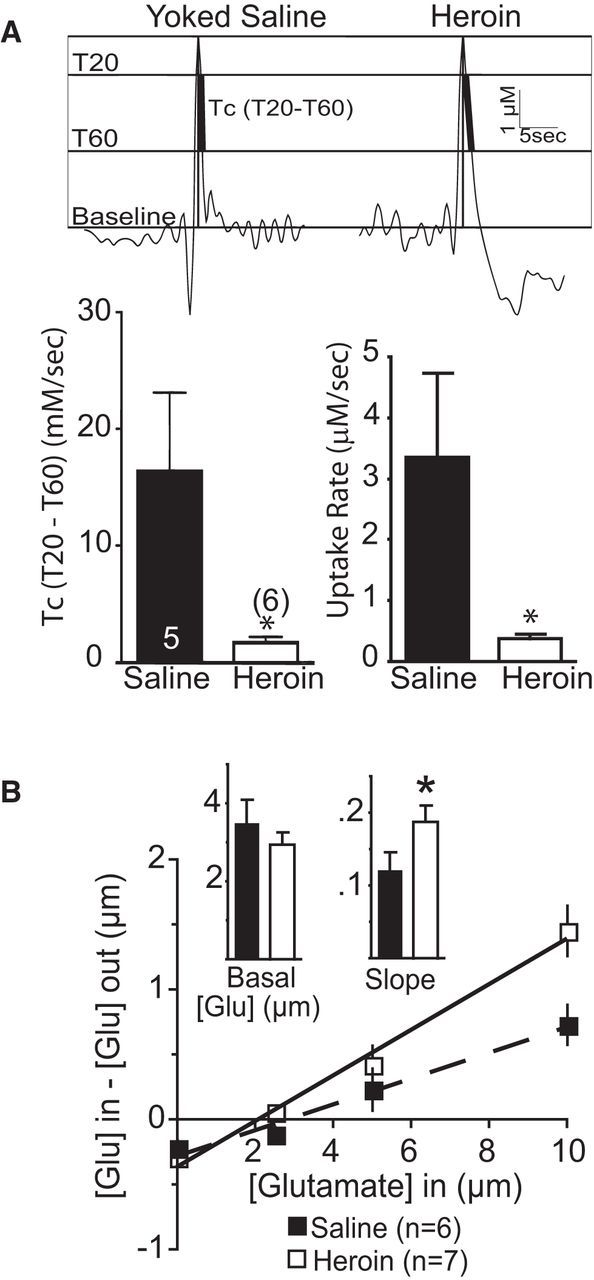
Slower rate of in vivo glutamate uptake in heroin rats. A, Top, Representative traces of K+-induced glutamate release showing where measurements were made at 20% and 60% of peak value. Bottom, Comparison between heroin and yoked-saline using two different estimates of glutamate uptake. B, No-net-flux microdialysis in the NAcore shows no difference in basal glutamate, but greater slope, indicating altered glutamate elimination. *p < 0.05 comparing heroin with yoked saline.
Heroin self-administration increases glutamate spillover onto NMDA receptors
To determine whether the heroin-induced downregulation of GLT-1 produced synaptic glutamate spillover, we made whole-cell patch recordings of NMDAR-mediated EPSCs from NAcore medium spiny neurons and took advantage of the fact that, when glutamate escapes the synaptic cleft, it gains access to distant extrasynaptic NMDAR that prolongs the EPSC decay time (Arnth-Jensen et al., 2002). To induce synaptic glutamate spillover, we triggered burst stimulation at 100 Hz (Fig. 3A) and showed an increased decay of NMDA EPSCs in drug-naive NAcore tissue slices (Fig. 3A,B). To mimic heroin-induced downregulation of GLT-1, we inhibited glutamate transport by bath applying the glutamate transport inhibitor TBOA. Inhibition of glutamate uptake did not change the amplitude of evoked NMDA EPSCs (two-way ANOVA revealed no effect of TBOA treatment F(1,6) = 0.254, p = 0.632 or an interaction between TBOA and number of pulses F(3,18) = 0.756, p = 0.533). Notably, TBOA significantly augmented the burst-stimulation-induced increase in EPSC decay time after 4 or 8 pulses (F(3,36) = 18.49, p < 0.001; Fig. 3C). Based on these findings, we used a change in NMDA EPSC decay time (t1/2) as an index of synaptic glutamate spillover resulting from reduced glutamate uptake and hypothesized that the reduced level and function of GLT-1 in the NAcore of heroin animals would manifest as increased NMDA EPSC decay t1/2 compared with yoked-saline rats. Unlike the TBOA experiment, heroin self-administration increased the NMDA decay t1/2 after the delivery of a single pulse (F(3,96) = 8.96, p < 0.001; Fig. 3D). This was likely due to heroin increasing the surface expression of GluN2B-containing NMDA receptors that convey increased opening time to NMDA channels (Shen et al., 2011). When the data were normalized to the net increase in t1/2 after a single pulse, heroin rats still showed a significant increase in t1/2 after 8 pulses (F(3,96) = 8.96, p < 0.001), indicating potentiated glutamate spillover compared with yoked-saline animals.
Figure 3.

Heroin self-administration increases spillover of synaptic glutamate. A, Stimulation protocols used for triggering NMDAR-mediated EPSC when NAcore spiny neurons were at +40 mV holding voltage. The interpulse and interburst intervals were 10 ms and 30 s, respectively. B, Representative original and normalized traces of evoked NMDA EPSC (1, 2, 4, and 8 pulses measured after AMPA current blockade) before and after TBOA incubation. The red line indicates the decay kinetics fit to a regression exponential curve. N is shown as #cells/#rats. C, Averaged t1/2 decay of NMDA EPSC and net increase of t1/2 decay of NMDA EPSC evoked by incremental pulse number was increased by blocking glutamate uptake with TBOA (10 μm). N is shown as #cells/#rats. D, Decay of NMDA EPSCs triggered by burst stimuli was slower in heroin compared with saline-yoked rats. Top, Example normalized traces of NMDA EPSCs. Bottom, Averaged t1/2 decay of NMDA EPSC and net increase of t1/2 decay. *p < 0.05 comparing heroin with yoked-saline, 2-way ANOVA and Bonferroni's test.
Restoring GLT-1 function prevented reinstated heroin seeking
We used daily ceftriaxone treatment to restore glutamate transport by GLT-1 in heroin animals (Rothstein et al., 2005). Figure 4A shows that NAcore tissue slices obtained from heroin-extinguished rats treated with daily ceftriaxone had elevated Na+-dependent 3H-glutamate uptake (t(9) = 2.43, p = 0.038) compared with heroin animals pretreated (6409 ± 236 cpm/mg protein/min) with daily saline (5608 ± 223 cpm/mg protein/min). Ceftriaxone did not alter Na+-independent uptake, indicating no change (t(9) = 0.388, p = 0.707) in cystine-glutamate exchange (253 ± 13 and 245 ± 14 cpm/mg protein/min; heron and saline group, respectively). Commensurate with restoring glutamate uptake, ceftriaxone abolished the increase in glutamate spillover seen in the NAcore of heroin compared with yoked-saline rats also injected with daily ceftriaxone (F(3,75) = 0.36, p = 0.784; Fig. 4B). In addition, if the data shown in Figure 4B are analyzed together with the data in Figure 3D, ceftriaxone did not alter the net increase in t1/2 decay in yoked saline animals at any pulse number, but significantly restored the net increase in t1/2 decay in heroin-extinguished rats at 8 pulses (two-way ANOVA with repeated measures over pulses, interaction F(9,228) = 3.01, p = 0.002, followed by a Tukey's test for multiple comparisons).
Figure 4.
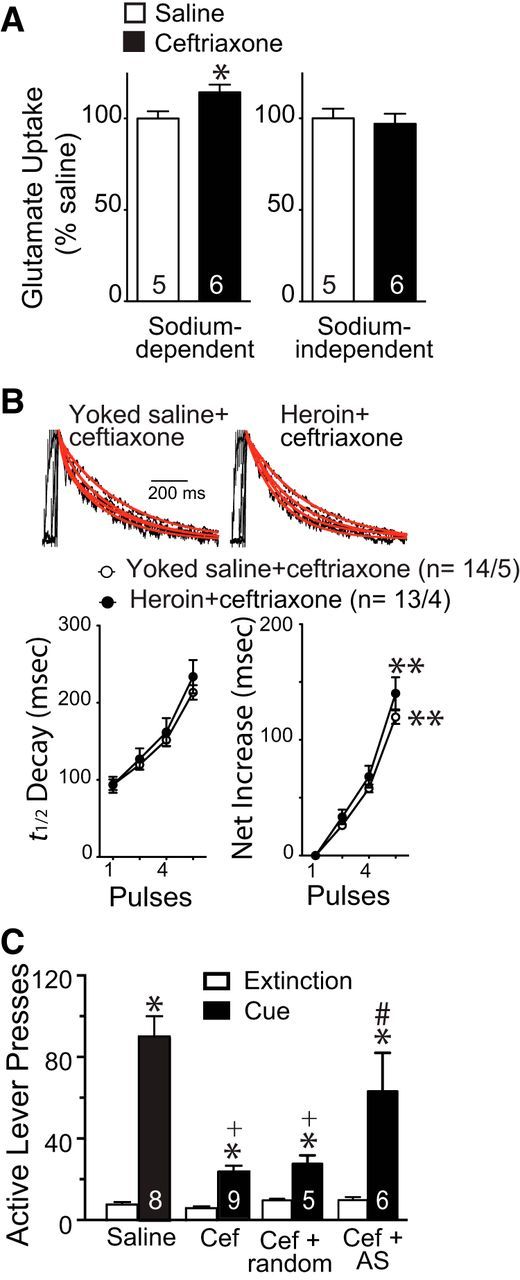
Ceftriaxone restores glutamate uptake and spillover and inhibits cue-induced reinstatement of heroin-seeking. A, Repeated ceftriaxone (7 daily injections of 100 mg/kg, i.p.) during extinction increased Na+-dependent glutamate uptake in heroin-trained rats (normalized to saline CPM). *p < 0.05 comparing ceftriaxone- with saline-treated heroin rats. B, No significant difference in the decay of NMDA EPSCs between yoked saline and heroin rats after ceftriaxone treatment. Top, Representative normalized traces of NMDA EPSCs. Bottom, Averaged t1/2 decay of NMDA EPSC and net increase of t1/2 decay. N is shown as #cells/#rats. **p < 0.05 comparing the heroin data in Figure 3D with the saline data in Figure 3D and the ceftriaxone data in Figure 4B, two-way ANOVA with repeated measures over pulses followed by a Tukey's test for multiple comparisons. C, Ceftriaxone (Cef) prevented cue-induced reinstatement of active lever pressing and antisense (AS) suppression of ceftriaxone-induced increases in GLT-1 prevented ceftriaxone-mediated inhibition of reinstated heroin seeking. *p < 0.05 comparing cue-induced active lever with extinction pressing using a 2-way ANOVA with repeated measures over extinction/cue followed by a Bonferroni's post hoc test; +p < 0.05 comparing Cef with saline; #p < 0.05 comparing Cef + AS with Cef + random.
Importantly, ceftriaxone markedly reduced the reinstatement of heroin seeking induced by the presentation of a light/tone cue that was previously paired with heroin infusions during self-administration training (Fig. 4C). Because ceftriaxone has biological actions in addition to increasing GLT-1 synthesis and expression (Rothstein et al., 2005), we used a morpholino-antisense strategy to inhibit the translation of GLT-1 mRNA that reduces GLT-1 protein by 56 ± 10% (n = 7; Reissner et al., 2012). Intra-NAcore administration of antisense targeting GLT-1 mRNA prevented ceftriaxone from reducing reinstated heroin seeking compared with treatment with a random-sequence oligonucleotide (Fig. 4C). The behavioral data shown in Figure 4C were analyzed using a 2-way ANOVA with repeated measures over extinction/cue (interaction F(3,24) = 9.68, p < 0.001), followed by a Bonferroni post hoc test.
Discussion
We show here that, after extinction from heroin self-administration, there is a significant reduction in the capacity of the NAcore to eliminate extracellular glutamate. The reduced uptake of glutamate via the glutamate transporter GLT-1 in heroin animals caused spillover of synaptically released glutamate into the nonsynaptic extracellular space that could be detected as a decrease in the rate of elimination of K+-induced release in vivo or by an increase in the decay of NMDA currents in vitro. Importantly, restoring GLT-1 and glutamate uptake with ceftriaxone inhibited glutamate spillover and the capacity of conditioned cues to reinstate heroin seeking, suggesting the downregulation of GLT-1 as a potentially critical pathogenic adaptation underpinning heroin relapse.
Downregulation of GLT-1 and glutamate spillover
Glutamate uptake via GLT-1 is strategically positioned on glial processes adjacent to the synaptic cleft to maximize the rapid elimination of synaptically released glutamate (Minelli et al., 2001; Cholet et al., 2002). Accordingly, downregulated GLT-1 in heroin-trained rats or by a pharmacological transporter antagonist reduces the elimination of synaptically released glutamate, thereby allowing glutamate to more freely diffuse into the adjacent nonsynaptic space. Using an animal model of heroin relapse, we show that the enduring downregulation of GLT-1 in the NAcore of rats extinguished from heroin self-administration was associated with increased synaptic spillover measured by an increased time constant of NMDA current decay. Supporting this mechanism, we showed that pharmacologically inhibiting GLT-1 with TBOA produced a similar increase in the duration of NMDA currents, and restoration of heroin-induced reductions in GLT-1 with ceftriaxone normalized NMDA currents. Interestingly, inhibiting glutamate uptake did not alter the amplitude of NMDA currents, likely due to the fact that the magnitude of NMDA current depends on the amount of synaptic NMDA receptors localized to terminal regions. In addition, a previous study indicated that pharmacological inhibition of GLT-1 preferentially affects the duration of glutamate uptake as opposed to the extent of glutamate release (Bortz et al., 2013). It is important to note that the paradigm used in our study always involved extinction training after heroin self-administration, making it possible that the changes in glutamate transport and spillover may involve both heroin use and extinction training. However, arguing that extinction training may not be relevant, it was recently shown that after 40 d of abstinence without extinction training from cocaine self-administration, GLT-1 was selectively downregulated in the NAcore (Fischer-Smith et al., 2012).
There is an emerging number of animal studies of other drugs of abuse, including nicotine, alcohol, and cocaine, that strongly points to downregulated glutamate uptake and the resulting increased extrasynaptic glutamate in NAcore as a shared, potentially critical drug-induced neuroadaptation in creating vulnerability to relapse (Madayag et al., 2007; Knackstedt et al., 2010; Sari et al., 2011; Gipson et al., 2013b). This is associated with spillover of synaptic glutamate into the extrasynaptic space during increased synaptic activity, as indicated by in vivo measurements of extrasynaptic glutamate made with microdialysis or biosensors (McFarland et al., 2003; Madayag et al., 2007; LaLumiere and Kalivas, 2008; Gass et al., 2011; Gipson et al., 2013b). In some studies, the synaptic origin of the in vivo rise in glutamate was validated by blocking the reinstatement-associated increase with tetrodotoxin or pharmacological inhibition of prefrontal cortex glutamatergic afferents to the NAcore (McFarland et al., 2003; LaLumiere and Kalivas, 2008). Our ex vivo data demonstrate for the first time that heroin self-administration reduced glutamate uptake via downregulating GLT-1 and that downregulated GLT-1 facilitated synaptic glutamate spillover that accessed extrasynaptic NMDA receptors, leading to prolonged NMDA current decay. Although the prolonged NMDA current decay is consistent with activating NMDA receptors more distant from the synaptic release site, it is possible that these receptors could be in adjacent synaptic release sites and in the extrasynaptic space. Regardless, augmented synaptic spillover in the NAcore of rats extinguished from heroin would be expected to increase the radius of activation of glutamate receptors from the release site compared with control animals.
Consequences of spillover of synaptic glutamate
The reduced clearance of glutamate in rats extinguished from heroin self-administration or other addictive drugs will prolong stimulation of NMDA and AMPA receptors within the cleft of the synaptic release site and also increase spillover of glutamate to stimulate receptors that are largely located outside of the synaptic cleft, including mGluR5 and NMDA receptors containing the GluN2B subunit (Fig. 5). Although prolonged activation of synaptic ionotropic glutamate receptors would be limited by receptor desensitization (Nahum-Levy et al., 2001; Hansen et al., 2007), an increased diffusion radius of glutamate outside of the synaptic cleft would be expected to recruit increased numbers of glutamate receptors, in particular the higher affinity mGlu and NMDA receptors. Consistent with a role for this latter process in drug relapse, antagonists of mGluR5 are widely reported to reduce the reinstatement of alcohol, cocaine, or nicotine seeking (Bäckström et al., 2004; Bespalov et al., 2005; Schroeder et al., 2008; Gass et al., 2009; Kumaresan et al., 2009; Sinclair et al., 2012; Wang et al., 2013). Although not as widely studied, there is also strong evidence that antagonizing GluN2B-containing NMDA receptors inhibits opioid, nicotine, and alcohol reinstatement (Ma et al., 2007; Wang et al., 2010; Shen et al., 2011; Gipson et al., 2013b). Moreover, reinstated cocaine, heroin, and nicotine seeking is paralleled by a marked transient potentiation of glutamatergic synapses in the NAcore (Anderson et al., 2008; Shen et al., 2011; Gipson et al., 2013a; Gipson et al., 2013b) and, for heroin-trained animals, this is prevented by pharmacological inhibition or genetic downregulation of GluN2B-containing NMDA receptors in the NAcore (Shen et al., 2011). Both mGluR5 and NMDA receptor stimulation increase intracellular concentrations of calcium that can signal synaptic potentiation (Gnegy, 2000; Yashiro and Philpot, 2008; Anwyl, 2009), posing a shared calcium signaling mechanism whereby the synaptic glutamate spillover arising from downregulated GLT-1 can potentiate excitatory neurotransmission and promote the reinstatement of cocaine, heroin, and nicotine seeking.
Clinical implications
The model in Figure 5 illustrates how the downregulation of GLT-1 promotes stimulation of extrasynaptic mGluR5 and/or GluN2B and thereby facilitates reinstatement. Figure 5 also offers a number of potential pharmacotherapeutic targets for treating addiction. Importantly, the literature outlined in the preceding paragraph indicates that this model may be applicable to animal models of relapse using a wide variety of chemical classes of addictive drug, including alcohol, cocaine, heroin, and nicotine. Three potentially druggable targets shown in Figure 5 are restoration of glutamate transport via increasing GLT-1 and antagonizing mGluR5 and/or GluN2B receptors. Here, we demonstrate that upregulating GLT-1 inhibits cue-induced reinstatement of heroin seeking, which has also been shown for alcohol, cocaine, and nicotine seeking (Sari et al., 2009; Knackstedt et al., 2010; Ramirez-Niño et al., 2013). There are currently drug discovery efforts for other diseases at all three of these molecular targets (Gogas, 2006; Berry-Kravis et al., 2009; Berry et al., 2013) and our data, combined with the literature, support that targeting these three pharmacotherapeutic mechanisms, either separately or in concert, might be useful to broadly treat substance use disorders.
Footnotes
This work was supported by the National Institutes of Health (Grants DA003906, DA012513, and DA015369 grants to P.W.K.) and the Natural Science Foundation of China (Grants 81271472 and 81221002 to S.H.W.). We thank Arthur Riegel for technical support and Charles Thomas for help with training animals.
The authors declare no competing financial interests.
References
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11:344–353. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735–740. doi: 10.1016/j.neuropharm.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Bäckström P, Bachteler D, Koch S, Hyytiä P, Spanagel R. mGluR5 antagonist MPEP reduces ethanol-seeking and relapse behavior. Neuropsychopharmacology. 2004;29:921–928. doi: 10.1038/sj.npp.1300381. [DOI] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, Felsenstein D, Krivickas L, David WS, Vriesendorp F, Pestronk A, Caress JB, Katz J, Simpson E, Rosenfeld J, Pascuzzi R, Glass J, Rezania K, Rothstein JD, Greenblatt DJ, Cudkowicz ME Northeast ALS Consortium. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One. 2013;8:e61177. doi: 10.1371/journal.pone.0061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bespalov AY, Dravolina OA, Sukhanov I, Zakharova E, Blokhina E, Zvartau E, Danysz W, van Heeke G, Markou A. Metabotropic glutamate receptor (mGluR5) antagonist MPEP attenuated cue- and schedule-induced reinstatement of nicotine self-administration behavior in rats. Neuropharmacology. 2005;49:167–178. doi: 10.1016/j.neuropharm.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Bortz DM, Mikkelsen JD, Bruno JP. Localized infusions of the partial alpha 7 nicotinic receptor agonist SSR180711 evoke rapid and transient increases in prefrontal glutamate release. Neuroscience. 2013;255:55–67. doi: 10.1016/j.neuroscience.2013.09.047. [DOI] [PubMed] [Google Scholar]
- Bridges R, Lutgen V, Lobner D, Baker DA. Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev. 2012;64:780–802. doi: 10.1124/pr.110.003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119:163–171. doi: 10.1016/S0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Cholet N, Pellerin L, Magistretti PJ, Hamel E. Similar perisynaptic glial localization for the Na+, K+-ATPase alpha 2 subunit and the glutamate transporters GLAST and GLT-1 in the rat somatosensory cortex. Cereb Cortex. 2002;12:515–525. doi: 10.1093/cercor/12.5.515. [DOI] [PubMed] [Google Scholar]
- Cousin MA, Nicholls DG, Pocock JM. Flunarizine inhibits both calcium-dependent and -independent release of glutamate from synaptosomes and cultured neurones. Brain Res. 1993;606:227–236. doi: 10.1016/0006-8993(93)90989-Z. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Day BK, Pomerleau F, Burmeister JJ, Huettl P, Gerhardt GA. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J Neurochem. 2006;96:1626–1635. doi: 10.1111/j.1471-4159.2006.03673.x. [DOI] [PubMed] [Google Scholar]
- Fischer KD, Houston AC, Rebec GV. Role of the major glutamate transporter GLT1 in nucleus accumbens core versus shell in cue-induced cocaine-seeking behavior. J Neurosci. 2013;33:9319–9327. doi: 10.1523/JNEUROSCI.3278-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Smith KD, Houston AC, Rebec GV. Differential effects of cocaine access and withdrawal on glutamate type 1 transporter expression in rat nucleus accumbens core and shell. Neuroscience. 2012;210:333–339. doi: 10.1016/j.neuroscience.2012.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, See RE. Basolateral amygdala inactivation abolishes conditioned stimulus- and heroin-induced reinstatement of extinguished heroin-seeking behavior in rats. Psychopharmacology (Berl) 2002;160:425–433. doi: 10.1007/s00213-001-0997-7. [DOI] [PubMed] [Google Scholar]
- Gass JT, Osborne MP, Watson NL, Brown JL, Olive MF. mGluR5 antagonism attenuates methamphetamine reinforcement and prevents reinstatement of methamphetamine-seeking behavior in rats. Neuropsychopharmacology. 2009;34:820–833. doi: 10.1038/npp.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JT, Sinclair CM, Cleva RM, Widholm JJ, Olive MF. Alcohol-seeking behavior is associated with increased glutamate transmission in basolateral amygdala and nucleus accumbens as measured by glutamate-oxidase-coated biosensors. Addiction Biology. 2011;16:215–228. doi: 10.1111/j.1369-1600.2010.00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW. Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron. 2013a;77:867–872. doi: 10.1016/j.neuron.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, Kalivas PW. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci U S A. 2013b;110:9124–9129. doi: 10.1073/pnas.1220591110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnegy ME. Ca2+/calmodulin signaling in NMDA-induced synaptic plasticity. Crit Rev Neurobiol. 2000;14:91–129. [PubMed] [Google Scholar]
- Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006;6:68–74. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yuan H, Traynelis SF. Structural aspects of AMPA receptor activation, desensitization and deactivation. Curr Opin Neurobiol. 2007;17:281–288. doi: 10.1016/j.conb.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Rutherford EC, Quintero JE, Day BK, Nickell JR, Pomerleau F, Huettl P, Burmeister JJ, Gerhardt GA. Second-by-second measures of L-glutamate and other neurotransmitters using enzyme-based microelectrode arrays. In: Michael AC, Borland LM, editors. electrochemical methods for neuroscience. Boca Raton, FL: CRC Press; 2007. [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kau KS, Madayag A, Mantsch JR, Grier MD, Abdulhameed O, Baker DA. Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience. 2008;155:530–537. doi: 10.1016/j.neuroscience.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, Markou A, Kalivas PW. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry. 2010;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaresan V, Yuan M, Yee J, Famous KR, Anderson SM, Schmidt HD, Pierce RC. Metabotropic glutamate receptor 5 (mGluR5) antagonists attenuate cocaine priming- and cue-induced reinstatement of cocaine seeking. Behav Brain Res. 2009;202:238–244. doi: 10.1016/j.bbr.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longuemare MC, Swanson RA. Net glutamate release from astrocytes is not induced by extracellular potassium concentrations attainable in brain. J Neurochem. 1997;69:879–882. doi: 10.1046/j.1471-4159.1997.69020879.x. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YY, Chu NN, Guo CY, Han JS, Cui CL. NR2B-containing NMDA receptor is required for morphine-but not stress-induced reinstatement. Exp Neurol. 2007;203:309–319. doi: 10.1016/j.expneurol.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, Grier MD, Baker DA. Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci. 2007;27:13968–13976. doi: 10.1523/JNEUROSCI.2808-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael AC, Borland LM. Electrochemical methods for neuroscience. Boca Raton, FL: CRC/Taylor and Francis; 2007. [PubMed] [Google Scholar]
- Minelli A, Barbaresi P, Reimer RJ, Edwards RH, Conti F. The glial glutamate transporter GLT-1 is localized both in the vicinity of and at distance from axon terminals in the rat cerebral cortex. Neuroscience. 2001;108:51–59. doi: 10.1016/S0306-4522(01)00375-X. [DOI] [PubMed] [Google Scholar]
- Nahum-Levy R, Lipinski D, Shavit S, Benveniste M. Desensitization of NMDA receptor channels is modulated by glutamate agonists. Biophys J. 2001;80:2152–2166. doi: 10.1016/S0006-3495(01)76188-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onifer SM, Quintero JE, Gerhardt GA. Cutaneous and electrically evoked glutamate signaling in the adult rat somatosensory system. J Neurosci Methods. 2012;208:146–154. doi: 10.1016/j.jneumeth.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Amsterdam: Elsevier Academic; 2005. [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Quintero JE, Pomerleau F, Huettl P, Johnson KW, Offord J, Gerhardt GA. Methodology for rapid measures of glutamate release in rat brain slices using ceramic-based microelectrode arrays: basic characterization and drug pharmacology. Brain Res. 2011;1401:1–9. doi: 10.1016/j.brainres.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Niño AM, D'Souza MS, Markou A. N-acetylcysteine decreased nicotine self-administration and cue-induced reinstatement of nicotine seeking in rats: comparison with the effects of N-acetylcysteine on food responding and food seeking. Psychopharmacology. 2013;225:473–482. doi: 10.1007/s00213-012-2837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, Sartor GC, Vazey EM, Dunn TE, Aston-Jones G, Kalivas PW. Use of vivo-morpholinos for control of protein expression in the adult rat brain. J Neurosci Methods. 2012;203:354–360. doi: 10.1016/j.jneumeth.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, Brown RM, Spencer S, Tran PK, Thomas CA, Kalivas PW. Chronic administration of the methylxanthine propentofylline impairs reinstatement to cocaine by a GLT-1-dependent mechanism. Neuropsychopharmacology. 2014;39:499–506. doi: 10.1038/npp.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Savtchenko LP, Zheng K, Henley JM. Shaping the synaptic signal: molecular mobility inside and outside the cleft. Trends Neurosci. 2011;34:359–369. doi: 10.1016/j.tins.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore MF, Davis RW, Arnold JC, Chotibut T. Transient striatal GLT-1 blockade increases EAAC1 expression, glutamate reuptake, and decreases tyrosine hydroxylase phosphorylation at ser(19) Exp Neurol. 2012;234:428–436. doi: 10.1016/j.expneurol.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Sari Y, Smith KD, Ali PK, Rebec GV. Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci. 2009;29:9239–9243. doi: 10.1523/JNEUROSCI.1746-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Sakai M, Weedman JM, Rebec GV, Bell RL. Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol and Alcoholism. 2011;46:239–246. doi: 10.1093/alcalc/agr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JP, Spanos M, Stevenson JR, Besheer J, Salling M, Hodge CW. Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: blockade by the mGluR5 antagonist MPEP. Neuropharmacology. 2008;55:546–554. doi: 10.1016/j.neuropharm.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci U S A. 2011;108:19407–19412. doi: 10.1073/pnas.1112052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair CM, Cleva RM, Hood LE, Olive MF, Gass JT. mGluR5 receptors in the basolateral amygdala and nucleus accumbens regulate cue-induced reinstatement of ethanol-seeking behavior. Pharmacol Biochem Behav. 2012;101:329–335. doi: 10.1016/j.pbb.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumlinski KK, Dehoff MH, Kang SH, Frys KA, Lominac KD, Klugmann M, Rohrer J, Griffin W, 3rd, Toda S, Champtiaux NP, Berry T, Tu JC, Shealy SE, During MJ, Middaugh LD, Worley PF, Kalivas PW. Homer proteins regulate sensitivity to cocaine. Neuron. 2004;43:401–413. doi: 10.1016/j.neuron.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, LaLumiere RT, Reissner KJ, Kalivas PW, Knackstedt LA. Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci. 2012;32:12406–12410. doi: 10.1523/JNEUROSCI.1976-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Moussawi K, Knackstedt L, Shen H, Kalivas PW. Role of mGluR5 neurotransmission in reinstated cocaine-seeking. Addiction Biology. 2013;18:40–49. doi: 10.1111/j.1369-1600.2011.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33:391–398. doi: 10.1016/j.tins.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang S, Jia L, Xu Z, Zeng Y. An electrochemical sensor for determination of calcium dobesilate based on PoPD/MWNTs composite film modified glassy carbon electrode. J Biochem Biophys Methods. 2008;70:1203–1209. doi: 10.1016/j.jbbm.2007.10.002. [DOI] [PubMed] [Google Scholar]