

Abstract
Recent studies have demonstrated that human immunodeficiency virus (HIV) protease inhibitors (PIs) exert inhibitory effects on erythrocytic stages of the human-malaria parasite Plasmodium falciparum in vitro and on erythrocytic stages of the rodent-malaria parasite Plasmodium chabaudi in vivo. Although it remains unclear how HIV PIs inhibit the parasite, the effect seen on parasite development in the erythrocytic stages is potent. The effect on preerythrocytic stages has not yet been investigated. Using the rodent parasite Plasmodium berghei, we screened a panel of HIV PIs in vitro for effects on the preerythrocytic stages. Our data indicated that the HIV PIs lopinavir and saquinavir affect preerythrocytic-stage parasite development in vitro. We then evaluated the effect of HIV PIs on preerythrocytic stages in vivo using the rodent parasite Plasmodium yoelii. We found that lopinavir/ritonavir had a dose-dependent effect on liver-stage parasite development. Given that sub-Saharan Africa is where the HIV/AIDS pandemic intersects with malaria, these results merit analysis in clinical settings.
The World Health Organization estimates that 300–500 million cases of malaria and 1–2 million deaths due to malaria occur annually, with most malaria-related morbidity and mortality occurring in children [1]. This malarial disease burden is compounded by the pandemic of HIV/AIDS, which is most prevalent in areas in which malaria is endemic, notably sub-Saharan Africa [2–4]. Moreover, when HIV and a malaria parasite are present as a coinfection, they enhance each other's pathogenicity [5, 6]. Clinical studies have shown that HIV burden is higher in patients with malaria than in control subjects without malaria [6–8]. It has even been hypothesized that the increased viral loads seen in HIV-positive patients with malaria may be an important factor in fueling the spread of HIV in sub-Saharan Africa [9]. Moreover, HIV-infected individuals with malaria are more likely to have recrudescence of malaria [10]. HIV affects the acquisition of malaria-specific immunity in individuals living in areas of higher and more stable transmission intensity, and this is especially true in patients with lower CD4+ cell counts and higher viral loads [11, 12]. In the absence of an effective antimalarial vaccine and with the intersection of the HIV/AIDS pandemic and malaria epidemics, the need to optimize treatment of patients with HIV infection while reducing the malarial disease burden becomes clearly urgent.
The 3 malarial life-cycle stages targeted for treatment and prevention strategies are the preerythrocytic stages (sporozoite stage and exoerythrocytic forms [EEFs]), the asexual blood stages, and the transmission stages (sexual blood stage and mosquito stage). Sporozoites are the infective form of the parasite and are injected into the mammalian host during blood feeding by infected Anopheles mosquitoes. From the injection site, sporozoites go to the liver, where they invade hepatocytes and develop into EEFs. Mature EEFs contain merozoites that exit the hepatocyte and invade erythrocytes, initiating the erythrocytic stages of disease [13].
Most antimalarial drugs being investigated or developed for Plasmodium species target the asexual blood stages, which are responsible for the clinical symptoms of disease. However, intervention during the preerythrocytic stages offers the ability to target the parasite while it is present in low numbers and before it initiates blood-stage infection [14].
Previous work demonstrated that HIV protease inhibitors (PIs) exert a potent antiplasmodial effect on erythrocytic-stage parasites [15–19]. Other classes of antiretrovirals tested for antimalarial effect, such as nonnucleoside reverse-transcriptase inhibitors, have not shown similar efficacy in vitro against Plasmodium falciparum erythrocytic-stage parasites [15, 19, 20]. Evaluation of the effect of HIV PIs on preerythrocytic stages has heretofore not been performed. Thus, using rodent models of malaria, we investigated the effect of HIV PIs on the preerythrocytic-stage parasite.
Methods
Mice. Female Swiss Webster mice, aged 4–6 weeks and weighing 20–25 g, were purchased from Taconic and were handled according to institutional guidelines.
Parasites and mosquitoes. Plasmodium berghei (NK65 and ANKA strains) and Plasmodium yoelii (17XNL) sporozoites were grown in Anopheles stephensi mosquitoes. Three to 5 days after emergence, mosquitoes were fed on P. yoelii-infected or P. berghei-infected Swiss Webster mice with abundant gametocyte-stage parasites. Salivary gland sporozoites were harvested by mosquito dissection on days 14 and 15 after the infective blood meal for P. yoelii and on days 18–20 after the infective blood meal for P. berghei. Sporozoites were counted using a hemocytometer.
Drug preparation. For in vitro assays, pure HIV PIs were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. Stocks (×1000) were prepared in DMSO as described elsewhere [16]. Final concentrations for experiments were prepared by diluting stock with culture medium. For all in vitro experiments, DMSO diluted in culture medium alone was added to the control wells (the DMSO concentration was ≤0.04% per well, a concentration equal to that added to treated wells).
For in vivo assays, pure saquinavir-mesylate (provided by Hoffmann-LaRoche) was prepared from stock solutions diluted in olive oil. Olive oil was used as a vehicle and control for the saquinavir experiments (high-fat, high-calorie meals enhance the pharmacokinetic profile of saquinavir [21]). Given the variability in store-bought olive oil, we maintained the same stock bottle for the duration of experimentation, and we used the same stock for control and treated animals. Only unboosted saquinavir was tested in vivo. Lopinavir/ritonavir and control vehicle (provided by Abbott Laboratories) were prepared using an optimized ratio of lopinavir to ritonavir (2:1) that was based on preclinical rodent pharmacokinetic data from Abbott [22].
Cytotoxicity assay. Hepa 1-6 cells (CRL-1830; ATCC) were grown at 37°C in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum and 1% l-glutamine (culture medium). Cells were plated at a density of 2.5×105 cells/well in Labtek chamber slides and cultured for 24 h. Cells were then incubated for 48 h in culture medium containing the appropriate drug concentration. The drug-containing medium was changed twice per day. At 24 and 48 h, cells were visualized for morphology with a standard light microscope. At 48 h, cells were washed with PBS, treated with trypsin, and counted using a hemocytometer.
Microscopic assay for quantification of EEF development in vitro. Hepa 1-6 cells were plated as outlined above, and 1×105P. berghei sporozoites were added to each well. Parasites were allowed to invade the Hepa 1-6 cells for 1 h, after which unattached sporozoites were washed away. HIV PI stock solutions, diluted in culture medium to the doses indicated in figure 1, were added to cells 1 h after the addition of sporozoites. After 46–48 h, cells were fixed with cold methanol, washed, and stained with monoclonal antibody (MAb) 2E6, which recognizes Plasmodium hsp70 [23], followed by anti-mouse immunoglobulin conjugated to rhodamine. Cell nuclei were stained with 1 µg/mL 3,3′-diaminobenzidine (Sigma). EEFs were counted using a Nikon Eclipse E600 fluorescence microscope; counting was done in 50 fields per well, and each point was plated in triplicate. Five randomly selected EEFs from each of 3 triplicate wells (with a total of 15 EEFs per assay per condition) were used to determine the average EEF size. The widest diameter of the EEF was measured using a Leica inverted laser-scanning confocal microscope (model TCS SP2 AOBS) with Leica LCSlite software (version 2.61, build 1538). Measurements were used to calculate means ± SDs. All in vitro development assays were performed with unboosted PIs (i.e., in the absence of ritonavir). Each experimental condition was run in triplicate. Differences between the mean number of EEFs per 20 high-powered fields in treated and control wells as well as differences in mean size (estimated by the widest diameter measurement) in treated and control wells were analyzed for statistical significance.
Figure 1.

Effect of saquinavir, lopinavir, atazanavir, amprenavir, and nelfinavir on the no. of developing exoerythrocytic forms (EEFs). Shown are the mean no. of EEFs per 20 high-powered fields (HPFs) (error bars show SEs), expressed as the percentage of control for the listed HIV protease inhibitors. Each bar represents data from 2 experiments, with each experiment run in triplicate wells. *P<.05 (statistically significant difference between the treated group and the control group).
Microscopic assay for quantification of sporozoite invasion in vitro. To determine effect of HIV PIs on sporozoite invasion, a double-staining assay that differentiates between intracellular and extracellular sporozoites was performed as described elsewhere, with a few modifications [24, 25]. The assay was performed using Hepa 1-6 cells, MAb 3D11 (antibody directed against the repeat region of circumsporozoite protein, the major sporozoite surface protein) [26], and secondary antibodies as detailed below. Hepa 1–6 cells were plated as outlined above, and P. berghei sporozoites, pretreated with 40 µmol/L saquinavir or 40 µmol/L lopinavir in culture medium for 30 min at 27°C, were washed and added to the cells. Control sporozoites were pretreated with 0.04% DMSO alone. Parasites were incubated with Hepa 1-6 cells for 1 h, after which unattached sporozoites were removed. Cells were then fixed with 4% paraformaldehyde and incubated with MAb 3D11 followed by goat anti-mouse immunoglobulin conjugated to fluorescein isothiocyanate (FITC). Cells were then permeabilized with methanol and incubated with MAb 3D11 followed by goat anti-mouse immunoglobulin conjugated to rhodamine. The number of extracellular sporozoites (FITC labeled) and total sporozoites (rhodamine labeled) per field were counted in 50 fields per well. Percent invasion was then calculated. All in vitro invasion assays were performed with unboosted PIs. The mean number of parasites invading (in treated and control wells run in triplicate for each experimental condition) was calculated and analyzed for statistically significant differences.
Assay for quantification of P. yoelii liver-stage development in vivo. P. yoelii was used for in vivo challenge because it is more infectious than P. berghei in vivo and better models the infectivity of the human-malaria parasite P. falciparum [27, 28]. The doses of HIV PIs indicated below were administered to mice by gavage. For once-a-day (QD) dosing regimens, doses were administered to mice 6 h before sporozoite inoculation, with 1 additional dose given 24 h later. For twice-a-day (BID) dosing regimens, doses were administered 6 h before sporozoite inoculation and every 12 h after the first dose for 2 more doses. Forty hours after sporozoite inoculation, mice were killed, livers were harvested, and total RNA was isolated using TRI reagent (Molecular Research Center), in accordance with the manufacturer's instructions. Parasite burden in the liver was quantified as outlined elsewhere [29]. Briefly, reverse transcription was performed using 1 µg of total RNA and random hexamers. After this, real-time polymerase chain reaction (PCR) was performed using primers that recognize P. yoelii-specific sequences within the 18S rRNA and the QuantiTect SYBR Green PCR Kit (Qiagen). Ten-fold dilutions of a plasmid construct containing the P. yoelii 18S rRNA gene were used to create a standard curve. Differences between the mean amounts of parasite 18S rRNA in the livers of treated and control mice were analyzed for statistical significance.
Assay for prepatent period (or assay for detection of parasites in blood). This assay was performed as described elsewhere [30], with a few modifications. To evaluate whether the development of EEFs was fully arrested with the HIV PI dosing regimens used, mice were administered 100 mg/kg lopinavir with 50 mg/kg ritonavir and then challenged with 1×102 sporozoites. Mice were injected intravenously with 1×102 P. yoelii sporozoites, and, beginning on day 3 after sporozoite injection, mice were monitored for the presence of erythrocytic-stage parasites by means of Giemsa-stained blood smears.
Pharmacodynamics of lopinavir/ritonavir and saquinavir in vivo. Groups of 5–6 mice were given the BID and QD HIV PI dosing regimens outlined above, and serum samples were obtained 6, 12, and/or 24 h after the last dose was administered. Blood was obtained by submandibular vein sampling and centrifuged; serum was removed, frozen at −80°C, and shipped overnight on dry ice to Abbott Laboratories for analysis. Analysis of plasma levels was performed by high-performance liquid chromatography at Abbott Laboratories.
Statistical analysis. One-way analysis of variance was used to compare mean parasite numbers in both in vitro and in vivo assays, taking into account multiple replicates. Tukey's method was used to identify statistically significant differences at the P<.05 level. Statistical analyses were done using Prism software (version 5; GraphPad).
Results
Assessment of drug-induced cell toxicity for in vitro assays. The maximum concentration of saquinavir, lopinavir, atazanavir, and amprenavir added to Hepa 1-6 cells at which cell growth was preserved was 40 µmol/L; for nelfinavir, this concentration was 10 µmol/L. At these concentrations, cell counts in control versus treated wells were not statistically significantly different, and morphology in drug-treated parasites (as assessed by light microscopy) was not different from the control (data not shown).
Inhibition of the development of P. berghei EEFs in vitro by saquinavir and lopinavir but not by atazanavir, amprenavir, and nelfinavir. Saquinavir treatment at 10 and 40 µmol/L reduced the number of EEFs by an average of 55% (P<.05) and 65% (P<.05), respectively. Lopinavir treatment at 10 and 40 µmol/L reduced the number of EEFs by an average of 38% (P<.05) and 90% (P<.05), respectively. No significant differences between treated and untreated samples were observed with atazanavir, amprenavir, or nelfinavir (figure 1). EEFs treated with saquinavir or lopinavir at 10 µmol/L were not significantly smaller than control EEFs, but treatment at 40 µmol/L resulted in a reduction in the size of EEFs by 42% for saquinavir and 75% for lopinavir (figure 2A and 2B). Additionally, EEFs treated with saquinavir and lopinavir had an altered appearance (figure 3A –3F).
Figure 2.
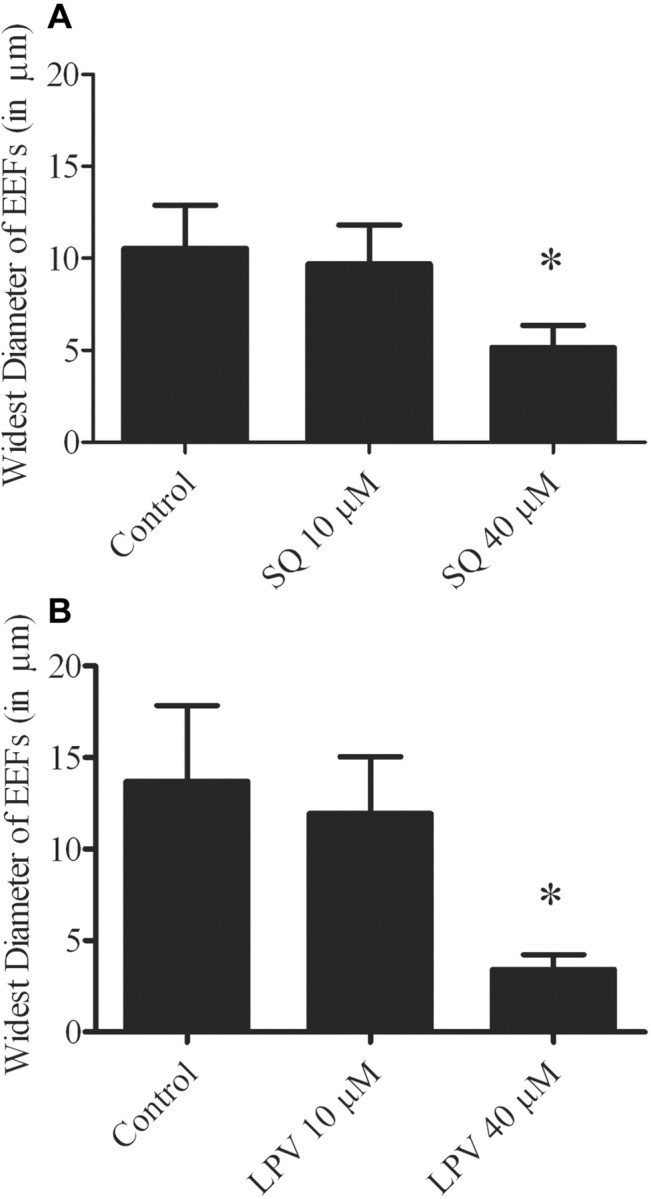
Effect of saquinavir (SQ) (A) and lopinavir (LPV) (B) on the size of developing exoerythrocytic forms (EEFs). Drugs were added to sporozoite-infected hepatocytes immediately after a 1-h sporozoite invasion period (after which unattached sporozoites were washed away). Shown is the average size of EEFs at 46–48 h (diameters in assays fixed at 46 h are slightly smaller). Means were calculated from 5 randomly selected EEFs from each of 3 triplicate wells; shown are means ± SDs. Results are representative of 2 experiments. *P<.05 (statistically significant difference between the treated group and the control group).
Figure 3.

Effect of saquinavir and lopinavir on exoerythrocytic form (EEF) morphology. Drugs were added to sporozoite-infected hepatocytes immediately after a 1-h sporozoite invasion period (after which unattached sporozoites were washed away); shown are confocal images of representative 46–48-h EEFs. A, Control with no drug. B, Saquinavir at 10 µmol/L. C, Saquinavir at 40 µmol/L. D, Control with no drug. E, Lopinavir at 10 µmol/L. F, Lopinavir at 40 µmol/L. Nuclear staining with 3,3'-diaminobenzidine (blue) shows the host cell nuclei. The bar indicates 6 microns. Diameters in assays fixed at 46 h are slightly smaller. Treatment at 40 µmol/L resulted in a reduction in EEF size by an average of 42% for saquinavir and 75% for lopinavir, compared with control.
Inhibition of P. berghei invasion in vitro only by high concentrations of saquinavir. Using P. berghei sporozoites preincubated with the PIs, we tested the effect of lopinavir and saquinavir on sporozoite invasion of Hepa 1-6 cells. We did not find an effect with lopinavir at 40 µmol/L, and saquinavir at 40 µmol/L had only a mild effect, with an average percent inhibition of invasion of 28% (P<.05) (figure 4).
Figure 4.

Effect of saquinavir (SQ) and lopinavir (LPV) on Plasmodium berghei invasion. Sporozoites were preincubated with the indicated concentrations of drug and then added to hepatocytes in the continued presence of the drug for a 1-h period. Extracellular sporozoites and total sporozoites per field were counted in 50 fields per well, and the percent invasion was then calculated. *P<.05 (statistically significant difference between the treated group and the control group).
Inhibition of P. yoelii development in vivo by lopnavir/ritonavir in a dose-dependent fashion. Lopinavir/ritonavir exerted a dose-dependent effect in reducing liver parasite burden. When mice were administered 25 mg/kg lopinavir with 12.5 mg/kg ritonavir or 100 mg/kg lopinavir with 50 mg/kg ritonavir 6 h before infection and then again 18 h after sporozoite inoculation, parasite liver burden was reduced by 45% and 93%, respectively (figure 5). With these regimens, drug was undectectable in serum 12 h after administration.
Figure 5.

Effect of lopinavir/ritonavir on Plasmodium yoelii liver burden. Mice were gavaged with lopinavir/ritonavir before being injected intravenously with 10,000 P. yoelii sporozoites. Mice were given vehicle (control), 100 mg/kg lopinavir with 50 mg/kg ritonavir once a day (100 mg/kg QD LPV/r), or 25 mg/kg lopinavir with 12.5 mg/kg ritonavir once a day (25 mg/kg QD LPV/r), the lowest dose that had a significant effect. All experiments were performed twice, with 5–6 mice per group per experiment. *P<.05 (statistically significant difference between the treated group and the control group).
Prolongation of the prepatent period by lopinavir/ritonavir. Treated mice had detectable parasites in blood 1–1.3 days later than control mice (table 1).
Table 1.

Prolongation of the prepatent period by lopinavir/ritonavir.
Lower serum levels of lopinavir/ritonavir in rodent serum sampling reduces liver parasite burden compared with levels achieved in humans receiving standard lopinavir/ritonavir. With BID dosing, 50 mg/kg lopinavir with 25 mg/kg ritonavir produced a mean ± SD peak level of 8.64±1.74 µg/mL (6-h serum level after 3 days of gavage dosing). With the same dose gavaged for 3 days, levels measured 12 h after the last dose had a mean ± SD of 2.7±3.6 µg/mL, and levels measured 24 h after the last dose were undetectable. (For reference, the mean ± SD peak plasma level for HIV-infected adults taking 400 mg of lopinavir with 100 mg of ritonavir BID for 3 weeks is 9.8±3.7 µg/mL, and the mean ± SD steady state trough concentratrion is 7.1±2.9 µg/mL [Abbott prescribing information]).
For 100 mg/kg lopinavir with 50 mg/kg ritonavir QD and for 50 mg/kg lopinavir with 25 mg/kg ritonavir QD gavaged for 3 days, levels at 12 and 24 h after last dose were all undetectable. (For reference, daily dosing of 800 mg of lopinavir with 200 mg of ritonavir [administered as part of a regimen that included 200 mg of emtricitabine and 300 mg of tenofovir disoproxil fumarate] generates a mean ± SD peak of 11.8±3.7 µg/mL and a mean ± SD trough of 3.2±2.1 µg/mL [Abbott prescribing information]).
All saquinavir levels at 12 h after 3 days of gavage dosing with unboosted doses as high as 200 mg/kg were undetectable.
No inhibition of the development of P. yoelii in vivo by saquinavir. We found that saquinavir alone in high doses did not have any effect on the liver stage of the parasite in vivo when administered before infection and during development. We tested doses as high as 200 mg/kg BID and parasite inocula as low as 1×102 sporozoites but still saw no effect (data not shown).
Discussion
We have described an inhibitory effect of HIV PIs on preerythrocytic stages of Plasmodium species in rodent models in vitro and in vivo. For the first time, our data show that lopinavir and saquinavir in vitro as well as lopinavir/ritonavir in vivo have a significant effect on the preerythrocytic development of Plasmodium parasites.
Our in vitro data indicate that both lopinavir and saquinavir decreased the number and size of EEFs, and the morphology of drug-treated EEFs was also altered. There was no demonstrable effect on invasion with lopinavir in vitro, even at high concentrations, suggesting that lopinavir works during the intrahepatic development of the parasite.
When tested in vivo, lopinavir/ritonavir significantly reduced liver burden by as much as 55% with a dose as low as 25 mg/kg lopinavir with 12.5 mg/kg ritonavir. Mice treated with lopinavir/ritonavir also tended to show a prolonged time to detectable parasitemia (i.e., a prolonged prepatent period), compared with control mice. This likely reflects the lower liver burden, which was seen in the quantitative PCR experiments. At the doses used, not all parasites were killed, however, because there was eventual detectable blood-stage parasitemia.
The doses of lopinavir/ritonavir used in our experiments are higher than doses used to achieve therapeutic efficacy for HIV treatment in humans, but serum-level analysis showed that the drug levels achieved in our mouse model are comparable to or lower than those achieved in humans receiving standard lopinavir/ritonavir dosing regimens. This is consistent with previous work showing the half-lives and steady-state levels of lopinavir and ritonavir to be shorter in mice than in humans, thus necessitating higher doses of these drugs in rodent models to achieve concentrations that may be compared with levels achieved in patients receiving standard HIV PI dosing [31].
There is an apparent effect with undetectable serum levels of lopinavir/ritonavir. This could be explained by previously published data in rats showing that tissue-to-plasma ratios are highest in organs of the gastrointestinal tract, including the liver, 4 h after oral administration of radioactively-labeled lopinavir/ritonavir [22]. This suggests that, even when plasma levels are low, levels in the liver (the site of parasite development) may be higher and thus may account for the antiparasite effect we observed.
Although we were not able to demonstrate an effect with saquinavir in unboosted form, the question as to whether boosted saquinavir would have an effect remains. Ritonavir, while exhibiting PI activity itself, is often used at subtherapeutic doses to boost the blood levels of other PIs by exploiting its inhibitory effect on liver cytochrome p450 enzymes [31]. Ritonavir use is critical to achieving therapeutic levels of lopinavir in vivo and is less so for saquinavir. Although it is possible that the use of lopinavir or saquinavir with ritonavir produces a synergistic antiparasite effect, it is also possible that the boosting effect of ritonavir enables the HIV PI to maintain therapeutic levels for a longer period of time, thus accounting for its enhanced effect.
Strikingly, our findings parallel previously published findings for an effect of HIV PIs on erythrocytic-stage Plasmodium parasites. Lopinavir and saquinavir were previously shown to be the most potent drugs affecting the erythrocytic-stage parasite in vitro with P. falciparum (lopinavir had the lowest IC50 for 4 different strains of P. falciparum) [16]. Considered together with our present findings, this suggests that HIV PIs may affect a common enzyme or pathway in these distinct Plasmodium life-cycle stages.
It remains unclear how HIV PIs function in hindering the development of the malaria parasite. HIV PIs target aspartyl protease [32], and Plasmodium species express a number of aspartyl proteases called “plasmepsins.” Ten predicted plasmepsin-encoding genes exist in P. falciparum [33]; 4 of them are food vacuole aspartyl proteases, which function in the blood stages for hemoglobin digestion [34]. Previous work has shown that HIV PIs are unlikely to inhibit the food vacuole plasmepsins of P. falciparum [35–37]. Plasmepsins are also expressed during the P. yoelii preerythrocyte stages [38], so these remain possible targets for the HIV PIs during this stage of the life cycle. Moreover, plasmepsins are present across Plasmodium rodent- and human-infecting species [18, 39]. However, whether HIV PIs affect only aspartyl proteases or are less selective and can act against other non-aspartyl protease targets in the malaria parasite remains to be determined. Interestingly, HIV PIs have also been shown to inhibit other apicomplexan parasites, including Toxoplasma and Cryptosporidium organisms [40].
Drugs that affect preerythrocytic stages are being investigated by a number of groups, given the potential to target the parasite when it is present in lower numbers. Atovaquone and primaquine are the only 2 drugs in practice with known activity against the liver stages. Atovaquone, however, does not act against the preerythrocytic dormant forms of the parasites (hypnozoite forms), which are exhibited in only 2 of the 4 Plasmodium species infecting humans—namely, P. vivax and P. ovale [41]. Atovaquone also can select resistant clones of erythrocytic-stage P. falciparum [42]. Primaquine does target the dormant forms of P. vivax and P. ovale but cannot be used in pregnant women or those with glucose-6-phosphate dehydrogenase deficiency. Other compounds have been experimentally shown to have an effect against the preerythrocytic stages of Plasmodium species [30, 43–48], but none are currently recommended for human malaria. With few exceptions, the World Health Organization recommends nevirapine-based regimens as first-line therapy and PI-based regimens as second-line therapy for HIV-infected patients in resource-poor settings [49]. If lopinavir/ritonavir regimens are shown to possess antimalarial activity in clinical settings, such knowledge may guide recommendations for HIV therapy in areas in which malaria is endemic. Initiation of lopinavir/ritonavir in young HIV-infected patients may offer secondary prophylactic benefits for modifying malarial infection.
Acknowledgments
We thank Dr. Brian M. Kirmse, Dr. Henry Pollack, Dr. Mona Rigaud, and Dr. Sunil Parikh for critical review of the manuscript. We especially thank Dr. Brian M. Kirmse for his support of and assistance with this project. We also thank Dr. Mona Rigaud for her assistance in obtaining the drugs for testing. We further thank the National Institutes of Health AIDS Reference and Reagent Program for provision of the drugs saquinavir (as freebase), lopinavir, amprenavir, atazanavir, and nelfinavir for in vitro assays. We thank Abbott Laboratories for its donation of lopinavir/ritonavir and vehicle, for helpful suggestions regarding dosing, and for helpful discussions (Kennan Marsh). We thank Lisa Hernandez and Edith McDonald for performing the high-performance liquid chromatography assays. We thank Roche Laboratories for its provision of saquinavir mesylate, and we thank Peter Larson and Malte Shultz at Roche Laboratories for helpful suggestions for optimizing saquinavir dosing. We thank Dabeiba Bernal-Rubio, Sandra Gonzalez, and Jean Nonon for their expert assistance with mosquito rearing. We thank Michelle Carrington and Helene Zinszner at the Center for AIDS Research for their assistance. We thank Dr. Fred Valentine at the Center for AIDS Research for his support of this project.
Footnotes
Potential conflicts of interest: K.M. is an employee of Abbott Laboratories. All other authors report no potential conflicts.
Presented in part: 56th American Society of Tropical Medicine and Hygiene Conference, 12–16 November 2006 (abstract 2546).
Financial support: National Institutes of Health (NIH; grant R01AI056840 to P.S., NIH Center for AIDS Research Grant P30 AI027742 to P.S. and C.V.H., NIH Institutional Grant T32 07382 to C.V.H. [noncontemporaneously with the previously mentioned grant P30 AI027742], and NIH Training Grant T32 AI07180 to T.V.).
References
- 1.Snow RW, Craig M, Deichmann U, Marsh K. Estimating mortality, morbidity and disability due to malaria among Africa's nonpregnant population. Bull World Health Organ. 1999;77:624–40. [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization, author. Malaria and HIV interactions and their implications for public health policy. [4 December 2007]. Available at: http://www.who.int/malaria/malaria_HIV/MalariaHIVinteractions_report.pdf.
- 3.Korenromp EL, Williams BG, de Vlas SJ, et al. Malaria attributable to the HIV-1 epidemic, sub-Saharan Africa. Emerg Infect Dis. 2005;11:1410–9. doi: 10.3201/eid1109.050337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.UNAIDS, author. Report on the global AIDS epidemic. [15 November 2007]. Available at: http://www.unaids.org/en/HIV_data/epi2006.
- 5.Slutsker L, Marston BJ. HIV and malaria: interactions and implications. Curr Opin Infect Dis. 2007;20:3–10. doi: 10.1097/QCO.0b013e328012c5cd. [DOI] [PubMed] [Google Scholar]
- 6.Kublin JG, Patnaik P, Jere CS, et al. Effect of Plasmodium falciparum malaria on concentration of HIV-1-RNA in the blood of adults in rural Malawi: a prospective cohort study. Lancet. 2005;365:233–40. doi: 10.1016/S0140-6736(05)17743-5. [DOI] [PubMed] [Google Scholar]
- 7.ter Kuile FO, Parise ME, Verhoeff FH, et al. The burden of co-infection with human immunodeficiency virus type 1 and malaria in pregnant women in sub-Saharan Africa. Am J Trop Med Hyg. 2004;71(Suppl 2):41–54. [PubMed] [Google Scholar]
- 8.Mwapasa V, Rogerson SJ, Molyneux ME, et al. The effect of Plasmodium falciparum malaria on peripheral and placental HIV-1 RNA concentrations in pregnant Malawian women. AIDS. 2004;18:1051–9. doi: 10.1097/00002030-200404300-00014. [DOI] [PubMed] [Google Scholar]
- 9.Abu-Raddad LJ, Patnaik P, Kublin J. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science. 2006;314:1603–6. doi: 10.1126/science.1132338. [DOI] [PubMed] [Google Scholar]
- 10.Shah SN, Smith EE, Obonyo CO, et al. HIV immunosuppression and antimalarial efficacy: sulfadoxine-pyrimethamine for treatment of uncomplicated malaria in HIV-infected adults in Siaya, Kenya. J Infect Dis. 2006;194:1519–28. doi: 10.1086/508892. [DOI] [PubMed] [Google Scholar]
- 11.Quigley MA, Hewitt K, Mayanja B, et al. Effect of HIV-1 and increasing immunosuppression on malaria parasitaemia and clinical episodes in adults in rural Uganda: a cohort study. Lancet. 2000;356:1051–6. doi: 10.1016/S0140-6736(00)02727-6. [DOI] [PubMed] [Google Scholar]
- 12.Patnaik P, Jere CS, Miller WC, et al. Effects of HIV-1 serostatus, HIV-1 RNA concentration, and CD4 cell count on the incidence of malaria infection in a cohort of adults in rural Malawi. J Infect Dis. 2005;192:984–91. doi: 10.1086/432730. [DOI] [PubMed] [Google Scholar]
- 13.Sinnis P, Coppi A. A long and winding road: the Plasmodium sporozoite's journey in the mammalian host. Parasitol Int. 2007;56:171–8. doi: 10.1016/j.parint.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikolajczak SA, Aly AS, Kappe SH. Pre-erythrocytic malaria vaccine development. Curr Opin Infect Dis. 2007;20:461–6. doi: 10.1097/QCO.0b013e3282ef6172. [DOI] [PubMed] [Google Scholar]
- 15.Skinner-Adams TS, McCarthy JS, Gardiner DL, Hilton PM, Andrews KT. Antiretrovirals as antimalarial agents. J Infect Dis. 2004;190:1998–2000. doi: 10.1086/425584. [DOI] [PubMed] [Google Scholar]
- 16.Parikh S, Gut J, Istvan E, Goldberg DE, Havlir DV, Rosenthal PJ. Antimalarial activity of human immunodeficiency virus type 1 protease inhibitors. Antimicrob Agents Chemother. 2005;49:2983–5. doi: 10.1128/AAC.49.7.2983-2985.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews KT, Fairlie DP, Madala PK, et al. Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria. Antimicrob Agents Chemother. 2006;50:639–48. doi: 10.1128/AAC.50.2.639-648.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins TM, Domingos A, Berry C, Wyatt DM. The activity and inhibition of the food vacuole plasmepsin from the rodent malaria parasite Plasmodium chabaudi. Acta Trop. 2006;97:212–8. doi: 10.1016/j.actatropica.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Redmond AM, Skinner-Adams T, Andrews KT, et al. Antimalarial activity of sera from subjects taking HIV protease inhibitors. AIDS. 2007;21:763–5. doi: 10.1097/QAD.0b013e328031f41a. [DOI] [PubMed] [Google Scholar]
- 20.Pillai DR, Rosenthal PJ, DeRisi JL. The antimalarial effect of HIV NRTIs on Plasmodium falciparum [abstract 999]. Abstracts of the 55th American Society of Tropical Medicine and Hygiene Conference (Atlanta) Am J Trop Med Hyg. 2006;75(Suppl 5):1–323. [Google Scholar]
- 21.Invirase product information. [4 November 2008]. Available at: http://www.rocheusa.com/products/invirase/pi.pdf.
- 22.Kumar GN, Jayanti VK, Johnson MK, et al. Metabolism and disposition of the HIV-1 protease inhibitor lopinavir (ABT-378) given in combination with ritonavir in rats, dogs, and humans. Pharm Res. 2004;21:1622–30. doi: 10.1023/b:pham.0000041457.64638.8d. [DOI] [PubMed] [Google Scholar]
- 23.Tsuji M, Mattei D, Nussenzweig RS, Eichinger D, Zavala F. Demonstration of heat-shock protein 70 in the sporozoite stage of malaria parasites. Parasitol Res. 1994;80:16–21. doi: 10.1007/BF00932618. [DOI] [PubMed] [Google Scholar]
- 24.Rénia L, Miltgen F, Charoenvit Y, et al. Malaria sporozoite penetration: a new approach by double staining. J Immunol Methods. 1988;112:201–5. doi: 10.1016/0022-1759(88)90358-4. [DOI] [PubMed] [Google Scholar]
- 25.Pinzon-Ortiz C, Friedman J, Esko J, Sinnis P. The binding of the circumsporozoite protein to cell surface heparan sulfate proteoglycans is required for Plasmodium sporozoite attachment to target cells. J Biol Chem. 2001;276:26784–91. doi: 10.1074/jbc.M104038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida N, Nussenzweig RS, Potocnjak P, Nussenzweig V, Aikawa M. Hybridoma produces protective antibodies directed against the sporozoite stage of malaria parasite. Science. 1980;207:71–3. doi: 10.1126/science.6985745. [DOI] [PubMed] [Google Scholar]
- 27.Khan ZM, Vanderberg JP. Role of host cellular response in differential susceptibility of nonimmunized BALB/c mice to Plasmodium berghei and Plasmodium yoelii sporozoites. Infect Immun. 1991;59:2529–34. doi: 10.1128/iai.59.8.2529-2534.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belmonte M, Jones TR, Lu M, et al. The infectivity of Plasmodium yoelii in different strains of mice. J Parasitol. 2003;89:602–3. doi: 10.1645/0022-3395(2003)089[0602:TIOPYI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 29.Bruña-Romero O, Hafalla JC, González-Aseguinolaza G, Sano G, Tsuji M, Zavala F. Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. Int J Parasitol. 2001;31:1499–502. doi: 10.1016/s0020-7519(01)00265-x. [DOI] [PubMed] [Google Scholar]
- 30.Gantt SM, Myung JM, Briones MR, et al. Proteasome inhibitors block development of Plasmodium spp. Antimicrob Agents Chemother. 1998;42:2731–8. doi: 10.1128/aac.42.10.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeldin RK, Petruschke RA. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J Antimicrob Chemother. 2004;53:4–9. doi: 10.1093/jac/dkh029. [DOI] [PubMed] [Google Scholar]
- 32.Pearl LH, Taylor WR. A structural model for the retroviral proteases. Nature. 1987;329:351–4. doi: 10.1038/329351a0. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y, Wang X, Liu X, Wang Y. Data-mining approaches reveal hidden families of proteases in the genome of malaria parasite. Genome Res. 2003;13:601–16. doi: 10.1101/gr.913403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci USA. 2002;99:990–5. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonilla JA, Bonilla TD, Yowell CA, Fujioka H, Dame JB. Critical roles for the digestive vacuole plasmepsins of Plasmodium falciparum in vacuolar function. Mol Microbiol. 2007;65:64–75. doi: 10.1111/j.1365-2958.2007.05768.x. [DOI] [PubMed] [Google Scholar]
- 36.Parikh S, Liu J, Sijwali P, Gut J, Goldberg DE, Rosenthal PJ. Antimalarial effects of human immunodeficiency virus type 1 protease inhibitors differ from those of the aspartic protease inhibitor pepstatin. Antimicrob Agents Chemother. 2006;50:2207–9. doi: 10.1128/AAC.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skinner-Adams TS, Andrews KT, Melville L, McCarthy J, Gardiner DL. Synergistic interactions of the antiretroviral protease inhibitors saquinavir and ritonavir with chloroquine and mefloquine against Plasmodium falciparum in vitro. Antimicrob Agents Chemother. 2007;51:759–62. doi: 10.1128/AAC.00840-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarun AS, Peng X, Dumpit RF, et al. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc Natl Acad Sci USA. 2008;105:305–10. doi: 10.1073/pnas.0710780104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dame JB, Yowell CA, Omara-Opyene L, Carlton JM, Cooper RA, Li T. Plasmepsin 4, the food vacuole aspartic proteinase found in all Plasmodium spp. infecting man. Mol Biochem Parasitol. 2003;130:1–12. doi: 10.1016/s0166-6851(03)00137-3. [DOI] [PubMed] [Google Scholar]
- 40.Pozio E, Morales MA. The impact of HIV-protease inhibitors on opportunistic parasites. Trends Parasitol. 2005;21:58–63. doi: 10.1016/j.pt.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Baird JK, Hoffman SL. Primaquine therapy for malaria. Clin Infect Dis. 2004;39:1336–45. doi: 10.1086/424663. [DOI] [PubMed] [Google Scholar]
- 42.Mahmoudi N, Garcia-Domenech R, Galvez J, et al. New active drugs against liver stages of Plasmodium predicted by molecular topology. Antimicrob Agents Chemother. 2008;52:1215–20. doi: 10.1128/AAC.01043-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carraz M, Jossang A, Franetich JF, et al. A plant-derived morphinan as a novel lead compound active against malaria liver stages. PLoS Med. 2006;3:e513. doi: 10.1371/journal.pmed.0030513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neerja J, Puri SK. Plasmodium yoelii: activity of azithromycin in combination with pyrimethamine or sulfadoxine against blood and sporozoite induced infections in Swiss mice. Exp Parasitol. 2004;107:120–4. doi: 10.1016/j.exppara.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 45.Singh N, Puri SK. Causal prophylactic activity of antihistaminic agents against Plasmodium yoelii nigeriensis infection in Swiss mice. Acta Trop. 1998;69:255–60. doi: 10.1016/s0001-706x(97)00138-1. [DOI] [PubMed] [Google Scholar]
- 46.Lindenthal C, Weich N, Chia YS, Heussler V, Klinkert MQ. The proteasome inhibitor MLN-273 blocks exoerythrocytic and erythrocytic development of Plasmodium parasites. Parasitology. 2005;131:37–44. doi: 10.1017/s003118200500747x. [DOI] [PubMed] [Google Scholar]
- 47.Coppi A, Cabinian M, Mirelman D, Sinnis P. Antimalarial activity of allicin, a biologically active compound from garlic cloves. Antimicrob Agents Chemother. 2006;50:1731–7. doi: 10.1128/AAC.50.5.1731-1737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coppi A, Pinzon-Ortiz C, Hutter C, Sinnis P. The Plasmodium circumsporozoite protein is proteolytically processed during cell invasion. J Exp Med. 2005;201:27–33. doi: 10.1084/jem.20040989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.World Health Organization, author. Antiretroviral therapy for HIV infection in infants and children: towards universal access. [1 November 2007]. Available at: http://www.who.int/hiv/pub/guidelines/paediatric020907.pdf. [PubMed] [Google Scholar]