

Abstract
Background. Nonspecific T-cell hyperactivation is the main driving force for human immunodeficiency virus (HIV)–1 disease progression, but the reasons why the excess immune response is not properly shut off are poorly defined.
Methods. Eighty-five HIV-1–infected individuals were enrolled to characterize B and T lymphocyte attenuator (BTLA) expression and function. Infection and blockade assays were used to dissect the factors that influenced BTLA signaling in vitro.
Results. BTLA expression on overall CD4+ and CD8+ T cells was progressively decreased in HIV-1 infection, which was directly correlated with disease progression and CD4+ T-cell differentiation and activation. BTLA+CD4+ T cells from HIV-1–infected patients also displayed an altered immune status, which was indicated by reduced expression of naive markers but increased activation and exhaustion markers. Cross-linking of BTLA can substantially decrease CD4+ T-cell activation in vitro. This responsiveness of CD4+ T cells to BTLA-mediated inhibitory signaling was further found to be impaired in HIV-1–infected patients. Furthermore, HIV-1 NL4-3 down-regulated BTLA expression on CD4+ T cells dependent on plasmacytoid dendritic cell (pDC)-derived interferon (IFN)-α. Blockade of IFN-α or depletion of pDCs prevents HIV-1-induced BTLA down-regulation.
Conclusions. HIV-1 infection potentially impairs BTLA-mediated signaling dependent on pDC-derived IFN-α, which may contribute to broad T-cell hyperactivation induced by chronic HIV-1 infection.
A chronic generalized immune activation is now increasingly being recognized to be the main driving force for T-cell depletion, loss of antiviral immunity, and disease progression during chronic human immunodeficiency virus (HIV)-1–infection [1, 2]. Clinical evidence has indicated that T-cell activation levels are predictive of an adverse prognosis for the HIV-1–infected patients [3, 4]. Additional evidence for this pathogenesis is the lack of disease progression in simian immunodeficiency virus (SIV)-infected monkeys that quickly down-regulate the initial inflammatory response upon SIV infection [5]. Additional studies indicated that the immune activation is driven directly by T cells that, in a cognate fashion, recognize HIV-1 proteins [6], by activation of plasmacytoid dendritic cells (pDCs) through Toll-like receptors (TLRs) [7] and by bacterial products translocated from gut to blood [8]. Thus, excess immune activation has been regarded as deleterious to the host; the interventions to temper immune hyper-activation, therefore, may prevent disease progression to AIDS. However, it is still unknown why this excess immune activation cannot be limited or properly shut off during HIV-1 infection.
Recently, some cell-surface costimulatory molecules have been demonstrated to have a substantial impact on the T-cell activation [9]. These cosignaling pathways play overlapping and distinct regulatory roles for each lymphocyte subset during their various stages of immune response, such that immune responses occur in the correct intensity and manner [9]. In particular, some immune-inhibitory molecules, such as PD-1 [10–13], CTLA-4 [14], and Tim-3 [15], have been implicated mediators of T-cell exhaustion during chronic HIV-1 infection. Other studies have further shown that exhausted T cells in fact express many coinhibitory molecules [16], and these coinhibitory receptors regulate T-cell responses through multiple, complex patterns [17]. Although these pioneering studies clearly indicate that coinhibitory signaling mediates T-cell exhaustion and partially leads to HIV-1 persistence, it is necessary to define whether these inhibitory pathways experience functional deficits that may be responsible for the immune hyperactivation in chronic HIV-1 infection.
B and T lymphocyte attenuator (BTLA, CD272), the most recently recognized member of the CD28 family, is primarily expressed by the majority of lymphocytes [18]. Interaction with its ligand, herpes virus entry mediator (HVEM) induces tyrosine phosphorylation of immunoreceptor tyrosine-based inhibition motifs [19] and attenuates T-cell activation [20]. In vitro, BTLA ligation can send a constitutive “off” signal to T cells and maintain T-cell tolerance [21–23]. BTLA-deficient mice generally develop exacerbated disease, suggesting that BTLA predominantly regulates immune responses negatively [24, 25]. In human diseases, BTLA was found to be persistently expressed by melanoma tumor antigen-specific CD8 T cells, thus inhibiting their anti-tumor function [26]. These studies have defined the inhibitory roles of BTLA in regulating immune responses; however, little is known regarding the functional role of BTLA in chronic HIV-1 infection.
Our previously preliminary data indicated that BTLA was down-regulated and associated with the decrease in CD4+ T-cell counts and the increase of plasma HIV-1 loads [27]. The present study greatly extends our previous data and further confirms that BTLA expression on T cells is progressively down-regulated during chronic HIV-1 infection. The BTLA down-regulation was dependent on pDC-derived interferon (IFN)-α induced by HIV-1 isolates in vitro and that it impaired BTLA-mediated inhibitory signaling on CD4+ T-cell activation, thus contributing to broad T-cell activation in chronic HIV-1 infection. Enhancing BTLA function may therefore present an alternative therapeutic strategy for overcoming aggressive immune activation in chronic HIV-1 infection.
SUBJECTS, MATERIALS, AND METHODS
Subjects
Eighty-five HIV-1–infected individuals were enrolled in our study. They were divided into 3 groups according to their infection status (Table 1.) [12, 28]. Twenty-six HIV-1–uninfected subjects were used as healthy control subjects. All HIV-1–infected subjects were divided into 3 groups, including a cohort of 14 long-term nonprogressors (LTNPs; defined as those who had a persistent peripheral CD4+ T-cell count > 500 cells/μL and plasma HIV-1 RNA level < 500 copies/mL, who had received no antiretroviral therapy, and who had experienced no clinical signs of disease for ≥10 years), 44 typical progressors (defined as those who exhibited a typical progressive disease with peripheral CD4+ T-cell counts >200 cells/μL and plasma HIV-1 RNA level >1000 copies/mL, without receiving antiviral treatment, and no AIDS-defining condition), and 27 patients with AIDS (defined as those who had an AIDS-defining condition, according to the World Health Organization classification, including a progressive decrease in peripheral CD4+ T-cell counts to < 200 cells/μL and a plasma viral RNA level > 1000 copies/mL, without receipt of antiviral treatment, and present or previous opportunistic infections). The majority of these individuals (89.4%) were paid blood donors who had become infected through illegal blood collection during the period 1994–1995, whereas other patients had been infected with HIV-1 via sexual transmission. The study protocol was approved by the ethics committee of our unit, and written informed consent was obtained from each subject.
Table 1.
Characteristics of Subjects in the Study
| Characteristic | Healthy control subjects | Long-term nonprogressors | Typical progressors | Patients with AIDS |
| No. of subjects | 26 | 14 | 44 | 27 |
| Age, median years (range) | 32 (19–31) | 36 (26–43) | 45 (32–56) | 49 (25–61) |
| Sex, no. of men/no. of women | 17/9 | 8/6 | 34/10 | 17/10 |
| CD4+ T-cell count, median cells/μL (range) | NA | 569 (507–1020)a | 339 (203–482)a | 112 (55–189) |
| HIV-1 load, median copies/ mL (range) | NA | <500a | 38,000 (1800–1,200,000)a | 43,000 (5500–8,500,000) |
| Infection route | ||||
| Blood transfusion | NA | 14 | 40 | 23 |
| Sex | NA | 1 | 4 | 4 |
NOTE. NA, not applicable
P < .05, compared with patients with AIDS.
Antibodies and Reagents
All antibodies were purchased from BD Biosciences, except for biotinylated anti-BTLA (clone MIH26); its isotype control antibody IgG2a-Biotin, allophycocyanin (APC)-, and phycoerythrin (PE)-conjugated streptavidin (eBioscience); and APC-conjugated major histocompatibility complex class I pentamers (ProImmune), which are reported to be frequently targeted by HIV-1–specific T cells (p17 gag; SLYNTVATL, SL9) [29], cytomegalovirus (CMV; pp65 495–503; NLVPMVATV, NV9), or influenza virus epitopes (matrix 58-66; GILGFVFTL, GL9) [30]. Anti-HLA-A2 Ab (Catalog Number: 551285, clone BB7.2; BD Pharmingen) was used to determine genotypes of the enrolled subjects. The pentamer analysis was limited to 36 of HLA-A2–positive subjects, including 12 healthy subjects, 7 LTNPs, and 17 typical progressors/patients with AIDS. All HIV-1 isolates were prepared according to previous described protocols [31]. Anti-HIV drug nevirapine, HIV-1 gp120 monoclonal antibody (IgG1 B12), and monoclonal antibodies against human IFN-α and IFN-β were obtained through the National Institutes of Health AIDS Research and Reference Reagent Program. Chloroquine was obtained commercially (Sigma-Aldrich). Cytokines IFN-α, interleukin (IL)–1β, IL-6, IL-17, IL-7, and IL-15 were purchased from Peprotec, and anti-IFN-α/β receptor chain 2 Ab (anti-IFN-αR) was purchased from R&D Systems.
Cell Isolation and Phenotypic Analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from freshly heparinized blood, and CD4+ T cells were purified by positive selection using the MiniMACS system (Miltenyi Biotech). In pDC depletion experiments, PBMCs were labeled with fluorescein isothiocyanate (FITC)-conjugated anti-BDCA-2 and APC-conjugated anti-CD123 (Miltenyi Biotech) and were then sorted using a FACSAria cell sorter (Becton Dickinson). Isolated memory CD4+ T cell and pDC populations were >95% in purity, whereas pDC-depleted PBMCs just contained few pDCs (<0.001% BDCA-2+CD123+ pDCs).
PBMCs were labeled with anti-CD3-peridin chlorophyll protein (PerCP), anti-CD4 or anti-CD8-FITC, anti-CD8 or pentamer-APC, and anti-BTLA-PE for measuring BTLA expression on total T-cells subsets or on virus-specific CD8+ T cells. For CD4+ T subset staining, PBMCs or purified CD4+ T cells were incubated with a cocktail of antibodies: CD4-PerCP, BTLA-PE, CD45RO-APC, and CD27-FITC. To characterize the phenotypes of BTLA+ and BTLA–CD4+ T cells, PBMCs were stained with anti-CD3-FITC, anti-CD4-PerCP, anti-BTLA-APC, and PE-conjugated anti-CD127, anti-CD38, anti-PD-1, anti-CD95, anti-ki67, and corresponding isotype antibodies. The cells were then washed, fixed, and analyzed using a FACSCalibur flow cytometer (Becton Dickinson) and FlowJo software (TreeStar).
CD4+ T-Cell Activation and Functional Analysis
Freshly isolated PBMCs were stimulated with anti-CD3/CD28 antibodies (each 1 μg/mL) in medium, plus either plate-coated anti-BTLA (10 μg/mL; clone MIH26, eBioscience, San Diego, CA) or isotype antibodies for 24 hours. The cells were then collected, and CD69, CD38 and CD25 expressions were analyzed by FACS. PBMCs were also stimulated with anti-CD3/CD28 antibodies (each 1 μg/mL) or PMA/ionomycin (50 ng/mL and 1 μg/mL, respectively), plus either plate-coated anti-BTLA (10 μg/mL) or isotype antibodies for 6 h. GolgiStop (BD PharMingen) was added into cells after 2 h of stimulation. The cells were then labeled with surface antibodies and intracellular anti-IFN-γ-FITC and anti-IL-2-APC, as previously described [12, 30].
Cytokine Stimulation and HIV-1 Infection Assay
PBMCs from healthy subjects were stimulated with IFN-α (1000 IU/mL), with or without anti-IFN-α/β receptor chain 2 (10 μg/mL), IL-7, IL-15, IL-1β, IL-6, and IL-17 for 5 days. The cells were then washed and were detected BTLA expression was detected on T cells. For the HIV-1 infection assay, fresh PBMCs (1 × 106 cells/mL) were incubated in 96-well plates with 100 μL mock or diluted HIV-1 R3A, R3B, and NL4-3 (50 ng/mL p24 antigen) virus supernatants, along with the anti-HIV-1 reagents anti-IFN-α/β (5 ng/mL), B12 IgG (10 μg/mL), nevirapine (5 μmol/L), and chloroquine (50 μmol/L) in the presence of IL-2 (20 U/mL) for 3 days. Alternatively, PBMCs, PBMCs-pDCs, CD4+ T cells, and CD4+ T cells plus pDCs (10:1) were incubated with NL4-3 isolates for 5 days. The cells were then collected for evaluation of BTLA expression on CD4 T cells with use of FACS-Calibur. 7-AAD was used to exclude dead cells. At least 10,000 live cells were acquired per run.
Semi-quantitative Real-time Polymerase Chain Reaction
Total RNA was extracted from the isolated total or CD4+ T-cell subsets using an RNAeasy Mini Kit (Qiagen). Total RNA was extracted and then reversed-transcribed to cDNA using oligo (dT) with avian myeloblastosis virus reverse transcriptase (Invitrogen) at 42°C for 30 min and 95°C for 5 min. The quantitative expressions of BTLA transcripts were subsequently determined using fluorogenic dye SYBR Green and primers. Glyceraldehyde 3-phosphate dehydrogenase was used to normalize the samples in each PCR reaction. Both melting-curve and gel-migration analyses were used to ensure the absence of nonspecific and primer-dimer products. The results are expressed as relative mRNA quantification calculated using the 2−ΔCt method.
Statistical Analysis
All data were analyzed using SPSS software (SPSS Inc). The Kruskal–Wallis H nonparametric test was performed for multiple comparisons among ≥3 groups. Statistical differences between 2 groups were determined by the Mann–Whitney nonparametric U test. Data from the same individuals were compared by the Wilcoxon matched-pairs t test. Correlations between variables were evaluated using the Spearman rank correlation test. For all tests, a P value of <.05 was considered statistically significant.
RESULTS
BTLA Expression Is Progressively Downregulated by All T Cells in Patients With Chronic HIV-1 Infection
We first determined BTLA expression profiles at cellular levels in healthy donors, and we found that BTLA was mainly expressed by B cells, CD4+ T cells and CD8+ T cells, NKT cells, and monocytes rather than on NK cells (data not shown). We further monitored BTLA expression on both CD4+ and CD8+ T cells in 85 HIV-1–infected individuals and 27 healthy subjects (Figure 1A). We found that BTLA expression on T cells was significantly down-regulated in HIV-1–infected subjects, compared with healthy subjects. Importantly, LTNPs exhibited higher levels of BTLA expression on CD4+ and CD8+ T cells than did typical progressors and patients with AIDS, while the lowest levels of BTLA expression on T cells were observed in patients with AIDS (Figure 1B and 1C). We also found that BTLA mRNA expression in purified CD4+ T cells was also significantly lower in HIV-1–infected patients than in healthy subjects (Figure 1D and 1E). The data clearly indicate that BTLA expression on T-cell subsets is progressively down-regulated during chronic HIV-1 infection.
Figure 1.

B and T lymphocyte attenuator (BTLA) expression on human T cells during chronic human immunodeficiency virus (HIV)-1–infection. A, Representative dot plots of BTLA staining in T cells isolated from healthy subjects and HIV-1–infected individuals. Values in the upper-left and upper-right quadrants represent the percentages of CD4+ T cells and CD8+ T cells that express BTLA, respectively. B and C, Statistical analysis of BTLA percentage and mean fluorescence intensity in CD4+ T cells (B) and CD8+ T cells (C) in healthy control subjects (HC; n = 26), long-term nonprogressors (LTNP; n = 14), typical progressors (TP; n = 44), and patients with AIDS (n = 27). D, Representative gel analysis of semi-quantitative real-time polymerase chain reaction products of BTLA mRNA transcripts extracted from purified CD4+ T cells from 2 HIV-1 infected TPs and 2 HCs. P, patient; H, healthy subject. E, Statistical analysis of BTLA mRNA expression in CD4+ T cells from HIV-1-infected TPs/patients with AIDS (n = 11) and HCs (n = 5). F, Representative dot plots of BTLA-expressing HIV-, cytomegalovirus (CMV)-, or influenza virus–specific CD8+ T cells from HIV-1-infected patients and HCs. Values in the upper-right quadrant represent the percentage of pentamer-positive CD8+ T cells that express BTLA. G, The frequency and mean fluorescence of BTLA expressed on HIV-, CMV-, or influenza virus–specific CD8+ T cells in the different study groups. In panels B, C, E, and G, each dot represents 1 individual, and horizontal bars represent the median values. Multiple comparisons were made using the Kruskal-Wallis H nonparametric test among the different groups. The Mann-Whitney U test was used to compare data from 2 different groups. The significant P values are shown.
Next, we found that BTLA expression was similar in HIV-1–specific versus CMV-specific and influenza-specific pentamer-positive CD8+ T cells in typical progressors/patients with AIDS (Figure 1F). As compared with healthy subjects, BTLA expression on both CMV-specific and influenza virus-specific pentamer-positive cells was significantly reduced in typical progressors/patients with AIDS (Figure 1F and 1G). These data indicate that BTLA expression is also progressively down-regulated in HIV-specific CD8+ T cells during HIV-1 infection.
BTLA Downregulation on CD4 T Cells Is Associated With Disease Progression and Immune Activation in Chronic HIV-1 Infection
We further examined the relationships between BTLA expression on T cells and plasma viral load, peripheral CD4+ T-cell counts and CD38 expression on CD8 T cells, which has been widely used as an activation marker predicting disease progression in HIV-1 infection [1, 3]. We found that the BTLA+ cell percentage in total CD4+ T cells was positively correlated with peripheral CD4+ T cell count but inversely correlated with plasma viral load and CD38 expression on CD8+ T cells (Figure 2A). Simultaneously, we also found that BTLA expression on total CD4+ T cells was negatively correlated with PD-1 [10, 12] and Ki67 [32] expression levels, but it was positively correlated with CD127 [33] expression in typical progressors/patients with AIDS (Figure 2B). These data indicate that BTLA down-regulation in CD4+ T cells can potentially serve as a marker of disease progression and immune hyperactivation in HIV-1 infection.
Figure 2.
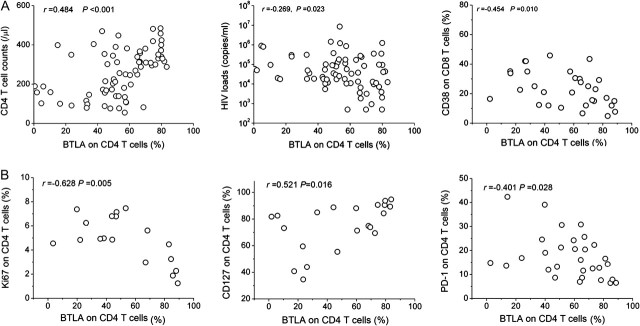
B and T lymphocyte attenuator (BTLA) down-regulation and human immunodeficiency virus (HIV)-1 disease progression. The correlation analysis is between BTLA expression on CD4+ T cells with CD4+ T-cell numbers (n = 71), HIV load (n = 71), and CD38 expression on CD8 T cells (n = 30; A) as well as the expression of Ki67 (n = 18), PD-1 expression (n = 30), and CD127 (n = 21; B) on CD4 T cells in HIV-1–infected patients. P values are shown. The Spearman rank correlation test was used to evaluate the correlations between variables.
BTLA Expression Is Downregulated in Various Differentiated CD4+ T-Cell Subsets in HIV-1 Infection
We then examined the distribution of BTLA expression in CD4+ naive T cell (Tn), central memory T cell (Tcm), and effector memory T cell (Tem) subsets on the basis of CD45RO and CD27 expression [34]. As in our previous report [12], chronic HIV-1 infection leads to a significant decrease in naive CD4+ T-cell subset levels but a continuous increase in memory CD4+ T-cell subset levels in HIV-1–infected typical progressors/patients with AIDS (Figure 3A, left). Importantly, we found that BTLA expression was more predominant in Tn cells in healthy subjects but gradually decreased in the Tn, Tcm, and Tem populations in HIV-1–infected individuals, regardless of disease status (Figure 3A, right). Pooled data further confirmed that BTLA expression on the 3 subsets was significantly decreased in HIV-1-infected subjects, compared with healthy subjects (Figure 3B). These data indicate that persistent HIV-1 infection leads to a significant loss of BTLA expression on both naive and memory CD4+ T-cell subsets.
Figure 3.
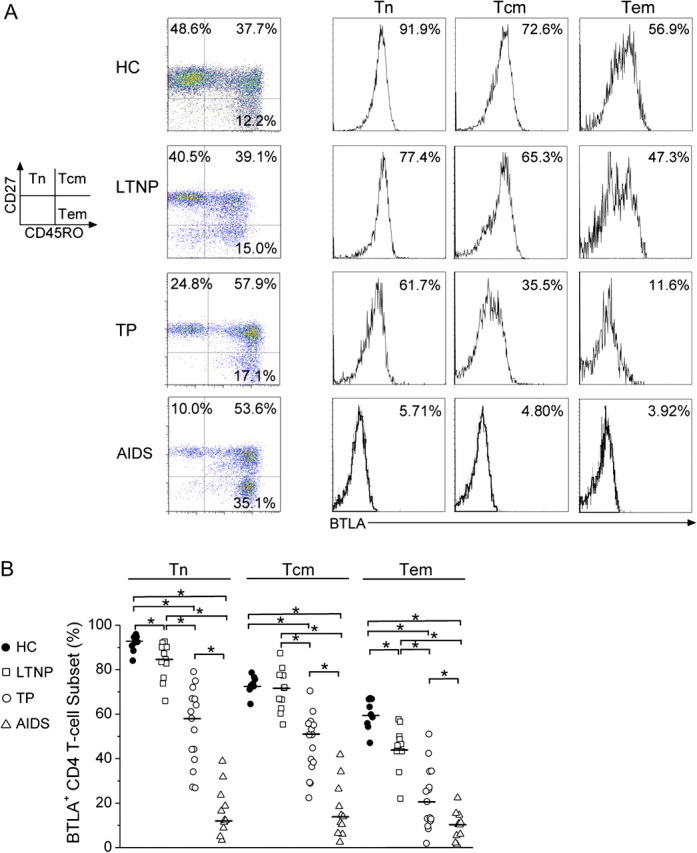
B and T lymphocyte attenuator (BTLA) down-regulation in CD4+ T-cell subsets in human immunodeficiency virus (HIV)-1–infected individuals. A, Representative BTLA expressions on CD4+ T-cell subsets from a healthy subject and an HIV-1-infected individual. The percentages of each CD4+ T-cell population are shown in each quadrant (left panel). The BTLA percentages in each CD4+ T-cell population are shown as histograms (right panel). B, Statistical analysis of the BTLA expression levels in CD4+ T-cell subsets in the different study groups. *P < .05. Multiple comparisons were made using the Kruskal-Wallis H nonparametric test among the different groups. The Mann-Whitney U test was used to compare data from 2 different groups. Tn, naive T cells; Tcm, central memory T cells; Tem, effector memory T cells.
BTLA+CD4+ T Cells Display an Altered Immune Status in HIV-1 Infection
We next determined whether HIV-1 infection influences BTLA+CD4+ T-cell status. In healthy subjects, BTLA+CD4+ T cells often displayed an immature profile, with 65% Tn, 30% Tcm, and 5% Tem subsets, whereas BTLA– CD4+ T cells displayed a relatively mature profile, with 25% Tn, 45% Tcm, and 30% Tem subsets (Figure 4A). This distribution was significantly altered in BTLA+ but not in BTLA– CD4+ T cells in HIV-1–infected subjects. Similar alterations were also occurred in CD38 (activation), CD95 (preapoptosis), PD-1 (exhaustion), Ki67 (proliferation), and perforin (cytolytic function) (Figure 4B). Compared with healthy subjects, patients with chronic HIV-1 infection had markedly increased CD38, CD95, PD-1, Ki67, and perforin expression and decreased CD127 expression in BTLA+ CD4+ T-cell subsets, but HIV-1 infection seldom influenced the expression of these markers in the BTLA– CD4+ T-cell population, except for CD38, which was also elevated in HIV-1–infected subjects. This comprehensive analysis suggests that HIV-1 infection significantly skews BTLA+ rather than BTLA– CD4+ T cells toward differentiation and maturation.
Figure 4.

B and T lymphocyte attenuator (BTLA)–positive CD4+ T cells and abnormal immune status in human immunodeficiency virus (HIV)–1 infection. A, The analysis of percentages of BTLA+ and BTLA– CD4+ T cells in naive T cell (Tn), central memory T cell (Tcm), and effector memory T-cell (Tem) subsets in healthy subjects and HIV-1–infected individuals. B, The phenotypic profiles of BTLA+ and BTLA– CD4+ T cells analyzed for CD38, CD95, CD127, perforin, PD-1, and Ki67 expression in healthy subjects (n = 16) and HIV-1-infected subjects (8 long-term nonprogressors 10 typical progressors, and 9 patients with AIDS). *P <.05, **P <.01. Multiple comparisons were made using the Kruskal-Wallis H nonparametric test among the different groups. The Mann-Whitney U test was used to compare data from 2 different groups.
HIV-1 Induces BTLA Downregulation on CD4+ T Cells Dependent on pDC-Derived IFN-α In Vitro
We investigated the impacts of IFN-α; inflammatory cytokines IL-1β, IL-6, and IL-17; and γ-chain cytokines IL-7 and IL-15 on BTLA expression in vitro. We found that IFN-α can significantly reduce BTLA expression on CD4+ T cells. Blockade of IFN-α pathway using anti-IFN-α receptor reversed the BTLA down-regulation. In contrast, IL-1β, IL-6, IL-17, IL-7, and IL-15 failed to decrease BTLA expression in vitro (Figure 5A). In addition, we found that various HIV-1 isolates can also directly down-regulate BTLA expression on CD4+ T cells in vitro, particularly NL4-3 isolate induced the most significant BTLA down-regulation compared with R3A and R3B (Figure 5B).
Figure 5.

B and T lymphocyte attenuator (BTLA) down-regulation on CD4+ T cells is dependent on pDC-derived interferon (IFN)-α induced by human immunodeficiency virus (HIV)–1 exposure. A, Representative histograms indicating the effects of various cytokines on BTLA expression on CD4 T cells from healthy subjects in vitro. Values represent the BTLA percentages and mean fluorescence intensity (MFI) on CD4+ T cells. The data are representative of 3 independent experiments. B, Representative histograms indicating the effects of various HIV-1 isolates on BTLA expression by CD4+ T cells in vitro. Values represent the BTLA percentages and MFI on CD4+ T cells. The data are representative of 4 independent experiments. C, Representative histograms indicating that the CD4 interaction (B12 antibody), endocytosis (chloroquine), and IFN-α (anti-IFN-α/β) but not reverse-transcriptase inhibitor (nevirapine) are required for NL4-3-induced BTLA down-regulation. Values represent the percentages of CD4+ T cells that express BTLA. D, Pooled data confirming that NL4-3 isolates down-regulated BTLA expression (percentages and MFI) dependening on IFN-α in vitro. Error bars indicate standard deviations. *P < .05. E, Representative histograms indicating that NL4-3 exposure induces BTLA down-regulation on CD4+ T cells in vitro depending on plasmacytoid dendritic cell -derived IFN-α. Values represent the percentages and MFI of CD4+ T cells that express BTLA.
More importantly, we found that anti-IFNα/β, B12 (the antagonist of viral gp120 binding to CD4+ T cells), and chloroquine (endocytosis inhibitor) but not nevirapine (which blocks viral replication) can efficiently prevent NL4-3-induced BTLA down-regulation (Figure 5C). Pooled data further confirmed these observations (Figure 5D). Interestingly, the depletion of pDCs from PBMCs partially blocked NL4-3-induced BTLA down-regulation on CD4+ T cells (Figure 5E, left). Notably, NL4-3 infection of purified CD4 T cells substantially reduces BTLA expression, whereas addition of the pDCs in vitro further decreased BTLA expression on CD4+ T cells. Blockade of the IFN-α-mediated pathway using both anti-IFN-α/β antibodies and anti-IFN-α receptor antibodies reversed the BTLA down-regulation on CD4+ T cells (Figure 5E, right). These lines of evidence indicate that HIV-1 can down-regulate BTLA expression on CD4+ T cells via induction of pDC-derived IFN-α, although some other alternative molecular pathways may be responsible for BTLA down-regulation in HIV-1 infection.
BTLA-Mediated Inhibition on CD4+ T-Cell Activation and Function Is Impaired in HIV-1 Infection
We further investigated the functional relevance of diminished BTLA expression on CD4+ T cells in HIV-1–infected subjects. Cross-linking of BTLA using an agonistic anti-BTLA monoclonal Ab (MIH26) substantially decreased CD69, CD38, and CD25 expression on CD4+ T cells in healthy subjects (Figure 6A). However, the inhibition of BTLA cross-linking on CD38, CD25, and CD69 expression was significantly reduced in typical progressors/patients with AIDS (Figure 6B). Similar results were also demonstrated in cytokine production by CD4 T cells. Anti-BTLA cross-linking significantly inhibits anti-CD3/CD28 and PMA/ionomycin-induced IL-2 and IFN-γ production by CD4+ T cells in healthy subjects (Figure 6C). This BTLA-mediated suppression of IL-2 and IFN-γ production was significantly reduced in typical progressors/patients with AIDS, compared with healthy subjects (Figure 6D). These data indicate that BTLA-mediated suppression of CD4+ T-cell activation and function is significantly impaired in chronic HIV-1 infected typical progressors/patients with AIDS.
Figure 6.
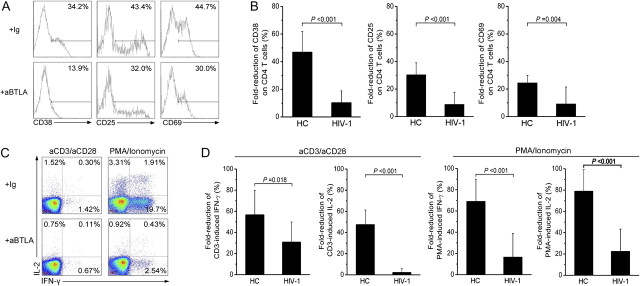
B and T lymphocyte attenuator (BTLA)–mediated inhibition on CD4+ T-cell activation during human immunodeficiency virus (HIV)–1 infection. A, Representative histograms depicting the expression of CD38, CD25, and CD69 in CD4+ T cells from a healthy subject. Values represent the percentage of CD4+ T cells that express CD38, CD25, and CD69. B, Pooled data showing the fold-inhibition of anti-BTLA cross-linking on anti-CD3-induced CD4+ T-cell activation in HIV-1–infected typical progressors/patients with AIDS (n = 26) and healthy subjects (n = 8). C, Representative dot plots show the effects of anti-BTLA cross-linking on interleukin (IL)-2 and interferon (IFN)–γ production of CD4+ T cells induced by anti-CD3 or PMA/ionomycin stimulation in a healthy subject. Values in the 3 quadrants represent the percentages of CD4+ T cells that express IFN-γ and IL-2. D, Pooled data showing the fold-inhibition of anti-BTLA cross-linking on IL-2 and IFN-γ production of CD4+ T cells induced by anti-CD3– and PMA/ionomycin–stimulation in HIV-infected typical progressors and patients with AIDS (n = 16) and healthy subjects (n = 8). In panels B and D, fold-inhibition was calculated as the differences between the baseline activation marker expression on isotype control antibody stimulation and anti-BTLA antibody stimulation were divided by the baseline activation marker expression on CD4+ T cells with isotype control antibody stimulation. The Mann-Whitney U test was used to compare data from 2 different groups. P values are shown.
DISCUSSION
Nonspecific T-cell hyperactivation is associated with HIV-1 disease progression, but its mechanisms are poorly defined. Here, we present several novel observations that may shed new light on the mechanism of immune activation in HIV-1 infection: (1) BTLA is progressively down-regulated in HIV-1–infected patients, and this loss of BTLA expression is associated with skewed CD4 T-cell activation and differentiation; (2) BTLA down-regulation is dependent on IFN-α from pDCs, which is induced by HIV-1 isolates; and (3) BTLA-mediated inhibitory signaling on T-cell activation and function is severely impaired in HIV-1–infected patients. These findings have unveiled the regulatory role of BTLA in HIV-1 infection.
We first observed that BTLA expression was progressively down-regulated by both CD4+ and CD8+ T cells in LTNPs, typical progressors, and patients with AIDS, strongly suggesting that BTLA expression may potentially serve as a surrogate marker for HIV-1 immune hyperactivation and disease progression. We further identified the BTLA expression patterns differing from the expression of other coinhibitory molecules, such as PD-1 and CTLA-4, during HIV-1 infection [10–13]. BTLA was highly constitutively expressed by T cells, but it was down-regulated by HIV-1 infection. By contrast, PD-1 and CTLA-4 were only slightly expressed by a small fraction of exhausted T cells, and are both up-regulated by HIV-1 infection [10–13]. In addition, BTLA was preferentially expressed by naive CD4+ T cells, and its expression significantly decreased with the T-cell differentiation naive T cells to memory T cells in HIV-1–infected patients; this pattern is similar to recent data for patients with melanoma [26] and in a mouse model [17]. Meanwhile, PD-1 and CTLA-4 are seldom expressed by resting CD4+ T cells, and levels are generally elevated in Tem subsets during HIV-1 infection [12, 14]. These findings open new avenues for studying the role of BTLA in HIV-1 infection.
BTLA also displayed its uniquely functional property differing from other coinhibitory molecules in HIV-1 infection. It is generally believed that PD-1 and CTLA-4 up-regulation during HIV-1 infection can lead to T-cell exhaustion [10–13]. We proposed that BTLA loss in CD4+ T cells may cause generalized immune activation during HIV-1 infection. The present study provides several lines of evidence in support of this notion: (1) BTLA expression on CD4 T cells was negatively associated with immune activation in HIV-1 infected patients; (2) enhancing BTLA-mediated inhibitory signaling significantly decreased TCR- and PMA-induced T-cell activation and cytokine production in vitro, but BTLA-mediated inhibitory function is largely impaired in HIV-1–infected patients, thus promoting generalized T cell immune activation; and (3) HIV isolate-induced IFN-α can decrease BTLA expression and increase T-cell activation. Indeed, other several studies have also demonstrated that BTLA ligation can send a constitutive “off” signal to T cells and maintain T-cell tolerance in vitro [21–23]. BTLA-deficient mice generally develop an exacerbated autoimmune disease symptom, suggesting that BTLA silence or down-regulation may increase the abnormal activation of immune cells, including T cells [24, 25]. Collectively, these findings strongly suggested that HIV-1 can down-regulate BTLA expression on T cells, which subsequently causes more nonspecific activation of T cells to allow for greater replication of HIV-1 in as a positive feedback loop. Therefore, HIV-1 may manipulate the BTLA inhibitory pathway for its own survival.
More important, we found that BTLA down-regulation depends on pDC-derived IFN-α induced by HIV-1 isolates; blockade of IFN-α-mediated pathway or depletion of pDCs partially blocked NL4-3–induced BTLA down-regulation in vitro. In addition, some anti-HIV reagents targeting HIV-1 entry (B12) and endocytosis of pDCs (chloroquine) can also efficiently rescue HIV-1–induced BTLA down-regulation. Specially, chloroquine can specially inhibit pDCs producing type I IFNs through blocking TLR endocytosis, and it can further decrease CD8 T-cell activation [35]. These findings, in combination with a recent study demonstrating that CpG can down-regulate BTLA expression on tumor-specific CD8 T cells in vivo [26], suggest that pDC-derived IFN-α is likely responsible for the BTLA down-regulation during HIV-1 infection. These data indicate that multiple virological factors (such as Vpr [36] and Nef [37]) and immunological factors (such as common γ-chain cytokines [38]) have the ability to regulate the expression of costimulatory molecules on T cells.
Taken together, this study emphasizes host-autologous immune regulatory mechanisms, and highlights BTLA down-regulation may link IFN-α production by pDCs to T-cell over-activation in HIV-1 infection [6, 39, 40]. Indeed, pDCs-derived IFN-α has been demonstrated closely with immune activation in HIV infection [39, 41]. These findings, therefore, shed a new light on the understanding of coinhibitory molecules in the view of protective response aiming to restrict damage by an out-of-control T-cell immune response in HIV-1 infection.
Funding
The National Natural Science Foundation of China (30801040), the National Key Basic Research Program of China (2006CB504205), and the National Grand Program on Key Infectious Disease (2008ZX10001-002, 2008ZX10001-006 and 2009ZX10005-017).
Acknowledgments
We express sincere thanks to all of the participants in this study. We also thank Prof Xue-Guang Zhang (Medical Biotechnology Institute, Soochow University, Suzhou, China) for his helpful assistance with anti-BTLA antibodies and Dr Nicholas Brown for his careful editing of our revision.
References
- 1.Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol. 2008;214:231–241. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- 2.Hazenberg MD, Hamann D, Schuitemaker H, Miedema F. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat Immunol. 2000;1:285–289. doi: 10.1038/79724. [DOI] [PubMed] [Google Scholar]
- 3.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179:859–870. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- 4.Deeks SG, Kitchen CM, Liu L, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104:942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 5.Silvestri G, Sodora DL, Koup RA, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18:441–452. doi: 10.1016/s1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 6.Simmons A, Aluvihare V, McMichael A. Nef triggers a transcriptional program in T cells imitating single-signal T cell activation and inducing HIV virulence mediators. Immunity. 2001;14:763–777. doi: 10.1016/s1074-7613(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 7.Beignon AS, McKenna K, Skoberne M, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 9.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 11.Petrovas C, Casazza JP, Brenchley JM, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang JY, Zhang Z, Wang X, et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood. 2007;109:4671–4678. doi: 10.1182/blood-2006-09-044826. [DOI] [PubMed] [Google Scholar]
- 13.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 14.Kaufmann DE, Kavanagh DG, Pereyra F, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8:1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 15.Jones RB, Ndhlovu LC, Barbour JD, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy KM, Nelson CA, Sedy JR. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol. 2006;6:671–681. doi: 10.1038/nri1917. [DOI] [PubMed] [Google Scholar]
- 19.Chemnitz JM, Lanfranco AR, Braunstein I, Riley JL. B and T lymphocyte attenuator-mediated signal transduction provides a potent inhibitory signal to primary human CD4 T cells that can be initiated by multiple phosphotyrosine motifs. J Immunol. 2006;176:6603–6614. doi: 10.4049/jimmunol.176.11.6603. [DOI] [PubMed] [Google Scholar]
- 20.Sedy JR, Gavrieli M, Potter KG, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 21.Otsuki N, Kamimura Y, Hashiguchi M, Azuma M. Expression and function of the B and T lymphocyte attenuator (BTLA/CD272) on human T cells. Biochem Biophys Res Commun. 2006;344:1121–1127. doi: 10.1016/j.bbrc.2006.03.242. [DOI] [PubMed] [Google Scholar]
- 22.Wang XF, Chen YJ, Wang Q, et al. Distinct expression and inhibitory function of B and T lymphocyte attenuator on human T cells. Tissue Antigens. 2007;69:145–153. doi: 10.1111/j.1399-0039.2006.00710.x. [DOI] [PubMed] [Google Scholar]
- 23.Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9:176–185. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe N, Gavrieli M, Sedy JR, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 25.Krieg C, Boyman O, Fu YX, Kaye J. B and T lymphocyte attenuator regulates CD8+ T cell-intrinsic homeostasis and memory cell generation. Nat Immunol. 2007;8:162–171. doi: 10.1038/ni1418. [DOI] [PubMed] [Google Scholar]
- 26.Derre L, Rivals JP, Jandus C, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010;120:157–167. doi: 10.1172/JCI40070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu XS, Zhang Z, Gu LL, Wang FS. BTLA characterization and its association with disease progression in patients with chronic HIV-1 infection. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2009;25:1158–1160. [PubMed] [Google Scholar]
- 28.Wang X, Zhang Z, Zhang S, et al. B7-H1 up-regulation impairs myeloid DC and correlates with disease progression in chronic HIV-1 infection. Eur J Immunol. 2008;38:3226–3236. doi: 10.1002/eji.200838285. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Fu J, Zhao Q, et al. Differential restoration of myeloid and plasmacytoid dendritic cells in HIV-1-infected children after treatment with highly active antiretroviral therapy. J Immunol. 2006;176:5644–5651. doi: 10.4049/jimmunol.176.9.5644. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Zhang JY, Wherry EJ, et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B. Gastroenterology. 2008;134:1938–1949. doi: 10.1053/j.gastro.2008.03.037. 1949 e1931–1933. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Kovalev GI, Su L. HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood. 2007;109:2978–2981. doi: 10.1182/blood-2006-07-033159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sieg SF, Bazdar DA, Lederman MM. Impaired TCR-mediated induction of Ki67 by naive CD4+ T cells is only occasionally corrected by exogenous IL-2 in HIV-1 infection. J Immunol. 2003;171:5208–5214. doi: 10.4049/jimmunol.171.10.5208. [DOI] [PubMed] [Google Scholar]
- 33.Koesters SA, Alimonti JB, Wachihi C, et al. IL-7Rα expression on CD4+ T lymphocytes decreases with HIV disease progression and inversely correlates with immune activation. Eur J Immunol. 2006;36:336–344. doi: 10.1002/eji.200535111. [DOI] [PubMed] [Google Scholar]
- 34.Ochsenbein AF, Riddell SR, Brown M, et al. CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J Exp Med. 2004;200:1407–1417. doi: 10.1084/jem.20040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinson JA, Montoya CJ, Usuga X, Ronquillo R, Landay AL, Desai SN. Chloroquine modulates HIV-1-induced plasmacytoid dendritic cell alpha interferon: implication for T-cell activation. Antimicrob Agents Chemother. 2010;54:871–881. doi: 10.1128/AAC.01246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkatachari NJ, Buchanan WG, Ayyavoo V. Human immunodeficiency virus (HIV-1) infection selectively downregulates PD-1 expression in infected cells and protects the cells from early apoptosis in vitro and in vivo. Virology. 2008;376:140–153. doi: 10.1016/j.virol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Muthumani K, Choo AY, Shedlock DJ, et al. Human immunodeficiency virus type 1 Nef induces programmed death 1 expression through a p38 mitogen-activated protein kinase-dependent mechanism. J Virol. 2008;82:11536–11544. doi: 10.1128/JVI.00485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinter AL, Godbout EJ, McNally JP, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008;181:6738–6746. doi: 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 39.Mandl JN, Barry AP, Vanderford TH, et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008;14:1077–1087. doi: 10.1038/nm.1871. [DOI] [PubMed] [Google Scholar]
- 40.Baenziger S, Heikenwalder M, Johansen P, et al. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood. 2009;113:377–388. doi: 10.1182/blood-2008-04-151712. [DOI] [PubMed] [Google Scholar]
- 41.Meier A, Chang JJ, Chan ES, et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med. 2009;15:955–959. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]